本发明涉及荧光染料领域,尤其涉及一种氟硼二吡咯荧光染料及其制备方法和应用。
背景技术:
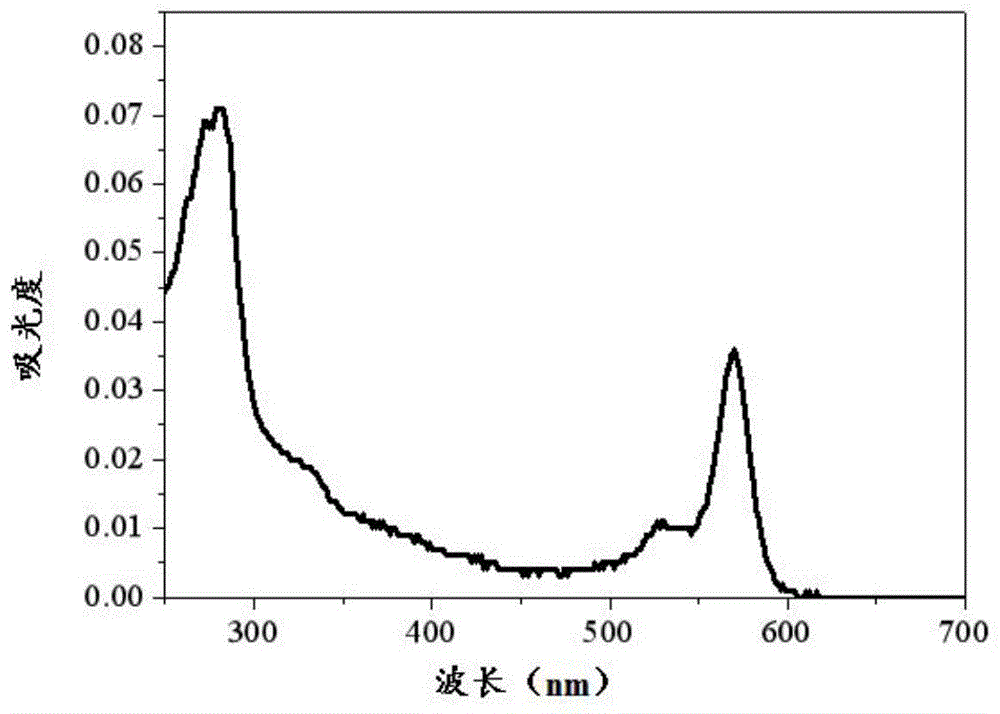
:荧光染料已经存在了近一个世纪的时间,吸引了多学科领域科学家的关注。荧光染料作为功能性材料在很多领域都有应用,例如光能转化、有机发光器件、生物成像和诊断治疗等,并且取得了长足的发展。现在已经发现了很多种高荧光的有机染料,氟硼二吡咯类荧光染料作为众多染料中的一种,具有较高的荧光量子产率和摩尔消光系数,同时还具有较强的光稳定性和化学稳定性,受到了众多科研工作者的关注。但是现有的氟硼二吡咯类荧光染料的红移较小。如现有技术“photophysicalpropertiesofborondipyrrometheneanaloguesinsolution”(j.phy.chem.a2005,109,7371-7384)在bodipy母核的8号位引入苯环,3号和5号位引入甲基,所得氟硼二吡咯衍生物在520nm处有较强的吸收峰,在525nm处有较强的发射峰,使其在绿光光敏剂和绿光荧光探针、有机发光二极管的发光层中的应用受限。技术实现要素:本发明的目的在于提供一种氟硼二吡咯荧光染料及其制备方法和应用,本发明所提供的氟硼二吡咯荧光染料具有较大的红移,在绿光范围内有着较强的吸收峰,在荧光发射光谱中在绿光范围内有着较强的发射峰。为了实现上述发明目的,本发明提供以下技术方案:本发明提供了一种氟硼二吡咯荧光染料,具有式ⅰ所示结构:本发明还提供了上述技术方案所述的氟硼二吡咯荧光染料的制备方法,包括如下步骤:将吡咯、对甲苯甲醛和三氟乙酸混合,在保护气氛下进行缩合反应,得到式ⅱ所示的中间体;在保护气氛下,将式ⅱ所示中间体的四氢呋喃溶液和n-溴代丁二酰亚胺在-80~-76℃混合,待n-溴代丁二酰亚胺溶解后,加入2,3-二氯-5,6-二氰基-1,4-苯醌的四氢呋喃溶液,然后在室温下进行溴化反应,得到式ⅲ所示的中间体;将所述式ⅲ所示的中间体和三乙胺、三氟化硼乙醚、二氯甲烷混合,进行氟硼化反应,得到式ⅳ所示的中间体;将式ⅳ所示的中间体与烯丙基硼酸频哪醇酯、碳酸钾、四三苯基膦钯、乙醇和甲苯混合,进行suzuki偶联反应,得到式ⅰ所示的氟硼二吡咯荧光染料;优选地,所述吡咯和对甲苯甲醛的摩尔比为18~22:1;所述对甲苯甲醛与三氟乙酸的摩尔比为1:0.08~0.12;所述缩合反应的时间为12~16h;所述缩合反应的温度为室温。优选地,所述缩合反应完成后,将所述缩合反应所得反应液与二氯甲烷、氢氧化钠溶液混合进行第一萃取,将所得的有机相依次经洗涤和干燥后,蒸馏去除吡咯,然后将所得残余物进行第一柱层析;所述二氯甲烷和氢氧化钠溶液的体积比为1.8~2.2:1;所述第一柱层析用洗脱剂为石油醚、乙酸乙酯和三乙胺按照体积比为8:2:1的比例混合得到的混合液。优选地,所述式ⅱ所示的中间体与n-溴代丁二酰亚胺的摩尔比为1:2~2.2;所述式ⅱ所示的中间体与2,3-二氯-5,6-二氰基-1,4-苯醌的摩尔比为1:1~1.2;所述溴化反应的时间为1.8~2.2h。