1.本发明涉及电磁屏蔽材料技术领域,尤其涉及一种石墨烯/碳纳米管气凝胶电磁屏蔽复合织物及其制备方法和应用。
背景技术:
2.随着社会和科学技术的不断发展和进步,电子设备不断进入我们的生产生活之中,这些设备在带给人们生活便利的同时也产生了一些消极的影响,例如,一些精密电子元器件会受到其他电磁波的干扰而无法正常工作,一些重要信息可能会发生电磁泄密,而过多的电磁波同样会对人体造成危害。为应对过量电磁辐射带来的问题,并且符合下一代便携式、可穿戴式电子设备的使用要求,关于柔性高效电磁屏蔽材料的研究势在必行。
3.电磁屏蔽材料是指通过反射或者吸收电磁波从而防止电磁波透射材料的一类材料,目前常用的金属纤维制成的电磁屏蔽材料虽然具有较好的电磁屏蔽效果,但是其质量大、加工困难、化学稳定性差等缺点限制了其进一步使用。为克服金属材料的缺点,石墨烯、碳纳米管、炭黑等具有优良导电性的碳材料也被应用于电磁屏蔽领域,相比于传统金属材料,碳材料不仅质量轻而且化学稳定性更好,但是纳米碳材料在解决上述问题的同时也存在一定缺陷,首先这些材料的小尺寸使得其具有极大的比表面积,而大比表面积带来的表面能使得材料极易团聚从而限制材料的使用效率并且材料的屏蔽性能依然较低,现有技术如“石墨烯/石墨单层涂层织物的电磁性能和力学性能的研究”中将石墨烯和石墨制成电磁屏蔽涂料并涂覆于织物表面,制得的涂层织物的电磁屏蔽效率最高仅达到22db,同时为使碳材料在复合材料中构建导电网络结构,往往需要较大的添加量,这也增加了材料的制备成本,而且目前的电磁屏蔽复合材料往往不具备柔性,难于应用于未来智能电子设备和可穿戴电子设备等领域。
4.因此,如何进一步提高材料的电磁屏蔽性能并使其具有柔性成为现有技术的难题。
技术实现要素:
5.本发明的目的在于提供一种石墨烯/碳纳米管气凝胶电磁屏蔽复合织物及其制备方法和应用。本发明提供的制备方法制备的石墨烯/碳纳米管气凝胶电磁屏蔽复合织物具有优异的电磁屏蔽效果且具有柔性。
6.为了实现上述发明目的,本发明提供以下技术方案:
7.本发明提供了一种石墨烯/碳纳米管气凝胶电磁屏蔽复合织物的制备方法,包括如下步骤:
8.(1)将氧化石墨烯、氧化碳纳米管、分散剂和水混合,得到氧化石墨烯/氧化碳纳米管分散液;
9.(2)将所述步骤(1)得到的氧化石墨烯/氧化碳纳米管分散液与还原剂混合,进行
还原反应,得到rgo/cnts分散液;
10.(3)将所述步骤(2)得到的rgo/cnts分散液进行交联反应后干燥,得到rgo/cnts气凝胶;
11.(4)将所述步骤(3)得到的rgo/cnts气凝胶贴合在织物上,得到石墨烯/碳纳米管气凝胶电磁屏蔽复合织物。
12.优选地,所述步骤(1)中氧化石墨烯和氧化碳纳米管的质量比为(1.5~9):1。
13.优选地,所述步骤(1)中氧化石墨烯和氧化碳纳米管的总质量与水的体积比为(2~10)mg:1ml。
14.优选地,所述步骤(1)中的分散剂与氧化石墨烯的质量比为(0.2~0.8):1。
15.优选地,所述步骤(2)中的还原剂包括抗坏血酸、水合肼或柠檬酸。
16.优选地,所述步骤(2)中还原剂与所述步骤(1)中氧化石墨烯的质量比为(3~8):1。
17.优选地,所述步骤(3)中交联反应的温度为50~70℃。
18.优选地,所述步骤(3)中交联反应的时间为5~7h。
19.本发明提供了上述技术方案所述制备方法制备的石墨烯/碳纳米管气凝胶电磁屏蔽复合织物,包括织物和贴合于所述织物表面的石墨烯/碳纳米管气凝胶。
20.本发明还提供了上述技术方案所述石墨烯/碳纳米管气凝胶电磁屏蔽复合织物在电磁屏蔽领域的应用。
