合肼。
9.在优选的实施例中,所述表面活性剂用量为铬盐用量的5wt%
‑
50wt%。
10.在优选的实施例中,所述铬盐溶液包括:硝酸铬、硫酸铬、氯化铬、高氯酸铬中的一 种或多种。
11.在优选的实施例中,所述溶剂为水。
12.在优选的实施例中,所述超重力反应器为旋转填充床。
13.在优选的实施例中,所述旋转填充床内设有疏水填料。
14.在优选的实施例中,所述疏水填料上形成有多个贯穿的孔道。
15.在优选的实施例中,所述孔道为微纳尺度。
16.本发明的有益效果:
17.本发明提供一种非晶态氢氧化铬分散体的制备方法,首先将铬盐溶液和沉淀剂溶液泵 入一超重力反应器循环设定时长得到氢氧化铬颗粒;所述超重力反应器运行时馈入超声; 然后采用表面活性剂对所述氢氧化铬颗粒进行表面改性;最后将改性后的所述氢氧化铬颗 粒和溶剂泵入所述超重力反应器,得到非晶态氢氧化铬分散体;本发明通过首先采用超声 耦合超重力场强化氢氧化铬成核过程的微观混合,制备出非晶态超小的氢氧化铬纳米颗粒, 然后通过表面活性剂进行表面改性,并使用超声场耦合超重力场对氢氧化铬进行再分散, 通过超重力旋转填充床的强剪切以及超声场对纳米颗粒的振动作用使得超小粒径的纳米颗 粒能够均匀再分散,从而制备出能够在溶剂中稳定分散的非晶态超小氢氧化铬纳米分散体, 该分散体的zeta电位的绝对值高,能够在水相中保持高稳定性,静置10个月内未发生沉降。
附图说明
18.为了更清楚地说明本发明实施方式或现有技术中的技术方案,下面将对实施方式或现 有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本 发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还 可以根据这些附图获得其他的附图。
19.图1为本发明实施例1的产物透射电子显微镜图(tem)。
20.图2为本发明实施例1制备产物的粒径分布统计图。
21.图3为本发明实施例1的产物x
‑
射线粉末衍射图案。
22.图4为本发明实施例2的产物的zeta电位随静置时间的变化趋势。
23.图5为本发明实施例1得到的非晶态氢氧化铬水相分散体电子照片。
24.图6为本发明实施例2的产物透射电子显微镜图(tem)。
25.图7为本发明实施例2制备产物的粒径分布统计图。
26.图8为本发明实施例2的产物x
‑
射线粉末衍射图案。
27.图9为本发明实施例2的产物的zeta电位随静置时间的变化趋势。
28.图10为本发明实施例2得到的非晶态氢氧化铬水相分散体电子照片。
29.图11为本发明对比例1的产物透射电子显微镜图(tem)。
30.图12为本发明对比例1的产物x
‑
射线粉末衍射图案。
31.图13为本发明对比例1得到的非晶态氢氧化铬水相分散体电子照片。
32.图14为实施例1产物催化高氯酸铵热分解的tg
‑
dsc曲线图。
33.图15为对比例1产物催化高氯酸铵热分解的tg
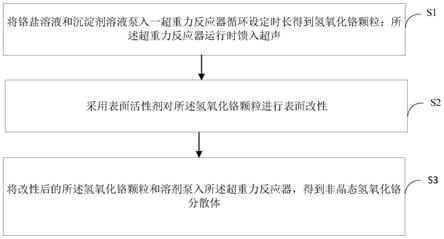
‑
dsc曲线图。
34.图16为本发明实施例中一种新的非晶态氢氧化铬分散体的制备方法的流程示意图。
具体实施方式
35.下面将结合本发明实施方式中的附图,对本发明实施方式中的技术方案进行清楚、完 整地描述,显然,所描述的实施方式仅仅是本发明一部分实施方式,而不是全部的实施方 式。基于本发明中的实施方式,本领域普通技术人员在没有做出创造性劳动前提下所获得 的所有其他实施方式,都属于本发明保护的范围。