优选地,所述式ⅱ所示的中间体与三乙胺的摩尔比为1:3.8~4.0;所述式ⅱ所示的中间体与三氟化硼乙醚的摩尔比为1:5~5.4;所述氟硼化反应的温度为室温,时间为1.8~2.2h。优选地,所述氟硼化反应完成后,将所述氟硼化反应所得反应液与二氯甲烷、水混合进行第二萃取,将所得的有机相依次经洗涤和干燥后,蒸馏去除溶剂,将所得残余物进行第二柱层析;所述二氯甲烷和水的体积比为1.8~2.2:1;所述第二柱层析用洗脱剂为石油醚和二氯甲烷按照体积比为3:1的比例混合得到的混合液。优选地,所述式ⅳ所示的中间体与烯丙基硼酸频哪醇酯的摩尔比为1:1.4~1.6;所述式ⅳ所示的中间体与碳酸钾的摩尔比为1:5.8~6.2;所述式ⅳ所示的中间体与四三苯基膦钯的摩尔比为1:0.06~0.08;所述乙醇和甲苯的体积比为1:4.8~5.2;所述suzuki偶联反应的温度为95~105℃,时间为10~14h。优选地,所述suzuki偶联反应完成后,将所述suzuki偶联反应所得反应液与二氯甲烷、水混合进行第三萃取,将所得的有机相依次经洗涤和干燥后,蒸馏去除二氯甲烷,将所得残余物进行第三柱层析;所述二氯甲烷和水的体积比为1.8~2.2:1;所述第三柱层析用洗脱剂为石油醚和二氯甲烷按照体积比为4:1的比例混合得到的混合液。本发明还提供了上述技术方案所述的氟硼二吡咯荧光染料在制备光敏剂或荧光探针或有机发光二极管的发光层中的应用。本发明提供了一种氟硼二吡咯荧光染料,具有式ⅰ所示结构。本发明以氟硼二吡咯(缩写为bodipy)为中心,在其8号位上引入甲苯,提高化合物的共轭程度,3号和5号位上引入丙烯,提高了吸电子能力,使所得氟硼二吡咯荧光染料的紫外吸收光谱和荧光发射光谱产生较大红移,其在紫外吸收光谱中在绿光范围内有着较强的吸收峰使其可以作为绿光光敏剂,在荧光发射光谱中在绿光范围内有着较强的发射峰因此其在绿光荧光探针和绿光有机发光二极管的发光层有着潜在的应用。此外,本发明所提供的bodipy-ak在不同溶剂中均有较高的量子产率。附图说明图1实施例1所得bodipy-ak的紫外-可见吸收光谱图;图2实施例1所得bodipy-ak的荧光法发射光谱图;图3实施例1所得bodipy-ak在不同溶剂中的归一化荧光发射光谱图。具体实施方式本发明提供了一种氟硼二吡咯荧光染料,具有式ⅰ所示结构:本发明还提供了上述技术方案所述氟硼二吡咯荧光染料的制备方法,包括如下步骤:将吡咯、对甲苯甲醛和三氟乙酸混合,在保护气氛下进行缩合反应,得到式ⅱ所示的中间体;在保护气氛下,将式ⅱ所示的中间体的四氢呋喃溶液和n-溴代丁二酰亚胺在-80~-76℃混合,待n-溴代丁二酰亚胺溶解后,加入2,3-二氯-5,6-二氰基-1,4-苯醌的四氢呋喃溶液,然后在室温下进行溴化反应,得到式ⅲ所示的中间体;将所述式ⅲ所示的中间体和三乙胺、三氟化硼乙醚、二氯甲烷混合,进行氟硼化反应,得到式ⅳ所示的中间体;将式ⅳ所示的中间体与烯丙基硼酸频哪醇酯、碳酸钾、四三苯基膦钯、乙醇和甲苯混合,进行suzuki偶联反应,得到式ⅰ所示的氟硼二吡咯荧光染料;在本发明中,如无特殊说明,所用有机溶剂均为经干燥的有机溶剂。