21.本发明提供了一种石墨烯/碳纳米管气凝胶电磁屏蔽复合织物的制备方法,包括将氧化石墨烯、氧化碳纳米管、分散剂和水混合,得到氧化石墨烯/氧化碳纳米管分散液;将得到的氧化石墨烯/氧化碳纳米管分散液与还原剂混合,进行还原反应,得到rgo/cnts分散液;将得到的rgo/cnts分散液进行交联反应后干燥,得到rgo/cnts气凝胶;将得到的rgo/cnts气凝胶贴合在织物上,得到石墨烯/碳纳米管气凝胶电磁屏蔽复合织物。本发明通过使用还原氧化石墨烯与碳纳米管共同构建气凝胶并将其与织物复合,利用具有三维多孔网络结构的气凝胶作为电磁屏蔽功能体,直接将其贴合于织物上,完整的保留了气凝胶原有的空间结构,提高了织物的电磁屏蔽性能。实施例的结果表明,本发明制备的石墨烯/碳纳米管气凝胶电磁屏蔽复合织物的电磁屏蔽效能达到34.5db。
附图说明
22.图1为本发明实施例1、实施例6和对比例1制备的气凝胶的扫描电镜图;
23.图2为本发明实施例1中原料石墨、制备的氧化石墨烯、气凝胶和热处理气凝胶的红外光谱图;
24.图3为本发明实施例1中原料石墨、制备的氧化石墨烯、气凝胶的xrd图;
25.图4为本发明实施例1制备的气凝胶的热重曲线;
26.图5为本发明实施例1制备的复合织物的扫描电镜图;
27.图6为本发明实施例1、实施例2和实施例4中的棉织物及制备的复合织物的热重曲线;
28.图7为本发明实施例1~5制备的复合织物的表面电阻率图;
29.图8为本发明实施例1~5制备的复合织物的电磁屏蔽效能图;
30.图9为本发明实施例1~5制备的复合织物的电磁屏蔽吸收效能和电磁屏蔽反射效能图;
31.图10为本发明实施例1、实施例7、对比例2~3制备的复合织物的拉伸强力、顶破强力和撕裂强力图;
32.图11为本发明实施例1、实施例7、对比例2~3制备的复合织物的柔性图;
33.图12为本发明实施例1、实施例7、对比例2~3制备的复合织物的透气率图;
34.图13为本发明实施例1、实施例7、对比例2~3制备的复合织物的质量保持率图;
35.图14为本发明实施例7、对比例2制备的复合织物超声振荡后的表面形态图。
具体实施方式
36.本发明提供了一种石墨烯/碳纳米管气凝胶电磁屏蔽复合织物的制备方法,包括如下步骤:
37.(1)将氧化石墨烯、氧化碳纳米管、分散剂和水混合,得到氧化石墨烯/氧化碳纳米管分散液;
38.(2)将所述步骤(1)得到的氧化石墨烯/氧化碳纳米管分散液与还原剂混合,进行还原反应,得到rgo/cnts分散液;
39.(3)将所述步骤(2)得到的rgo/cnts分散液进行交联反应后干燥,得到rgo/cnts气凝胶;
40.(4)将所述步骤(3)得到的rgo/cnts气凝胶贴合在织物上,得到石墨烯/碳纳米管气凝胶电磁屏蔽复合织物。
41.如无特殊说明,本发明对所述各组分的来源没有特殊的限定,采用本领域技术人员熟知的市售产品或常规制备方法制备的产品即可。
42.本发明将氧化石墨烯、氧化碳纳米管、分散剂和水混合,得到氧化石墨烯/氧化碳纳米管分散液。
43.在本发明中,所述氧化石墨烯和氧化碳纳米管的质量比优选为(1.5~9):1,进一步优选为(2~8):1,更优选为(3~7):1,最优选为(4~6):1。本发明将氧化石墨烯和氧化碳纳米管的质量比限定在上述范围内,能够使氧化碳纳米管较为均匀的吸附在氧化石墨烯表面,避免氧化碳纳米管过多对氧化石墨烯表面进行完整的包覆,更有利于形成结构规整、致密的三维多孔气凝胶,进一步提高产物的电磁屏蔽性能。
44.本发明对所述氧化石墨烯的来源没有特殊的限定,采用本领域技术人员熟知的制备方法制备的产品即可。在本发明中,所述氧化石墨烯的制备方法优选为采用改进hummers法制备:在0~5℃的条件下,向100ml浓硫酸(质量浓度98%)中分别加入2g石墨(500目)和2g硝酸钠,之后分6次加入高锰酸钾共12g(每次2g、每次间隔5min),添加完毕后继续磁力搅拌1h,然后利用磁力加热套将体系温度升温至35℃并继续搅拌2h,逐滴加入去离子水100ml,随后将体系置于提前加热好的恒温水浴锅内在92℃下反应15min,加入去离子水50ml,冷却至室温后加入过氧化氢溶液(质量浓度为30%)10ml,静置24h后利用高速离心机(gt10
‑
1型)用稀盐酸(质量浓度为5%)洗涤3次,得到氧化石墨烯泥浆,再利用真空烘箱(dzf
‑
6020)70℃干燥得到氧化石墨烯。
45.在本发明中,所述石墨经过氧化处理后,石墨片层间距增加产生石墨烯,同时在强