36.本发明发明人研究发现,常规方法无法制备出粒径小并且分散性能好的氢氧化铬颗粒, 业内仍然没有较好的解决方式,有关方面的研究进展一直停滞不前。
37.经过发明人的实验探究,本发明尝试采用分步法进行氢氧化铬分散体的装备,本发明 提供一种新的非晶态氢氧化铬分散体的制备方法,如图16所示,其具体包括:
38.s1:将铬盐溶液和沉淀剂溶液泵入一超重力反应器循环设定时长得到氢氧化铬颗粒; 所述超重力反应器运行时馈入超声;
39.s2:采用表面活性剂对所述氢氧化铬颗粒进行表面改性;
40.s3:将改性后的所述氢氧化铬颗粒和溶剂泵入所述超重力反应器,得到非晶态氢氧化 铬分散体。
41.本发明提供一种非晶态氢氧化铬分散体的制备方法,首先将铬盐溶液和沉淀剂溶液泵 入一超重力反应器循环设定时长得到氢氧化铬颗粒;所述超重力反应器运行时馈入超声; 然后采用表面活性剂对所述氢氧化铬颗粒进行表面改性;最后将改性后的所述氢氧化铬颗 粒和溶剂泵入所述超重力反应器,得到非晶态氢氧化铬分散体;本发明通过首先采用超声 耦合超重力场强化氢氧化铬成核过程的微观混合,制备出非晶态超小的氢氧化铬纳米颗粒, 然后通过表面活性剂进行表面改性,并使用超声场耦合超重力场对氢氧化铬进行再分散, 通过超重力旋转填充床的强剪切以及超声场对纳米颗粒的振动作用使得超小粒径的纳米颗 粒能够均匀再分散,从而制备出能够在溶剂中稳定分散的非晶态超小氢氧化铬纳米分散体, 该分散体的zeta电位的绝对值高,能够在水相中保持高稳定性,静置10个月内未发生沉降。
42.在优选的实施例中,所述表面活性剂包括:柠檬酸钠、酒石酸钠、十六烷基三甲基溴 化铵、聚乙烯吡咯烷酮、十六烷基三甲基氯化铵、聚乙二醇中的一种或多种。
43.在优选的实施例中,所述沉淀剂包括:氢氧化钠、氢氧化钾、氨水、硼氢化钠以及水 合肼。
44.在优选的实施例中,所述表面活性剂用量为铬盐用量的5wt%
‑
50wt%。
45.在优选的实施例中,所述铬盐溶液包括:硝酸铬、硫酸铬、氯化铬、高氯酸铬中的一 种或多种。
46.在优选的实施例中,所述溶剂为水。
47.在优选的实施例中,所述超重力反应器为旋转填充床。
48.具体而言,本发明提供的工艺流程如下:
49.步骤一、将可溶性三价铬盐溶于水中,得到铬盐溶液;将沉淀剂溶于水中,得到沉淀 剂溶液;
50.步骤二、开启超重力旋转填充床,将铬盐溶液和沉淀剂溶液通过输送泵泵入到超重力 旋转填充床中,铬盐和沉淀剂在超重力旋转填充床中反应并均匀成核,得到含有氢氧化铬 晶核的反应液经超重力旋转填充床的液体出口流出;
51.步骤三、将含有氢氧化铬晶核的反应液再通过输送泵回到超重力旋转填充床中,使反 应液在超重力旋转填充床中循环一段时间,以使得氢氧化铬晶核在超重力环境下均匀生长, 得到非晶态的超小氢氧化铬;
52.步骤四、将步骤三中得到的非晶态的超小氢氧化铬离心并用水洗涤至电导率低于200 μs/cm;
53.步骤五、将洗涤后的非晶态氢氧化铬继续分散到水中,再向洗涤后的非晶态氢氧化铬 中加入适量的表面活性剂,并在一定温度下搅拌一段时间;
54.步骤六、将步骤五得到的含有表面活性剂的非晶态氢氧化铬离心,再用水洗涤,得到 柠檬酸钠改性的非晶态超小氢氧化铬;
55.步骤七、向步骤六中得到的洗涤后得柠檬酸钠改性的非晶态超小氢氧化铬中加入一定 量的水,然后开启超重力反应器,再用输送泵将柠檬酸钠改性的氢氧化铬浆料泵回到反应 器中,并在反应器中循环一段时间,以实现纳米颗粒的再分散,最终即可得到非晶态超小 氢氧化铬水相分散体。
56.进一步的,优选的,步骤一所述的铬盐浓度为0.05mol/l~1mol/l。
57.优选的,步骤一中的沉淀剂液与铬盐的摩尔比为1:1~1:10。
