1.本发明属于化合物合成领域,具体涉及一种利用连续流电化学氧化合成邻位三氟甲基取代苯胺的方法。
背景技术:
2.向有机分子中引入三氟甲基基团后,因其强偶极作用会显著改变分子的理化性质和生物活性,如改变分子极性,提高分子的脂溶性、代谢稳定性和化学稳定性等,在药物研发领域具有极高的应用价值。据报道,25%的农药和20%的医药药物分子含有三氟甲基基团。此外,在材料以及染料领域,含三氟甲基基团化合物也发挥着越来越重要的作用。传统的引入三氟甲基基团的方法通常是在高温高压下利用氟化氢、氟单质或者金属氟化物(sbf5等)直接氟化,该方法条件极为剧烈且官能团兼容性较差,并会带来环境危害,不符合绿色化学以及可持续发展的需要。随着越来越多的有机合成工作者致力于开发三氟甲基化的新方法,逐渐形成了高经济利用性、环境友好性和工业应用性的良好发展。
3.值得注意的是,邻位三氟甲基取代苯胺作为多种药物及材料的核心组成部分,如何绿色经济高效的合成邻位三氟甲基取代苯胺一直是合成化学的热门研究方向。基于过渡金属催化的碳氢键直接活化实现苯胺邻位三氟甲基化逐渐成为了研究热点,但是昂贵的过渡金属催化剂以及强氧化剂的使用限制了其工业化生产的前景。
4.有机电化学作为新兴的合成方法,采用单电子转移进行氧化还原反应,避免强氧化剂和过渡金属的使用,符合绿色化学的发展理念。釜式电解槽合成因为存在电极间距、反应不均匀、时间长易分解等问题,在应用上存在诸多限制及。
技术实现要素:
5.发明目的:本发明的目的在于提供一种反应条件温和的利用连续流电化学氧化合成邻位三氟甲基取代苯胺的方法,无需使用金属催化剂、有毒试剂、以及氧化剂等,而是使用电催化氧化,更加绿色环保,符合绿色化学合成的发展方向。
6.技术方案:为实现上述目的,本发明提出一种利用连续流电化学氧化合成邻位三氟甲基取代苯胺的方法,包括以下步骤:
7.(1)将如式(i)所示的苯胺类化合物、如式(ii)所示的三氟甲基亚磺酸钠以及电解质溶解在混合溶剂中混合,得到反应溶液;
8.(2)将反应溶液泵入设有电极的微通道反应装置中进行电解反应,收集流出液,即得含有式iii所示邻位三氟甲基化的苯胺类化合物的溶液;
[0009][0010]
其中:
[0011]
r选自c1
‑
c5烷基、3
‑
6元环烷基、5
‑
6元芳杂环基、萘环基、非取代或取代的苯基,所述取代的苯基是被c1
‑
c5烷基、卤素、
‑
cf3、
‑
cn或c1
‑
c5烷氧基取代的苯基;
[0012]
r1选自氢、c1
‑
c5烷基、卤素、
‑
cf3或c1
‑
c5烷氧基。
[0013]
优选的:
[0014]
r选自c1
‑
c3烷基、5
‑
6元环烷基、5
‑
6元芳杂环基、萘环基、非取代或取代的苯基,所述取代的苯基是被甲基、乙基、异丙基、卤素、
‑
cf3、
‑
cn或甲氧基取代的苯基,取代基为单取代或双取代;
[0015]
r1选自氢、c1
‑
c3烷基、卤素、
‑
cf3或c1
‑
c3烷氧基。
[0016]
优选的:
[0017]
r选自甲基、环己烷基、呋喃基、非取代或取代的苯基,所述取代的苯基是被甲基、卤素、
‑
cf3、
‑
cn或甲氧基取代的苯基,取代基为单取代或双取代;
[0018]
r1选自氢或甲基。
[0019]
优选的,步骤(1)中,所述电解质为高氯酸四乙基铵、高氯酸四丁基铵、四乙基四氟硼酸铵中的任意一种,所述的电解质与式(i)所示的苯胺类化合物的摩尔比为1:4~2:1,优选至1:2。电解质优选为高氯酸四乙基铵。
[0020]
优选的,步骤(1)中,式(ii)所示的三氟甲基亚磺酸钠与式(i)所示的苯胺类化合物的摩尔比为1:1~3:1,优选至2:1。
[0021]
优选的,步骤(1)中,所述混合溶剂选自乙腈与水的混合物,其中,乙腈与水的体积比为1:2~3:1,优选地,乙腈与水的体积比为2:1。如式(i)所示的苯胺类化合物在反应溶液中的浓度为0.02~0.06mol/l,优选为0.033mol/l。