氧化剂的作用下,使得石墨烯的边缘及缺陷位置等活性较高的位置反应产生了羟基和羧基等含氧官能团,从而大大提高了氧化石墨烯在水中的分散性,进而有利于制备水凝胶。
46.本发明对所述氧化碳纳米管的来源没有特殊的限定,采用本领域技术人员熟知的制备方法制备的产品即可。在本发明中,所述氧化碳纳米管的制备方法优选为:将浓硫酸(质量浓度98%)和浓硝酸(质量浓度70%)按照体积比为3:1的比例配置100ml混酸溶液,将1g碳纳米管加入混酸溶液后利用磁力搅拌器和加热套以800rpm在90℃的条件下搅拌90min,使用高速离心机用去离子水洗涤3次后过滤,将洗涤后的碳纳米管利用真空烘箱60℃干燥12h,得到氧化碳纳米管。
47.在本发明中,所述碳纳米管优选为多壁碳纳米管;所述多壁碳纳米管的外径优选为10~20nm,所述多壁碳纳米管的长度优选为10~30μm。
48.在本发明中,所述氧化处理能够提高碳纳米管中含氧官能团的含量,有利于其在水中分散和通过静电引力与分子间作用力吸附于氧化石墨烯表面。
49.在本发明中,所述分散剂优选包括聚乙烯吡咯烷酮、聚乙烯醇或聚乙烯胺,更优选为聚乙烯吡咯烷酮。在本发明中,所述分散剂与氧化石墨烯的质量比优选为(0.2~0.8):1,进一步优选为(0.3~0.7):1,更优选为(0.4~0.6):1,最优选为0.5:1。本发明将分散剂与氧化石墨烯的质量比限定在上述范围内,能够使氧化石墨烯和氧化碳纳米管充分分散,避免团聚,进一步提高产物的性能。
50.在本发明中,所述水优选为去离子水。在本发明中,所述氧化石墨烯和氧化碳纳米管的总质量与水的体积比优选为(2~10)mg:1ml,进一步优选为(3~9)mg:1ml,更优选为(4~8)mg:1ml,最优选为(5~7)mg:1ml。本发明将氧化石墨烯和氧化碳纳米管的总质量与水的体积比限定在上述范围内,能够使氧化石墨烯和氧化碳纳米管更加均匀的分散。
51.本发明对所述氧化石墨烯、氧化碳纳米管、分散剂和水的混合的操作没有特殊的限定,采用本领域技术人员熟知的物料混合的技术方案即可。在本发明中,所述氧化石墨烯、氧化碳纳米管、分散剂和水的混合优选为将氧化石墨烯和氧化碳纳米管与水混合,再加入分散剂。
52.在本发明中,所述氧化石墨烯、氧化碳纳米管、分散剂和水的混合优选首先在搅拌条件下混合然后再进行超声混合;所述搅拌优选为磁力搅拌;所述搅拌的转速优选为700~900rpm,更优选为800rpm;所述搅拌的时间优选为3~5h,更优选为4h;所述搅拌的温度优选为20~30℃,更优选为25℃;所述超声的功率优选为360~480w,更优选为360w;所述超声的时间优选为1~3h,更优选为2h。采用本发明的混合方式,能够使各组分混合的更加均匀和分散。
53.得到氧化石墨烯/氧化碳纳米管分散液后,本发明将所述氧化石墨烯/氧化碳纳米管分散液与还原剂混合,进行还原反应,得到rgo/cnts分散液。
54.在本发明中,所述还原剂优选包括抗坏血酸、水合肼或柠檬酸,更优选为抗坏血酸。在本发明中,所述还原剂与氧化石墨烯的质量比优选为(3~8):1,更优选为(4~7):1,最优选为(5~6):1。在本发明中,所述还原剂用于还原氧化石墨烯和氧化碳纳米管中的含氧官能团。本发明将还原剂与氧化石墨烯的质量比限定在上述范围内,能够使氧化石墨烯与氧化碳纳米管充分被还原以提高导电性,进一步提高织物的电磁屏蔽性能。
55.本发明对所述氧化石墨烯/氧化碳纳米管分散液与还原剂的混合的操作没有特殊
的限定,采用本领域技术人员熟知的物料混合的技术方案即可。
56.在本发明中,所述还原反应优选在搅拌条件下进行;所述搅拌优选为磁力搅拌;所述搅拌的转速优选为700~900rpm,更优选为800rpm;所述搅拌的时间优选为3~5h,更优选为4h;所述搅拌的温度优选为20~30℃,更优选为25℃。
57.得到rgo/cnts分散液后,本发明将所述rgo/cnts分散液进行交联反应后干燥,得到rgo/cnts气凝胶。