58.优选的,步骤二中所述的超重力的旋转床的转速为300~3000r/min,超声波的功率为 5
‑
50kw。
59.优选的,步骤三中所述的循环时间为0.5
‑
3h。
60.优选的,步骤五中所述的一定温度为25
‑
90℃,所述的一段时间为1h~10h。
61.优选的,步骤七中所述的循环时间为0.5~5h。
62.本发明发明人发现铬离子与氢氧根反应生成氢氧化铬的过程为一快速反应过程,该过 程生成晶核的诱导时间通常低于1ms,因此若想制备粒径均匀分布的超小粒径的氢氧化铬纳 米颗粒,需大大强化氢氧化铬成核前铬离子与氢氧根之间的微观混合。鉴于此,本专利采 用超声波耦合超重力场强化氢氧化铬成核过程的微观混合,制备出非晶态超小的氢氧化铬 纳米颗粒。此外,通过表面活性剂进行表面改性,并使用超声场耦合超重力场对氢氧化铬 进行再分散,通过超重力旋转填充床的强剪切以及超声场对纳米颗粒的振动作用使得超小 粒径的纳米颗粒能够在水相中均匀再分散,从而制备出能够在水相中稳定分散的非晶态超 小氢氧化铬纳米分散体。
63.进一步的,本发明为了使得不易发生堵塞现象,所述超重力反应器为旋转填充床,所 述旋转填充床内设有疏水填料。
64.可以理解,超重力技术一种典型过程强化技术,目前已经被成功应用于各种强化传质、 传热和微观混合的工业过程中并取得了优异的成果,其特点是设备占地面积小,停留时间 短,传质效率高,反应快速高效。
65.本发明将超重力技术与超声技术结合,将之应用于非晶态氢氧化铬分散体的制备
工艺 中,首先将铬盐溶液和沉淀剂溶液泵入一超重力反应器循环设定时长得到氢氧化铬颗粒; 所述超重力反应器运行时馈入超声;然后采用表面活性剂对所述氢氧化铬颗粒进行表面改 性;最后将改性后的所述氢氧化铬颗粒和溶剂泵入所述超重力反应器,得到非晶态氢氧化 铬分散体;本发明通过首先采用超声耦合超重力场强化氢氧化铬成核过程的微观混合,制 备出非晶态超小的氢氧化铬纳米颗粒,然后通过表面活性剂进行表面改性,并使用超声场 耦合超重力场对氢氧化铬进行再分散,通过超重力旋转填充床的强剪切以及超声场对纳米 颗粒的振动作用使得超小粒径的纳米颗粒能够均匀再分散,从而制备出能够在溶剂中稳定 分散的非晶态超小氢氧化铬纳米分散体,该分散体的zeta电位的绝对值高,能够在水相中 保持高稳定性,静置10个月内未发生沉降。
66.下面以具体场景案例对本发明进行举例。
67.场景例1
68.一种制备非晶态超小氢氧化铬水相分散体的方法,包括如下步骤:
69.步骤一、将6g硝酸铬(cr(no3)3
·
9h2o)溶于100ml水中,得到0.15mol/l的铬盐 溶液;将6g水合肼(n2h4
·
h2o,>98%)溶于100ml水中,得到1.2mol/l水合肼溶液;
70.步骤二、开启超重力旋转填充床以1及超声波,设置转速为2000r/min,超声波的频率 为50khz,再将步骤一得到的硝酸铬溶液和水合肼溶液通过输送泵同时泵入到超声波耦合 超重力旋转填充床中,铬盐和水合肼在超重力和超声波的耦合场下反应并均匀成核,得到 含有氢氧化铬晶核的反应液经超重力旋转填充床的液体出口流出;
71.步骤三、仅关闭超声波,然后通过输送泵将步骤二得到的含有氢氧化铬晶核的反应液 再泵回到超重力反应器中,使反应液在超重力反应器中循环1h,氢氧化铬晶核在超重力场 强化下继续生长,得到非晶态超小氢氧化铬浆料;
72.步骤四、将步骤三中得到的非晶态超小氢氧化铬浆料离心并用水洗涤至电导率低于200 μs/cm;
73.步骤五、将洗涤后的非晶态超小氢氧化铬继续分散到200ml水中,并向其中加入1g 柠檬酸钠,并在50℃搅拌6h;