[0022]
优选的,步骤(2)中,进行电解反应的恒定电流为6~15ma,优选的恒定电流为9ma。反应的恒定温度为23~27℃,反应时间为2
‑
15min。
[0023]
优选的,步骤(2)中,所述的反应溶液泵入反应器的流速为0.015~0.1ml/min,优选为0.025ml/min。
[0024]
优选的,步骤(2)中,所述设有电极的微通道反应装置包括注射泵、微通道反应器、负极片、正极片和接收器;其中,微通道反应器的两侧分别设有阴极片和阳极片;注射泵、微通道反应器和接收器以串联的方式连接;所述的连接为管道连接。
[0025]
进一步优选,所述正极片选用碳片电极,负极片选用镀铂电极。
[0026]
优选的,所述微通道反应装置中,微通道反应器的反应体积为200
‑
250μl。
[0027]
有益效果:与现有技术相比,本发明无需传统的金属催化剂、氧化剂,创造性地开发一种连续流电催化氧化合成邻位三氟甲基取代苯胺类化合物的新方法,具有后处理方便、环境友好等特点,符合绿色化学合成的发展方向。
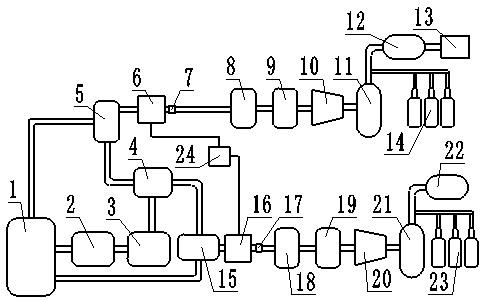
附图说明
[0028]
图1为本发明反应流程及装置示意图。
具体实施方式
[0029]
下面结合具体实施例对本发明做进一步详细说明。给出了详细的实施方式和具体的操作过程,实施例将有助于理解本发明,但是本发明的保护范围不限于下述的实施例。
[0030]
本发明设有电极的微通道反应装置包括注射泵、微通道反应器、负极片、正极片和接收器;其中,微通道反应器的两侧分别设有阴极片和阳极片;注射泵、微通道反应器和接收器以串联的方式连接;所述的连接为管道连接。
[0031]
以下实施例中,微通道反应装置中的微通道反应器模块,选自从syrris购买的电化学流动合成仪asia flux模块,其中,正极选用平板镀铂电极(5.0
×
4.0cm),负极选用微槽碳片电极(碳填充pps,5.0
×
4.0cm)。微通道反应器的反应体积为225μl。
[0032]
实施例1
[0033]
0.2mmol的原料苯甲酰苯胺,0.4mmol的三氟甲基亚磺酸钠,0.1mmol的高氯酸四乙基铵溶于4ml乙腈和2ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟以助溶。在环境温度,6ma的恒定电流下,以0.025ml/min的流速注入碳片电极做正极,镀铂电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以88%收率得到产品邻位三氟甲基取代苯胺产物1。对其进行了核磁共振氢谱、碳谱和高分辨质谱hrms(esi)标准进行验证。
[0034][0035]
n
‑
(2
‑
(三氟甲基)苯基)苯甲酰胺(1)
[0036]
white solid,80%yield.1h nmr(400mhz,cdcl3)δ8.44(d,j=8.3hz,1h),8.24(br,1h),7.91
–
7.86(m,2h),7.68
–
7.50(m,5h),7.27(t,j=7.6hz,1h).
13
c nmr(100mhz,cdcl3)δ165.5,135.5,134.3,133.1,132.3,129.0,127.0,126.2(q,j=5.3hz),124.5,124.3(d,j=271hz),124.1,120.1(q,j=29.7hz).
19
f nmr(376mhz,cdcl3)δ
‑
60.42.hrms(tof)m/z[m h]
calcd for c
14
h
11
f3no 266.0787 found 266.0790.