本发明将吡咯、对甲苯甲醛和三氟乙酸混合,在保护气氛下进行缩合反应,得到式ⅱ所示的中间体。在本发明中,所述吡咯、对甲苯甲醛和三氟乙酸的混合优选为将吡咯和对甲苯甲醛混合,在保护气氛中搅拌均匀,然后加入三氟乙酸。在本发明中,如无特殊说明,所述保护气氛均优选为氮气或惰性气体氛围。在本发明中,所述吡咯和对甲苯甲醛的摩尔比优选为18~22:1,更优选为20:1;所述对甲苯甲醛与三氟乙酸的摩尔比优选为1:0.08~0.12,更优选为1:0.1;所述吡咯和对甲苯甲醛混合后搅拌的间优选为13~17min,更优选为15min;所述缩合反应的时间优选为12~16h;所述缩合反应的温度优选为室温,即不需要额外的加热或冷却。缩合反应完成后,本发明优选将所述缩合反应所得反应液与二氯甲烷、氢氧化钠溶液混合进行第一萃取,将所得的有机相依次经洗涤和干燥后,蒸馏去除吡咯,然后将所得残余物进行第一柱层析;所述二氯甲烷和氢氧化钠溶液的体积比优选为1.8~2.2:1,更优选为2:1,所述氢氧化钠溶液的浓度优选为0.1m,所述氢氧化钠溶液的作用为中和反应中的三氟乙酸,本发明对所述第一萃取用二氯甲烷和氢氧化钠溶液的用量没有特殊限定,在本发明实施例中,所述对甲苯甲醛和第一萃取用二氯甲烷、氢氧化钠溶液的用量比为20mmol:100ml:50ml;本发明对第一萃取所得有机相的洗涤和干燥的方法没有特殊限定,采用常规的有机相洗涤方法(如水洗)和干燥方法(如干燥剂干燥)即可;所述第一柱层析用洗脱剂优选为石油醚、乙酸乙酯和三乙胺按照体积比为8:2:1的比例混合得到的混合液。第一柱层析完成后,本发明优选将所得含有式ⅱ所示的中间体的流分进行真空干燥,得到式ⅱ所示的中间体。本发明对所述真空干燥的具体参数没有特殊限定,能够得到恒重的产品即可。得到式ⅱ所示的中间体后,本发明在保护气氛下,将式ⅱ所示的中间体的四氢呋喃溶液和n-溴代丁二酰亚胺在-80~-76℃混合,待n-溴代丁二酰亚胺溶解后,加入2,3-二氯-5,6-二氰基-1,4-苯醌的四氢呋喃溶液,然后在室温下进行溴化反应,得到式ⅲ所示的中间体。在本发明中,所述溴化反应的产物不稳定性较强,因此,在低温进行溴化反应。在本发明中,所述式ⅱ所示的中间体与n-溴代丁二酰亚胺的摩尔比优选为1:2~2.2,更优选为1:2.1;所述式ⅱ所示的中间体与2,3-二氯-5,6-二氰基-1,4-苯醌的摩尔比优选为1:1~1.2;所述溴化反应的时间优选为1.8~2.2h,更优选为2h。在本发明中,所述式ⅱ所示的中间体的四氢呋喃溶液和n-溴代丁二酰亚胺在-80~-76℃混合优选为将所述n-溴代丁二酰亚胺优选分批次加入至式ⅱ所示的中间体的四氢呋喃溶液中,更优选为在1h内分两次加入至式ⅱ所示的中间体的四氢呋喃溶液。