58.在本发明中,所述交联反应的温度优选为50~70℃,更优选为60℃;所述交联反应的时间优选为5~7h,更优选为6h。在本发明中,所述交联反应过程中还原氧化石墨烯与还原氧化石墨烯、还原氧化石墨烯与碳纳米管之间发生自组装交联反应,从而得到三维网状结构的水凝胶。本发明将交联反应的温度和时间限定在上述范围内,能够使交联反应充分进行,又能防止水凝胶在成形过程中的体积收缩,提高水凝胶的规整性和致密度,进一步提高其性能。
59.在本发明中,所述交联反应优选在玻璃培养皿中进行。本发明采用玻璃培养皿进行反应能够使形成的水凝胶的形状尽量扁平,有利于其形成的气凝胶更好的贴合在织物上。
60.在本发明中,所述交联反应优选在密封条件下进行;所述密封优选采用保鲜膜密封。在本发明中,所述密封能够避免还原反应过程中水分的蒸发。
61.交联反应完成后,本发明优选将所述交联反应得到的水凝胶浸渍于乙醇和水的混合溶液(乙醇与水的体积比为1:9)中透析24h后进行冷冻再进行干燥,得到rgo/cnts气凝胶。
62.在本发明中,所述透析能够使水凝胶内部被水醇溶液浸润,从而避免冷冻过程中冰晶尺寸过大而造成水凝胶结构被破坏。
63.在本发明中,所述冷冻的温度优选为
‑
10~
‑
30℃,更优选为
‑
20℃;所述冷冻的时间优选为5~7h,更优选为6h。在本发明中,所述冷冻能够使水凝胶内部的水醇溶液冷冻结晶,避免后续冷冻干燥过程中内部结构的破坏。
64.在本发明中,所述干燥优选为冷冻干燥;所述干燥的温度优选为
‑
10~
‑
30℃,更优选为
‑
20℃;所述干燥的时间优选为20~28h,更优选为24h。本发明将冷冻干燥的温度和时间限定在上述范围内,能够使水凝胶完全冻干,得到结构完整的气凝胶。
65.得到rgo/cnts气凝胶后,本发明将所述rgo/cnts气凝胶贴合在织物上,得到石墨烯/碳纳米管气凝胶电磁屏蔽复合织物。
66.在本发明中,所述织物优选为棉织物。
67.在本发明中,所述织物在贴合前优选进行预处理。在本发明中,所述预处理优选为:将织物裁剪后浸泡在柔软整理液中,60~80℃加热2~4h后,蒸馏水洗涤,50~70℃烘干,得到预处理后的织物。
68.本发明对裁剪后织物的尺寸没有特殊的限定,根据实际需要选择即可。
69.在本发明中,所述柔软整理液的配置方法优选为:将32g naoh和脂肪醇聚氧乙烯醚(jfc低泡渗透剂)10ml,加入800ml蒸馏水中,搅拌即可。
70.在本发明中,所述预处理能够增强织物的柔韧性,去除织物表面的蜡质,便于后期气凝胶的贴合。
71.在本发明中,所述贴合优选采用水性聚氨酯进行贴合。在本发明中,所述贴合优选为将水性聚氨酯均匀涂覆于预处理后的织物表面,之后将气凝胶与涂覆有水性聚氨酯的一面贴合,用压辊来回碾压气凝胶表面,然后在气凝胶表面再次涂覆水性聚氨酯,最后固化,得到石墨烯/碳纳米管气凝胶电磁屏蔽复合织物。
72.在本发明中,所述第一次涂覆的水性聚氨酯的体积与织物的面积之比优选为20~25ml:254.34cm2。本发明将第一次涂覆的水性聚氨酯的体积与织物的面积之比限定在上述范围内,能够使气凝胶具有较高的结合牢度并使得织物具有优异的力学性能。
73.在本发明中,所述将气凝胶与涂覆有水性聚氨酯的一面贴合时优选快速进行。在本发明中,所述快速进行能够防止水性聚氨酯固化造成粘度下降而降低气凝胶与织物间的结合牢度。
74.在本发明中,所述碾压能够进一步提高气凝胶与织物间的结合牢度。
75.在本发明中,所述再次涂覆水性聚氨酯能够使水性聚氨酯在气凝胶表面固化成膜,起到保护气凝胶的作用。
76.在本发明中,所述再次涂覆的水性聚氨酯的体积与织物的面积之比优选为10ml:254.34cm2。
77.在本发明中,所述固化优选先进行真空固化再进行常压固化。在本发明中,所述真空固化的真空度优选为2
×
104pa;所述真空固化的时间优选为12h;所述真空固化的温度优选为20~30℃;所述常压固化的时间优选为48h;所述常压固化的温度优选为20~30℃。采用本发明的固化方式能够使固化充分完全。