[0037]
实施例2
[0038]
0.2mmol的原料苯甲酰苯胺,0.4mmol的三氟甲基亚磺酸钠,0.1mmol的高氯酸四乙基铵溶于4ml乙腈和2ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟以助溶。在环境温度,9ma的恒定电流下,以0.025ml/min的流速注入碳片电极做正极,镀铂电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以90%收率得到产品邻位三氟甲基取代苯胺产物1。
[0039]
实施例3
[0040]
0.2mmol的原料苯甲酰苯胺,0.4mmol的三氟甲基亚磺酸钠,0.1mmol的高氯酸四乙基铵溶于4ml乙腈和2ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟以助溶。在环境温度,12ma的恒定电流下,以0.025ml/min的流速注入碳片电极做正极,镀铂电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以87%收率得到产品邻位三氟甲基取代苯胺产物1。
[0041]
实施例4
[0042]
0.2mmol的原料苯甲酰苯胺,0.4mmol的三氟甲基亚磺酸钠,0.1mmol的高氯酸四乙基铵溶于4ml乙腈和2ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟以助溶。在环境温度,15ma的恒定电流下,以0.025ml/min的流速注入碳片电极做正极,镀铂电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以86%收率得到产品邻位三氟甲基取代苯胺产物1。
[0043]
实施例5
[0044]
0.2mmol的原料苯甲酰苯胺,0.4mmol的三氟甲基亚磺酸钠,0.1mmol的高氯酸四乙基铵溶于4ml乙腈和2ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟以助溶。在环境温度,9ma的恒定电流下,以0.015ml/min的流速注入碳片电极做正极,镀铂电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以83%收率得到产品邻位三氟甲基取代苯胺产物1。
[0045]
实施例6
[0046]
0.2mmol的原料苯甲酰苯胺,0.4mmol的三氟甲基亚磺酸钠,0.1mmol的高氯酸四乙基铵溶于4ml乙腈和2ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟以助溶。在环境温度,9ma的恒定电流下,以0.05ml/min的流速注入碳片电极做正极,镀铂电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以85%收率得到产品邻位三氟甲基取代苯胺产物1。
[0047]
实施例7
[0048]
0.2mmol的原料苯甲酰苯胺,0.4mmol的三氟甲基亚磺酸钠,0.1mmol的高氯酸四乙基铵溶于4ml乙腈和2ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟以助溶。在环境温度,9ma的恒定电流下,以0.075ml/min的流速注入碳片电极做正极,镀铂电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以81%收率得到产品邻位三氟甲基取代苯胺产物1。
[0049]
实施例8
[0050]
0.2mmol的原料苯甲酰苯胺,0.4mmol的三氟甲基亚磺酸钠,0.1mmol的高氯酸四乙基铵溶于4ml乙腈和2ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟以助溶。在环境温度,9ma的恒定电流下,以0.1ml/min的流速注入碳片电极做正极,镀铂电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以79%收率得到产品邻位三氟甲基取代苯胺产物1。
[0051]
实施例9
[0052]
0.2mmol的原料苯甲酰苯胺,0.4mmol的三氟甲基亚磺酸钠,0.1mmol的高氯酸四乙基铵溶于2ml乙腈和4ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟以助溶。在环境温度,9ma的恒定电流下,以0.025ml/min的流速注入碳片电极做正极,镀铂电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,
有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以86%收率得到产品邻位三氟甲基取代苯胺产物1。
[0053]
实施例10
[0054]
0.2mmol的原料苯甲酰苯胺,0.4mmol的三氟甲基亚磺酸钠,0.1mmol的高氯酸四乙基铵溶于3ml乙腈和3ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟以助溶。在环境温度,9ma的恒定电流下,以0.025ml/min的流速注入碳片电极做正极,镀铂电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以83%收率得到产品邻位三氟甲基取代苯胺产物1。
[0055]
实施例11
[0056]