本发明对所述式ⅱ所示的中间体的四氢呋喃溶液和2,3-二氯-5,6-二氰基-1,4-苯醌的四氢呋喃溶液的浓度没有特殊限定,能够保证反应顺利进行即可,在本发明实施例中,所述式ⅱ所示的中间体的四氢呋喃溶液的浓度优选为0.0564mmol/ml,所述2,3-二氯-5,6-二氰基-1,4-苯醌的四氢呋喃溶液的浓度优选为0.215mmol/ml。溴化反应完成后,本发明优选将所得反应液中的四氢呋喃去除,得到式ⅲ所示的中间体。本发明对所述四氢呋喃的去除方法没有特殊限定,在本发明实施例中,优选采用真空蒸馏的方法去除四氢呋喃。得到式ⅲ所示的中间体后,本发明将所述式ⅲ所示的中间体和三乙胺、三氟化硼乙醚、二氯甲烷混合,进行氟硼化反应,得到式ⅳ所示的中间体。在本发明中,所述三乙胺为缚酸剂,可促进氟硼化反应进行。在本发明中,所述式ⅱ所示的中间体与三乙胺的摩尔比优选为1:3.8~4.0,更优选为1:3.9;所述式ⅱ所示的中间体与三氟化硼乙醚的摩尔比为1:5~5.4;所述氟硼化反应的温度优选为室温,时间优选为1.8~2.2h。本发明对氟硼化反应所用二氯甲烷的量没有特殊限定,能够保证反应顺利进行即可,在本发明实施例中,所述三氟化硼乙醚和二氯甲烷的用量比优选为22.1mmol:20ml。氟硼化反应完成后,本发明优选将所述氟硼化反应所得反应液与二氯甲烷、水混合进行第二萃取,将所得的有机相依次经洗涤和干燥后,蒸馏去除溶剂,将所得残余物进行第二柱层析;所述二氯甲烷和水的体积比为1.8~2.2:1,更优选为2:1,本发明对所述第二萃取用二氯甲烷和水的用量没有特殊限定,在本发明实施例中,所述式ⅱ所示的中间体与第二萃取用二氯甲烷、水的用量比优选为4.23mmol:100ml:50ml;本发明对第二萃取所得有机相的洗涤和干燥的方法没有特殊限定,采用常规的有机相洗涤方法(如水洗)和干燥方法(如干燥剂干燥)即可;所述第二柱层析用洗脱剂优选为石油醚和二氯甲烷按照体积比为3:1的比例混合得到的混合液。第二柱层析完成后,本发明优选将所得含有式ⅳ所示的中间体的流分去除溶剂,得到式ⅳ所示的中间体。本发明对所述去除溶剂的具体方法没有特殊限定,能够得到恒重的产品即可,在本发明实施例中,所述去除溶剂的方法优选为减压蒸馏,所述减压蒸馏的温度优选为40℃。得到式ⅳ所示的中间体后,本发明将式ⅳ所示的中间体与烯丙基硼酸频哪醇酯、碳酸钾、四三苯基膦钯、乙醇和甲苯混合,进行suzuki偶联反应,得到式ⅰ所示的氟硼二吡咯荧光染料。在本发明中,所述碳酸钾为反应提供碱性环境,四三苯基膦钯作为催化剂使用,乙醇和甲苯作为混合溶剂,为反应提供合适的极性环境。