78.本发明通过使用还原氧化石墨烯与碳纳米管共同构建气凝胶并将其与织物复合,控制各组分用量、反应温度、时间等工艺参数,利用具有三维多孔网络结构的气凝胶作为电磁屏蔽功能体,直接将其贴合于织物上,完整的保留了气凝胶原有的空间结构,提高了织物的电磁屏蔽性能。
79.本发明提供了上述技术方案所述制备方法制备的石墨烯/碳纳米管气凝胶电磁屏蔽复合织物,包括织物和贴合于所述织物表面的石墨烯/碳纳米管气凝胶。
80.本发明制备的石墨烯/碳纳米管气凝胶电磁屏蔽复合织物具有优异的电磁屏蔽效果、力学性能且具有柔性。
81.本发明还提供了上述技术方案所述石墨烯/碳纳米管气凝胶电磁屏蔽复合织物在电磁屏蔽领域的应用。
82.本发明对所述石墨烯/碳纳米管气凝胶电磁屏蔽复合织物在电磁屏蔽领域的应用没有特殊的限定,采用本领域技术人员熟知的石墨烯/碳纳米管气凝胶电磁屏蔽复合织物在电磁屏蔽领域的应用的技术方案即可。
83.下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
84.实施例1
85.(1)采用改进hummers法制备氧化石墨烯,在0~5℃的条件下,向100ml浓硫酸(质量浓度98%)中分别加入2g石墨(500目)和2g硝酸钠,之后分6次加入高锰酸钾共12g(每次
2g、每次间隔5min),添加完毕后继续磁力搅拌1h,然后利用磁力加热套将体系温度升温至35℃并继续搅拌2h,逐滴加入去离子水100ml,随后将体系置于提前加热好的恒温水浴锅内在92℃下反应15min,加入去离子水50ml,冷却至室温后加入过氧化氢溶液(质量浓度为30%)10ml,静置24h后利用高速离心机(gt10
‑
1型)用稀盐酸(质量浓度为5%)洗涤3次,得到氧化石墨烯泥浆,再利用真空烘箱(dzf
‑
6020)70℃干燥得到氧化石墨烯;
86.(2)制备氧化碳纳米管:将浓硫酸(质量浓度98%)和浓硝酸(质量浓度70%)按照体积比为3:1的比例配置100ml混酸溶液,将1g碳纳米管加入混酸溶液后利用磁力搅拌器和加热套以800rpm在90℃的条件下搅拌90min,使用高速离心机用去离子水洗涤3次后过滤,将洗涤后的碳纳米管利用真空烘箱60℃干燥12h,得到氧化碳纳米管;
87.(3)将210mg氧化石墨烯和90mg氧化碳纳米管加入到60ml去离子水中(氧化石墨烯和氧化碳纳米管的质量比为7:3,氧化石墨烯和氧化碳纳米管的总质量与去离子水的体积比为5mg:1ml),加入0.1g聚乙烯吡咯烷酮(聚乙烯吡咯烷酮与氧化石墨烯的质量比为0.48:1),使用磁力搅拌器在25℃以800rpm搅拌4h后使用超声清洗机360w超声2h,得到氧化石墨烯/氧化碳纳米管分散液;
88.(4)向氧化石墨烯/氧化碳纳米管分散液中加入1g抗坏血酸(抗坏血酸与氧化石墨烯的质量比为4.76:1),在25℃以800rpm搅拌4h,得到rgo/cnts分散液;
89.(5)将rgo/cnts分散液倒入直径18cm的玻璃培养皿中并用保鲜膜密封,放入烘箱中60℃加热6h后取出,将成形好的水凝胶浸于乙醇和水的混合溶液(乙醇和水的体积比为1:9)中透析24h,置于冰箱的冷冻室中
‑
20℃冷冻6h,然后放入提前预冷好的冷冻干燥机中
‑
20℃冷冻干燥24h,得到rgo/cnts气凝胶;
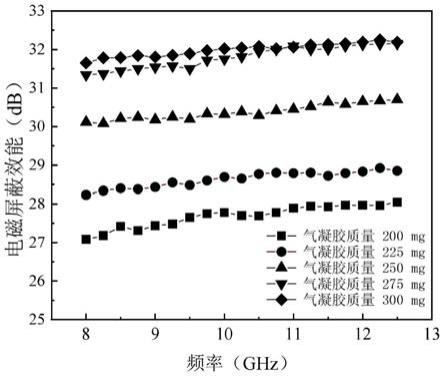
90.(6)将棉织物(经密:150根/10cm,纬密:100根/10cm,质量:295g/m2)裁剪成20cm
×
20cm的长方形布块备用,称取32gnaoh和10ml脂肪醇聚氧乙烯醚(jfc低泡渗透剂)加入800ml去离子水中搅拌均匀,得到柔软整理液;将裁剪后的棉织物浸泡在柔软整理液中70℃加热3h,然后用蒸馏水洗涤3次后60℃烘干,得到预处理后的织物;
91.(7)将预处理后的织物裁剪成直径18cm的圆形,将25ml水性聚氨酯均匀涂覆在织物表面,之后迅速将气凝胶与涂覆水性聚氨酯的一面贴合,并用压辊来回碾压气凝胶表面,在气凝胶表面再次涂覆10ml水性聚氨酯,然后在2