0.2mmol的原料苯甲酰苯胺,0.4mmol的三氟甲基亚磺酸钠,0.1mmol的高氯酸四丁基铵溶于4ml乙腈和2ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟以助溶。在环境温度,9ma的恒定电流下,以0.025ml/min的流速注入碳片电极做正极,镀铂电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以87%收率得到产品邻位三氟甲基取代苯胺产物1。
[0057]
实施例12
[0058]
0.2mmol的原料苯甲酰苯胺,0.4mmol的三氟甲基亚磺酸钠,0.1mmol的四乙基四氟硼酸铵溶于4ml乙腈和2ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟以助溶。在环境温度,9ma的恒定电流下,以0.025ml/min的流速注入碳片电极做正极,镀铂电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以86%收率得到产品邻位三氟甲基取代苯胺产物1。
[0059]
实施例13
[0060]
0.2mmol的原料苯甲酰苯胺,0.4mmol的三氟甲基亚磺酸钠,0.05mmol的高氯酸四乙基铵溶于4ml乙腈和2ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟以助溶。在环境温度,9ma的恒定电流下,以0.025ml/min的流速注入碳片电极做正极,镀铂电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以78%收率得到产品邻位三氟甲基取代苯胺产物1。
[0061]
实施例14
[0062]
0.2mmol的原料苯甲酰苯胺,0.4mmol的三氟甲基亚磺酸钠,0.2mmol的高氯酸四乙基铵溶于4ml乙腈和2ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟以助溶。在环境温度,9ma的恒定电流下,以0.025ml/min的流速注入碳片电极做正极,镀铂电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以85%收率得到产品邻位三氟甲基取代苯胺产物1。
[0063]
实施例15
[0064]
0.2mmol的原料苯甲酰苯胺,0.4mmol的三氟甲基亚磺酸钠,0.4mmol的高氯酸四乙
基铵溶于4ml乙腈和2ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟以助溶。在环境温度,9ma的恒定电流下,以0.025ml/min的流速注入碳片电极做正极,镀铂电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以87%收率得到产品邻位三氟甲基取代苯胺产物1。
[0065]
实施例16
[0066]
0.2mmol的原料苯甲酰苯胺,0.2mmol的三氟甲基亚磺酸钠,0.1mmol的高氯酸四乙基铵溶于4ml乙腈和2ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟以助溶。在环境温度,9ma的恒定电流下,以0.025ml/min的流速注入碳片电极做正极,镀铂电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以78%收率得到产品邻位三氟甲基取代苯胺产物1。
[0067]
实施例17
[0068]
0.2mmol的原料苯甲酰苯胺,0.6mmol的三氟甲基亚磺酸钠,0.1mmol的高氯酸四乙基铵溶于4ml乙腈和2ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟以助溶。在环境温度,9ma的恒定电流下,以0.025ml/min的流速注入碳片电极做正极,镀铂电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以91%收率得到产品邻位三氟甲基取代苯胺产物1。
[0069]
实施例18
[0070]
0.2mmol的原料4
‑
甲基
‑
n
‑
苯基苯甲酰胺,0.4mmol的三氟甲基亚磺酸钠,0.1mmol的高氯酸四乙基铵溶于4ml乙腈和2ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟以助溶。在环境温度,9ma的恒定电流下,以0.025ml/min的流速注入碳片电极做正极,镀铂电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以90%收率得到产品邻位三氟甲基取代苯胺产物2。对其进行了核磁共振氢谱、碳谱和高分辨质谱hrms(esi)标准进行验证。
[0071][0072]4‑
甲基
‑
n
‑
(2
‑
(三氟甲基)苯基)苯甲酰胺(2)
[0073]
white solid,82%yield.1h nmr(400mhz,cdcl3)δ8.39(d,j=8.3hz,1h),8.28(br,1h),7.80
–
7.75(m,2h),7.62(d,j=8.0hz,1h),7.56(t,j=7.9hz,1h),7.28(d,j=7.9hz,2h),7.23(t,j=7.7hz,1h),2.41(s,3h).
13
c nmr(100mhz,cdcl3)δ165.5,142.9,135.7(q,j=2.2hz),133.0,131.4,129.6,127.1,126.1(q,j=5.2hz),124.4,124.4,124.3(q,j=273.1hz),120.2(q,j=29.7hz),21.5.
19
f nmr(376mhz,cdcl3)δ
‑
60.45.hrms(tof)m/z[m h]
calcd for c
15
h
13
f3no 280.0944 found 280.0941.