在本发明中,所述式ⅳ所示的中间体与烯丙基硼酸频哪醇酯的摩尔比优选为1:1.4~1.6,更优选为1:1.5;所述式ⅳ所示的中间体与碳酸钾的摩尔比优选为1:5.8~6.2,更优选为1:6;所述式ⅳ所示的中间体与四三苯基膦钯的摩尔比优选为1:0.06~0.08,更优选为1:0.07;所述乙醇和甲苯的体积比优选为1:4.8~5.2,更优选为1:5,本发明对所述乙醇和甲苯的总用量没有特殊限定,能够保证反应顺利进行即可,在本发明实施例中,所述乙醇和甲苯的总用量与式ⅳ所示的中间体的用量比为20ml:0.68mmol;所述suzuki偶联反应的温度优选为95~105℃,更优选为100℃,时间优选为10~14h,更优选为12h,所述suzuki偶联反应优选在保护气氛下进行。suzuki偶联反应完成后,本发明优选将所述suzuki偶联反应所得反应液与二氯甲烷、水混合进行第三萃取,将所得的有机相依次经洗涤和干燥后,蒸馏去除二氯甲烷,将所得残余物进行第三柱层析;所述第三萃取中,所述二氯甲烷和水的体积比优选为1.8~2.2:1,更优选为2:1,本发明对所述第三萃取用二氯甲烷和水的用量没有特殊限定,在本发明实施例中,所述式ⅳ所示的中间体与第三萃取用二氯甲烷、水的用量比优选为0.68mmol:100ml:50ml,本发明对所述第三萃取所得有机相的洗涤和干燥的方法没有特殊限定,采用常规的有机相洗涤方法(如水洗、饱和食盐水洗涤)和干燥方法(如干燥剂干燥)即可;所述第三柱层析用洗脱剂为石油醚和二氯甲烷按照体积比为4:1的比例混合得到的混合液。第三柱层析完成后,本发明优选将所得含有式ⅰ所示的氟硼二吡咯荧光染料的流分进行真空干燥,得到式ⅰ所示的氟硼二吡咯荧光染料。本发明对所述真空干燥的具体参数没有特殊限定,能够得到恒重的产品即可。本发明还提供了上述技术方案所述的氟硼二吡咯荧光染料在制备光敏剂或荧光探针或有机发光二极管的发光层中的应用。下面结合实施例对本发明提供的一种氟硼二吡咯荧光染料及其制备方法和应用进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。实施例1(1)式ⅱ所示的中间体的制备将吡咯(27.8ml,400mmol)与对甲苯甲醛(2.36ml,20mmol)混合,在氮气氛围下搅拌反应15min,然后加入三氟乙酸(0.15ml,2mmol),搅拌反应14h,然后将所得反应液与50ml浓度为0.1m的氢氧化钠溶液、100ml二氯甲烷混合,进行第一萃取,将所得有机相用水洗涤两次,每次用水量为50ml,然后依次进行饱和食盐水洗涤和无水硫酸镁干燥,再进行真空蒸馏除去未反应的吡咯,将所得残余物进行第一柱层析,所用洗脱剂为石油醚、乙酸乙酯和三乙胺按照体积比为8:2:1的比例混合得到的混合液,将所得含有目标产物的流分进行真空干燥,得到式ⅱ所示的中间体,为红褐色固体,经计算产率为85%;将该产物进行核磁表征,结果如下:1hnmr(chloroform-d)δ7.92(br,2h),7.12(s,4h),6.69(m,2h),6.16(m,2h),5.93(s,2h),2.35(s,3h);由上述表征数据可知,本步骤所得产物具有式ⅱ所示结构;(2)式ⅳ所示的中间体的制备将式ⅱ所示的中间体(1.0g,4.23mmol)与75ml四氢