74.步骤六、将步骤五得到的含有柠檬酸钠的非晶态超小氢氧化铬浆料离心,再用等量的 水洗涤2次,得到柠檬酸钠改性的非晶态超小氢氧化铬。
75.步骤七、向步骤六得到的洗涤后得柠檬酸钠改性的非晶态超小氢氧化铬中加入40ml 水,然后开启超重力反应器以及超声波(转速为2000r/min,超声波的频率为50khz),再 用输送泵将柠檬酸钠改性的非晶态超小氢氧化铬浆料泵回到反应器中,并在反应器中循环 2h,以实现再分散,最终即可得到非晶态超小氢氧化铬水相分散体,其固含量经测定约为 4wt%。
76.说明书附图中图一为实施例1的产物tem图,可以看出氢氧化铬形貌为不规则颗粒, 图2为产物的粒径分布统计图,氢氧化铬尺寸分布在4
‑
11nm,平均粒径约为7nm。图3为 实施例1产物的x射线粉末衍射图案,图中没有尖锐的衍射特征峰,表明实施例1所得产 物的为非晶态。图4为实施例1得到的非晶态氢氧化铬水相分散体在静置10个月后的照片, 没有观察到分层现象,表明分散体具有良好的稳定性。图4为本发明实施例1的所得非晶 态氢氧化铬水相分散体的zeta电位值随静置时间的变化趋势图,在静置10个月后,非晶态 氢氧化铬水相分散体的zeta电位值为
‑
43mv,表明氢氧化铬颗粒在水相中仍能稳定分散。
77.场景例2
78.在与实施例1相同的制备的条件下,仅将超声波耦合超重力旋转填充床换成超重力反 应器,仍然可以制备得到无定型超小氢氧化铬分散体。
79.说明书附图中图6为实施例2的产物tem图,可以看出氢氧化铬形貌为不规则颗粒, 图7为实施例2产物的粒径分布统计图,图中可以看出实施例2制备的氢氧化铬颗粒尺寸 分布在10~30nm,平均粒径约为17nm。图8为实施例2制备的氢氧化铬产物的x射线粉末 衍射图案,图中依然没有尖锐的衍射特征峰,表明实施例2所得氢氧化铬产物也为非晶态。 图9为实施例2得到的非晶态氢氧化铬水相分散体在静置10个月后的照片,也没有观察到 分层现象,表明氢氧化铬分散体具有良好的稳定性。图10为本发明实施例2的所得非晶态 氢氧化铬水相分散体的zeta电位值随静置时间的变化趋势图,在静置10个月后,非晶态氢 氧化铬水相分散体的zeta电位值为
‑
38.7mv,表明氢氧化铬颗粒在水相中仍能稳定分散。
80.场景例1与场景例2的结果相比可知:
81.(1)实施例1制备的氢氧化铬颗粒尺寸小于实施例2制备的氢氧化铬颗粒,使用超 重力旋转填充床能够合成非晶态的超小氢氧化铬纳米颗粒,而使用超声波耦合超重力场来 强化氢氧化铬成核过程能够制得尺寸更小的氢氧化铬;
82.(2)经过柠檬酸钠改性后,该非晶态氢氧化铬纳米颗粒能够在水相中稳定分散,而 实施例1制备的非晶态氢氧化铬水相分散体的zeta电位值的绝对值更低,这表明超声波和 超重力的耦合场相比于单独的超重力场更有利于实现氢氧化铬纳米颗粒在水相中的再分散。
83.对比例1
84.在于场景例1相同的实验条件下,在烧杯中搅拌进行反应沉淀,得到尺寸分布在 30~150nm的晶态氢氧化铬。
85.说明书附图中图13为对比例1产物的tem图,可观察到在烧杯中反应沉淀得到的氢 氧化铬形貌为六棱柱,尺寸分布在30~150nm。图14为对比例1产物的x射线粉末衍射图 案,表明在烧杯中反应沉淀得到的氢氧化铬结晶度高,与标准卡片完全吻合,产物为晶态 氢氧化铬。图15为对比例1制得的晶态氢氧化铬水相分散体静置5个月后的照片,图中可 观察到明显的分层现象,表明该颗粒尺寸更大的晶态氢氧化铬水相分散体与超重力反应沉 淀得到的非晶态超小氢氧化铬水相分散体相比稳定性更差。