[0074]
实施例19
[0075]
0.2mmol的原料4
‑
氰基
‑
n
‑
苯基苯甲酰胺,0.4mmol的三氟甲基亚磺酸钠,0.1mmol
的高氯酸四乙基铵溶于4ml乙腈和2ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟以助溶。在环境温度,9ma的恒定电流下,以0.025ml/min的流速注入碳片电极做正极,镀铂电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以90%收率得到产品邻位三氟甲基取代苯胺产物3。对其进行了核磁共振氢谱、碳谱和高分辨质谱hrms(esi)标准进行验证。
[0076][0077]4‑
氰基
‑
n
‑
(2
‑
(三氟甲基)苯基)苯甲酰胺(3)
[0078]
white solid,26%yield.1h nmr(400mhz,cdcl3)δ8.29(d,j=8.3hz,1h),8.21(br,1h),7.98
–
7.94(m,2h),7.82
–
7.78(m,2h),7.67(d,j=7.9hz,1h),7.62(t,j=7.9hz,1h),7.32(t,j=7.7hz,1h).
13
c nmr(100mhz,cdcl3)δ163.9,138.1,134.7,133.2,132.8,127.8,126.4(q,j=5.3hz),125.4,124.8,124.1(q,j=273.0hz),120.9(q,j=29.3.0hz),117.8,115.9.
19
f nmr(376mhz,cdcl3)δ
‑
60.30.hrms(tof)m/z[m h]
calcd for c
15
h
10
f3n2o 291.0740 found 291.0744.
[0079]
实施例20
[0080]
0.2mmol的原料3
‑
三氟甲基
‑
n
‑
苯基苯甲酰胺,0.4mmol的三氟甲基亚磺酸钠,0.1mmol的高氯酸四乙基铵溶于4ml乙腈和2ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟以助溶。在环境温度,9ma的恒定电流下,以0.025ml/min的流速注入碳片电极做正极,镀铂电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以90%收率得到产品邻位三氟甲基取代苯胺产物4。对其进行了核磁共振氢谱、碳谱和高分辨质谱hrms(esi)标准进行验证。
[0081][0082]3‑
(三氟甲基)
‑
n
‑
(2
‑
(三氟甲基)苯基)苯甲酰胺(4)
[0083]
white solid,57%yield.1h nmr(400mhz,cdcl3)δ8.30(d,j=8.2hz,1h),8.25(br,1h),8.17(s,1h),8.01(d,j=7.9hz,1h),7.83(d,j=7.9hz,1h),7.68
–
7.59(m,3h),7.30(t,j=7.7hz,1h).
13
c nmr(100mhz,cdcl3)δ164.2,135.1,135.0,133.1,131.7(q,j=33.3hz),129.9,129.6,128.9(q,j=3.6hz),126.3(q,j=5.3hz),125.2,124.8,124.5(q,j=3.7hz),124.2(q,j=273.1hz),123.6(q,j=272.5hz),120.9(q,j=29.7hz).
19
f nmr(376mhz,cdcl3)δ
‑
60.45,
‑
62.94.hrms(tof)m/z[m h]
calcd for c
15
h
10
f6no 334.0661 found 334.0653.
[0084]
实施例21
[0085]
0.2mmol的原料2
‑
氯
‑
n
‑
苯基苯甲酰胺,0.4mmol的三氟甲基亚磺酸钠,0.1mmol的高氯酸四乙基铵溶于4ml乙腈和2ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟
以助溶。在环境温度,9ma的恒定电流下,以0.025ml/min的流速注入碳片电极做正极,镀铂电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以90%收率得到产品邻位三氟甲基取代苯胺产物5。对其进行了核磁共振氢谱、碳谱和高分辨质谱hrms(esi)标准进行验证。
[0086][0087]2‑
氯
‑
n
‑
(2
‑
(三氟甲基)苯基)苯甲酰胺(5)
[0088]
white solid,73%yield.1h nmr(400mhz,cdcl3)δ8.38(d,j=8.3hz,1h),8.19(br,1h),7.76(d,j=7.4hz,1h),7.66(d,j=7.9hz,1h),7.62(t,j=7.9hz,1h),7.49
–
7.37(m,3h),7.29(t,j=7.7hz,1h).
13
c nmr(100mhz,cdcl3)δ164.9,134.9,134.7,133.0,132.1,130.9,130.6,130.2,127.4,126.2(q,j=5.4hz),125.1,124.9,124.0(q,j=273.1hz),120.9(d,j=30.3hz).