×
104pa下25℃固化12h,然后在常压25℃固化48h,得到石墨烯/碳纳米管气凝胶电磁屏蔽复合织物。
92.实施例2
93.将实施例1步骤(3)中氧化石墨烯的质量替换为140mg,氧化碳纳米管的质量替换为60mg,去离子水的体积替换为40ml,此时聚乙烯吡咯烷酮与氧化石墨烯的质量比为0.71:1,抗坏血酸与氧化石墨烯的质量比为7.14:1,其他参数与实施例1相同。
94.实施例3
95.将实施例1步骤(3)中氧化石墨烯的质量替换为157.5mg,氧化碳纳米管的质量替换为67.5mg,去离子水的体积替换为45ml,此时聚乙烯吡咯烷酮与氧化石墨烯的质量比为0.63:1,抗坏血酸与氧化石墨烯的质量比为6.35:1,其他参数与实施例1相同。
96.实施例4
97.将实施例1步骤(3)中氧化石墨烯的质量替换为175mg,氧化碳纳米管的质量替换为75mg,去离子水的体积替换为50ml,此时聚乙烯吡咯烷酮与氧化石墨烯的质量比为0.57:
1,抗坏血酸与氧化石墨烯的质量比为5.71:1,其他参数与实施例1相同。
98.实施例5
99.将实施例1步骤(3)中氧化石墨烯的质量替换为192.5mg,氧化碳纳米管的质量替换为82.5mg,去离子水的体积替换为55ml,此时聚乙烯吡咯烷酮与氧化石墨烯的质量比为0.52:1,抗坏血酸与氧化石墨烯的质量比为5.19:1,其他参数与实施例1相同。
100.实施例6
101.将实施例1步骤(3)中氧化石墨烯的质量替换为270mg,氧化碳纳米管的质量替换为30mg,此时,氧化石墨烯和氧化碳纳米管的质量比为9:1,聚乙烯吡咯烷酮与氧化石墨烯的质量比为0.37:1,抗坏血酸与氧化石墨烯的质量比为3.7:1,其他参数与实施例1相同。
102.实施例7
103.将实施例1步骤(7)中第一次涂覆的水性聚氨酯的体积替换为20ml,其他参数与实施例1相同。
104.对比例1
105.将实施例1步骤(3)中氧化石墨烯的质量替换为150mg,氧化碳纳米管的质量替换为150mg,此时,氧化石墨烯和氧化碳纳米管的质量比为1:1,其他参数与实施例1相同。
106.对比例2
107.将实施例1步骤(7)中第一次涂覆的水性聚氨酯的体积替换为10ml,其他参数与实施例1相同。
108.对比例3
109.将实施例1步骤(7)中第一次涂覆的水性聚氨酯的体积替换为15ml,其他参数与实施例1相同。
110.测试实施例1、实施例6和对比例1制备的气凝胶的扫描电镜图,结果如图1所示。图1中(a)为实施例6制备的气凝胶的sem图,(b)为实施例1制备的气凝胶的sem图,(c)为对比例1制备的气凝胶的sem图,(d)为(c)图方框部分的放大图。从图1中可以看出,实施例1和实施例6制备的气凝胶具有三维多孔结构,结构较为规整和致密,且随着碳纳米管含量的增加,气凝胶的孔径减小;而对比例1制备的气凝胶中碳纳米管将还原氧化石墨烯非常完整的包覆,制备的气凝胶的结构较为松散;说明碳纳米管的添加量不宜过高。
111.将实施例1得到的气凝胶置于管式炉中,在氮气保护下,以10℃/min的速率升温至800℃保持1h,然后以10℃/min的速率降温至室温,得到热处理气凝胶。测试实施例1中原料石墨、制备的氧化石墨烯、气凝胶和热处理气凝胶的红外光谱,结果如图2所示。从图2中可以看出,原料石墨的红外光谱曲线较为平直,特征峰不明显,这是因为石墨是由碳原子以sp2杂化形成的片状多层材料,石墨中并不含有官能团,而氧化石墨烯的红外光谱在多个位置出现了特征峰,如在413cm
‑1的位置出现了较宽的
‑
oh和
‑
cooh的吸收带,在1630cm
‑1的位置出现了苯环骨架的伸缩振动区,这是由于石墨经强氧化作用后一方面石墨片层间距增加产生石墨烯,另一方面在石墨烯的边缘及缺陷位置等活性较高的位置反应产生了羟基和羧基等含氧官能团,使得氧化石墨烯在水中的分散性大大提高;气凝胶经过了抗坏血酸的还原作用,由于抗坏血酸中同样存在
‑
ch3、
‑
oh、
‑