86.场景例1与对比例1的结果对比可知:
87.(1)通过超重力耦合超声波场强化氢氧化铬的成核与生长过程,能够制备出粒径超 小(平均粒径7nm),粒径分布范围更窄(粒径分布在4
‑
11nm)的非晶态氢氧化铬;
88.(2)经表面活性剂改性后,使用超声波耦合超重力场进行对纳米氢氧化铬进行再分 散的效果更好,zeta电位的绝对值更大,在水相中的分散更稳定。
89.将场景例1与对比例1所制得的氢氧化铬分散体用于催化高氯酸铵热分解。首先制备 高氯酸铵/氢氧化铬复合物,具体步骤如下:将1.6g高氯酸铵溶解于5ml固含量为13mg/ml 的实施例1或对比例1制备的氢氧化铬分散体中,加热至50oc使之完全溶解。再在搅拌 的条件下将溶解了高氯酸铵的氢氧化铬分散体滴加到10ml的正丁醇中,使氢氧化铬颗粒随 着高氯酸铵重结晶析出,将沉淀物抽滤、烘干即可得到高氯酸铵/氢氧化铬复合物。通过同 步热分析仪测试复合物的热分解特性,测试条件为:升温速率为10oc/min,气氛为氩气, 气
流量为60ml/min。测试结果见图14和图15。从图14和图15中分析可知,高氯酸铵复合 实施例1中制备的氢氧化铬分散体后,高氯酸铵分解峰温为323℃,分解终点温度为384℃。 而高氯酸铵复合了对比例1中制备的氢氧化铬分散体后,高氯酸铵分解峰温为387℃,分解 终点温度为401℃。更低的分解峰温和分解终点温度表明,复合了实施例1中制备的氢氧化 铬分散体的高氯酸铵更容易分解,催化活性更高。
90.可以看出,本发明具有如下效果:
91.1.本发明通过超重力耦合超声场进行反应沉淀结合表面改性的方法制备出能在水中长 期稳定分散的非晶态超小氢氧化铬水相分散体(小于10nm)。通过超重力耦合超声波场进 行反应沉淀得到的非晶态氢氧化铬经过表面活性剂改性再分散后,分散体的zeta电位的绝 对值高,能够在水相中保持高稳定性,静置10个月内未发生沉降。
92.2.本发明制备的非晶态氢氧化铬颗粒尺寸小,粒径分布范围窄。产品的透射电子显微镜 图可以看出,在优选条件下氢氧化铬的颗粒直径分布在4
‑
11nm范围内,平均粒径7nm。
93.3.本发明制备非晶态氢氧化铬水相分散体工艺流程简单,由于超重力场能够极大强化氢 氧化铬成核过程的混合效率,制备过程无明显放大效应,易于规模化放大生产。
94.本说明书中的各个实施方式均采用递进的方式描述,各个实施方式之间相同相似的部 分互相参见即可,每个实施方式重点说明的都是与其他实施方式的不同之处。尤其,对于 系统实施方式而言,由于其基本相似于方法实施方式,所以描述的比较简单,相关之处参 见方法实施方式的部分说明即可。
95.在本说明书的描述中,参考术语“一个实施方式”、“一些实施方式”、“示例”、“具体 示例”、或“一些示例”等的描述意指结合该实施方式或示例描述的具体特征、结构、材料 或者特点包含于本说明书实施方式的至少一个实施方式或示例中。在本说明书中,对上述 术语的示意性表述不必须针对的是相同的实施方式或示例。
96.此外,在不相互矛盾的情况下,本领域的技术人员可以将本说明书中描述的不同实施 方式或示例以及不同实施方式或示例的特征进行结合和组合。以上所述仅为本说明书实施 方式的实施方式而已,并不用于限制本说明书实施方式。对于本领域技术人员来说,本说 明书实施方式可以有各种更改和变化。凡在本说明书实施方式的精神和原理之内所作的任 何修改、等同替换、改进等,均应包含在本说明书实施方式的权利要求范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。