19
f nmr(376mhz,cdcl3)δ
‑
60.38.hrms(tof)m/z[m h]
calcd for c
14
h
10
clf3no 300.0398 found 300.0406.
[0089]
实施例22
[0090]
0.2mmol的原料3,4
‑
二甲氧基
‑
n
‑
苯基苯甲酰胺,0.4mmol的三氟甲基亚磺酸钠,0.1mmol的高氯酸四乙基铵溶于4ml乙腈和2ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟以助溶。在环境温度,9ma的恒定电流下,以0.025ml/min的流速注入碳片电极做正极,镀铂电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以90%收率得到产品邻位三氟甲基取代苯胺产物6。对其进行了核磁共振氢谱、碳谱和高分辨质谱hrms(esi)标准进行验证。
[0091][0092]
3,4
‑
二甲氧基
‑
n
‑
(2
‑
(三氟甲基)苯基)苯甲酰胺(6)
[0093]
white solid,65%yield.1h nmr(400mhz,cdcl3)δ8.41(d,j=8.3hz,1h),8.20(br,1h),7.63(d,j=7.9hz,1h),7.59(t,j=7.9hz,1h),7.50(d,j=2.1hz,1h),7.38(dd,j=8.3,2.2hz,1h),7.24(t,j=7.9hz,1h),6.94(d,j=8.4hz,1h),3.95(s,3h),3.94(s,3h).
13
c nmr(100mhz,cdcl3)δ164.7,153.1,149.4,135.8,133.0,126.9,126.1(q,j=5.2hz),124.4(q,j=273.2hz),124.3,124.0,119.8(q,j=30.7,29.4hz),119.5,110.7,110.6,56.1,56.0.
19
f nmr(376mhz,cdcl3)δ
‑
60.43.hrms(tof)m/z[m h]
calcd for c
16
h
15
f3no3326.0999 found 326.1001.
[0094]
实施例23
[0095]
0.2mmol的原料n
‑
(对甲苯基)苯甲酰胺,0.4mmol的三氟甲基亚磺酸钠,0.1mmol的高氯酸四乙基铵溶于4ml乙腈和2ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟以助溶。在环境温度,9ma的恒定电流下,以0.025ml/min的流速注入碳片电极做正极,镀铂
电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以90%收率得到产品邻位三氟甲基取代苯胺产物7。对其进行了核磁共振氢谱、碳谱和高分辨质谱hrms(esi)标准进行验证。
[0096][0097]
n
‑
(4
‑
甲基
‑2‑
(三氟甲基)苯基)苯甲酰胺(7)
[0098]
white solid,73%yield.1h nmr(400mhz,cdcl3)δ8.24(d,j=8.4hz,1h),8.14(br,1h),7.90
–
7.85(m,2h),7.60
–
7.56(m,1h),7.54
–
7.49(m,2h),7.45(s,1h),7.41(d,j=8.5hz,1h),2.40(s,3h).
13
c nmr(100mhz,cdcl3)δ165.5,134.7,134.4,133.5,132.8,132.2,129.0,127.0,126.5(q,j=5.1hz),124.5,124.3(q,j=271.5hz),120.3(d,j=29.3hz),20.9.
19
f nmr(376mhz,cdcl3)δ
‑
60.38.hrms(tof)m/z[m h]
calcd for c
15
h
13
f3no 280.0944 found 280.0944.
[0099]
实施例24
[0100]
0.2mmol的原料乙酰苯胺,0.4mmol的三氟甲基亚磺酸钠,0.1mmol的高氯酸四乙基铵溶于4ml乙腈和2ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟以助溶。在环境温度,9ma的恒定电流下,以0.025ml/min的流速注入碳片电极做正极,镀铂电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以90%收率得到产品邻位三氟甲基取代苯胺产物8。对其进行了核磁共振氢谱、碳谱和高分辨质谱hrms(esi)标准进行验证。
[0101][0102]
n
‑
(2
‑
(三氟甲基)苯基)乙酰胺(8)
[0103]
white solid,29%yield.1h nmr(400mhz,cdcl3)δ8.15(d,j=8.3hz,1h),7.61(d,j=7.9hz,1h),7.54(t,j=7.9hz,1h),7.42(br,1h),7.23(t,j=7.8hz,1h),2.22(s,3h).