cooh以及c=c等化学结构,因此红外光谱中的特征峰在多个位置都有所增强。
112.测试实施例1中原料石墨、制备的氧化石墨烯、气凝胶的xrd图,结果如图3所示。从
图3中可以看出,原料石墨在2θ=29.02
°
处出现了一个尖锐的特征峰,说明石墨结晶度较大且石墨的层间距较大,氧化石墨烯的主峰出现左移位于2θ=14.08
°
的位置,且宽度明显变大,这是由于石墨在强氧化的反应过程中,石墨片层表面含有大量的
‑
oh、
‑
cooh、
‑
co等官能团,使得原本石墨中石墨片层与层之间的范德华力被破坏,层与层之间的结合力被大幅弱化,由布拉格公式nλ=2dsinθ(λ为x射线波长,n为衍射级数,2θ为衍射角,d为晶面间距)计算得到氧化石墨烯的层间距已经从石墨的0.33nm增加到了1nm左右,说明氧化石墨烯制备成功;制备的气凝胶的特征峰变宽且变矮,这是因为氧化石墨烯被还原后,表面含氧基团减少,分子间作用力增强使得一部分石墨烯片层间的距离再次缩小,另一方面碳纳米管的加入使得纳米颗粒的平均尺寸进一步减小,由谢乐公式(k为sherrer常数,d为垂直晶面方向的厚度,b为衍射峰积分宽度时k=1)可知,晶粒尺寸的减小会导致衍射峰宽化程度的增加。
113.对实施例1制备的气凝胶进行热重测试,结果如图4所示。从图4中可以看出,在0~800℃的温度范围内,气凝胶的质量损失率不足10%,且在0~550℃的范围内质量下降率为0.6%/100℃,而550~800℃的范围内质量下降率增加至2.09%/100℃。气凝胶在800℃高温下质量损失较小是因为,还原氧化石墨烯和碳纳米管均具有良好的稳定性,其熔点均在3000℃以上,其质量损失主要是由还原氧化石墨烯和碳纳米管表面的有机官能团的热分解造成的。
114.测试实施例1制备的复合织物的扫描电镜图,结果如图5所示。图5中(a)为复合织物的表面sem图,(b)为大倍率下复合织物的表面sem图,(c)为复合织物的横截面sem图,(d)为大倍率下复合织物的横截面sem图。从图5中可以看出,织物上方气凝胶的表面已经被水性聚氨酯完全包覆,气凝胶原有的多孔网络结构因被表面的水性聚氨酯所覆盖以及浸透填充而无法被观察到,气凝胶层较为规整和致密,保留了原有气凝胶的结构,从图中可以看到气凝胶的孔洞边界以及被覆盖了水性聚氨酯后的石墨烯轮廓,说明聚氨酯只是对气凝胶的结构进行了填充和保护而并没有对气凝胶的内部结构产生破坏。
115.测试实施例1、实施例2和实施例4中的棉织物及制备的复合织物的热重曲线,结果如图6所示,图6中气凝胶复合棉织物(200mg)为实施例2制备的复合织物,气凝胶复合棉织物(250mg)为实施例4制备的复合织物,气凝胶复合棉织物(300mg)为实施例1制备的复合织物。从图6中可以看出,随着温度的升高,织物的质量先是保持稳定出现了一个平台,然后在300~400℃出现了质量的大幅下降,是由于织物内纤维素在高温下的热分解造成的。
116.测试实施例1~5制备的复合织物的表面电阻率,依照gb
‑
t 22042
‑
2008服装防静电性能、表面电阻率实验方法进行测试和计算,将复合织物裁成尺寸为2cm
×
1cm的矩形,并在样条两端贴上宽度为0.5mm的导电胶电极,用高压源表对材料的电阻进行测试,并根据以下公式计算材料的电阻率:
117.ρ=k
×
r
118.式中:
119.ρ——计算出的表面电阻率,单位为欧姆
·
米(ω
·
m);
120.r——测定的电阻值,单位为欧姆(ω)
121.k——电极的几何因子,对于本电极,此因子为19.8。
122.结果如图7所示。图7中碳材料用量为氧化石墨烯和氧化碳纳米管的总质量。从图7中可以看出,随着氧化石墨烯和氧化碳纳米管总质量的增加,复合织物的表面电阻率逐渐降低,这是因为氧化石墨烯和氧化碳纳米管总质量的增加增加了气凝胶的厚度,即增加了材料的导电通路,并且有利于提高电磁波在气凝胶内部反射的次数和概率。