13
c nmr(100mhz,cdcl3)δ168.5,135.2,132.9,126.1(q,j=5.4hz),124.1(q,j=259.3hz),124.8,124.6,120.1(q,j=26.0hz),24.6.
19
f nmr(376mhz,cdcl3)δ
‑
60.64.hrms(tof)m/z[m h]
calcd for c9h9f3no 204.0631 found 204.0637.
[0104]
实施例25
[0105]
0.2mmol的原料n
‑
苯基环己烷甲酰胺,0.4mmol的三氟甲基亚磺酸钠,0.1mmol的高氯酸四乙基铵溶于4ml乙腈和2ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟以助溶。在环境温度,9ma的恒定电流下,以0.025ml/min的流速注入碳片电极做正极,镀铂电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以90%收率得到产品邻位三氟甲基取代苯胺产物9。对其进行了核磁共振氢谱、碳
谱和高分辨质谱hrms(esi)标准进行验证。
[0106][0107]
n
‑
(2
‑
(三氟甲基)苯基)环己烷甲酰胺(9)
[0108]
white solid,78%yield.1h nmr(400mhz,cdcl3)δ8.17(d,j=8.3hz,1h),7.59
–
7.48(m,3h),7.18(t,j=7.7hz,1h),2.27(tt,j=11.7,3.5hz,1h),1.98(d,j=13.4hz,2h),1.86
–
1.79(m,2h),1.73
–
1.66(m,1h),1.55
–
1.45(m,2h),1.36
–
1.21(m,3h).
13
c nmr(100mhz,cdcl3)δ174.4,135.5,132.8,126.0(q,j=5.2hz),124.7,124.3,124.2(q,j=273.1hz),120.2(q,j=29.3hz),46.4,29.4,25.6,25.6.
19
f nmr(376mhz,cdcl3)δ
‑
60.66.hrms(tof)m/z[m h]
calcd for c
14
h
17
f3no 272.1257 found 272.1273.
[0109]
实施例26
[0110]
0.2mmol的原料n
‑
苯基呋喃甲酰胺,0.4mmol的三氟甲基亚磺酸钠,0.1mmol的高氯酸四乙基铵溶于4ml乙腈和2ml水的混合溶液中。反应混合物于25℃下超声震荡30分钟以助溶。在环境温度,9ma的恒定电流下,以0.025ml/min的流速注入碳片电极做正极,镀铂电极做负极的微通道反应器中。反应液收集后以50ml饱和碳酸氢钠溶液洗涤,50ml乙酸乙酯萃取3次,有机相经无水硫酸钠干燥,滤液减压浓缩,柱层析(石油醚:乙酸乙酯=8:1体积比)分离以90%收率得到产品邻位三氟甲基取代苯胺产物10。对其进行了核磁共振氢谱、碳谱和高分辨质谱hrms(esi)标准进行验证。
[0111][0112]
n
‑
(2
‑
(三氟甲基)苯基)呋喃
‑2‑
羧酰胺(10)
[0113]
white solid,76%yield.1h nmr(400mhz,cdcl3)δ8.52(br,1h),8.39(d,j=8.3hz,1h),7.60(d,j=8.0hz,1h),7.54(t,j=7.7hz,1h),7.51(dd,j=1.8,0.8hz,1h),7.23(dd,j=3.5,0.9hz,1h),7.20(t,j=7.7hz,1h),6.53(dd,j=3.5,1.8hz,1h).
13
c nmr(100mhz,cdcl3)δ156.0,147.3,144.9,135.0(d,j=2.0hz),132.9,126.1(q,j=5.2hz),124.4,124.2(q,j=271.3hz),123.8,119.9(q,j=29.6hz),115.9,112.6.
19
f nmr(376mhz,cdcl3)δ
‑
60.63.hrms(tof)m/z[m h]
calcd for c
12
h9f3no2256.0580 found 256.0651.
[0114]
本发明提供了一种利用连续流电化学氧化合成邻位三氟甲基取代苯胺类化合物的思路及方法,具体实现该技术方案的方法和途径很多,以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。本实施例中未明确的各组成部分均可用现有技术加以实现。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。