123.测试实施例1~5制备的复合织物的电磁屏蔽效能,结果如图8所示。图8中气凝胶质量200mg为实施例2制备的复合织物,气凝胶质量225mg为实施例3制备的复合织物,气凝胶质量250mg为实施例4制备的复合织物,气凝胶质量275mg为实施例5制备的复合织物,气凝胶质量300mg为实施例1制备的复合织物。从图8中可以看出,随着氧化石墨烯和氧化碳纳米管总质量的增加复合织物的电磁屏蔽效能逐渐增加,实施例1和5制备的复合织物的电磁屏蔽效能超过34db。
124.测试实施例1~5制备的复合织物的电磁屏蔽吸收效能和电池屏蔽反射效能,结果如图9所示。图9中气凝胶质量200mg为实施例2制备的复合织物,气凝胶质量225mg为实施例3制备的复合织物,气凝胶质量250mg为实施例4制备的复合织物,气凝胶质量275mg为实施例5制备的复合织物,气凝胶质量300mg为实施例1制备的复合织物。从图9中可以看出,复合织物的电磁屏蔽效能主要以电磁吸收为主,也说明了气凝胶表面及内部的聚氨酯树脂并未对气凝胶的电磁屏蔽功能产生消极影响。
125.测试实施例1、实施例7、对比例2~3制备的复合织物的拉伸强力:将复合织物裁成17cm
×
5cm的矩形(纬向长度为17cm)后利用强力测试仪对织物的拉伸断裂强力进行测试;顶破强力:将复合织物裁成直径为8cm的圆,将拉伸强力测试仪的上下夹头用一对支架取代后测试织物的顶破强力;撕裂强力:利用落锤式织物撕裂仪以双缝法对织物进行撕裂强力测试,结果如图10所示。图10中横坐标聚氨酯的用量为两次聚氨酯的总量。从图10中可以看出,本发明制备的复合织物的拉伸强力和顶破强力随着水性聚氨酯用量的增加逐渐增加,而撕裂强力随着水性聚氨酯用量的增加逐渐降低,复合织物具有较好的力学性能。
126.测试实施例1、实施例7、对比例2~3制备的复合织物的柔性,使用斜面法对织物的刚柔性进行测试,将织物样品剪成15cm
×
2cm的尺寸放在梯形木块上,以匀速将织物样条推出,由滑出长度l0和斜面角度可求出抗弯刚度c(cm):
[0127][0128]
在本实验中θ设为45
°
,由公式得c=0.487l0;
[0129]
织物的弯曲刚度b和弯曲弹性模量e
b
可由下式表示:
[0130]
b=9.8ω(0.487l0)3×
10
‑5[0131][0132]
式中ω为织物平方米重(g/m2);t
f
为织物厚度(mm)。结果如图11所示。图11中横坐标聚氨酯的用量为两次聚氨酯的总量。从图11中可以看出,随着水性聚氨酯用量的增加,复合织物的柔性下降,复合织物呈现典型的双层结构,水性聚氨酯的连接使得复合织物在发
生形变时两层相互制约,提高了复合织物的刚度。
[0133]
测试实施例1、实施例7、对比例2~3制备的复合织物的透气率,结果如图12所示。图12中横坐标聚氨酯的用量为两次聚氨酯的总量。从图12中可以看出,随着水性聚氨酯用量的增加复合织物的透气率逐渐降低,这是因为在织物表面除了有一层聚氨酯树脂外,还存在一层气凝胶层,由于内部填充了聚氨酯高分子,起到了对空气的隔绝作用,使得复合织物的透气率下降。
[0134]
测试实施例1、实施例7、对比例2~3制备的复合织物的质量保持率:将复合织物放入50℃,360w的超声振荡器中振荡2h并测试复合织物测试前后的质量变化间接表达聚氨酯对复合织物的结合牢度,结果如图13所示。图13中横坐标聚氨酯的用量为两次聚氨酯的总量。从图13中可以看出,本发明制备的复合织物经超声振荡后具有较高的质量保持率,具有较好的结合牢度,且随着水性聚氨酯用量的增加,复合织物的结合牢度逐渐增加。
[0135]
观察超声振荡后复合织物的表面形态,结果如图14所示。图14(a)是对比例2制备的复合织物,(b)为实施例7制备的复合织物。从图14中可以看出,实施例7制备的复合织物在超声振荡后表面无明显变化,具有较好的结合牢度,而对比例2制备的复合织物在超声振荡后出现了明显的脱落。
[0136]
综上,本发明制备的复合织物具有优异的电磁屏蔽效能,且具有较好的力学性能和结合牢度。
[0137]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。