呋喃混合,在氮气下冷却至-78℃,然后在1小时内将n-溴代丁二酰亚胺(1.6g,9.0mmol)分两次加入反应液中,待n-溴代丁二酰亚胺完全溶解后,用恒压滴液漏斗逐滴加入20ml浓度为0.215mmol/ml的2,3-二氯-5,6-二氰基-1,4-苯醌的四氢呋喃溶液,然后将所得反应液在室温下搅拌反应2h;反应完成后,进行真空蒸馏除去四氢呋喃,得到式ⅲ所示的中间体;将三乙胺(2.14g,16.57mmol)、三氟化硼乙醚(3.14g,22.1mmol)、二氯甲烷(20ml)和式ⅲ所示的中间体混合,室温下搅拌反应2h;然后将所得反应液与100ml二氯甲烷、50ml水混合,进行第二萃取;将第二萃取所得有机相用水洗涤两次,每次用水量为50ml,然后依次进行盐水洗涤和无水硫酸镁干燥,再进行真空蒸馏除去二氯甲烷,将所得残余物进行第二柱层析,第二柱层析所用洗脱剂为石油醚和二氯甲烷按照体积比为3:1的比例混合得到的混合液,将所得含有目标产物的溶液在40℃下减压蒸除溶剂,得到式ⅳ所示的中间体,为红色固体,产率80%;将本步骤所得产物进行核磁表征,结果如下:1hnmr(400mhz,cdcl3)δ7.39(d,j=8.1hz,2h),7.32(d,j=7.9hz,2h),6.81(d,j=4.0hz,2h),6.53(d,j=4.3hz,2h),2.47(s,3h);由上述表征数据分析可知,本步骤所得产物具有式ⅳ所示结构;(3)式ⅰ所示的氟硼二吡咯荧光染料的制备将式ⅳ所示的中间体(0.30g,0.68mmol)、烯丙基硼酸频哪醇酯(0.17g,1.03mmol)、碳酸钾(0.56g,4.08mmol)、四三苯基膦钯(0.056mg,0.05mmol)、乙醇(4ml)和甲苯(20ml)混合,在氮气氛围下加热到100℃搅拌反应12h;反应完成后冷却到室温,然后将所得反应液与100ml二氯甲烷、水(50ml)混合进行过第三萃取;将第三萃取所得有机相用水洗涤两次,每次洗涤所用水的量为50ml,然后将所得有机相依次进行盐水洗涤和无水硫酸镁干燥,然后进行真空蒸馏除去二氯甲烷,将所得残余物进行第三柱层析,第三柱层析所用洗脱剂为石油醚和二氯甲烷按照体积比为4:1的比例混合得到的混合液,然后将所得含有目标产物的流分进行真空干燥,得到黑色固体,经计算产率65%;将步骤(3)所得黑色固体进行核磁表征,结果如下:1hnmr(400mhz,cdcl3)δ7.38(d,j=5.4hz2h),7.28(d,j=6.6hz,2h),7.06(d,j=16.4hz,2h),6.75(d,j=4.4hz,2h),6.69(s,2h),6.57(m,2h),2.45(s,3h),2.03(d,j=1.7hz,3h),2.01(d,j=1.7hz,3h);对上述数据分析可知,所得黑色固体为式ⅰ所示的氟硼二吡咯荧光染料,记为bodipy-ak。紫外-可见吸收光谱的测试:在室温条件下,使用agilent8453型紫外可见光光度计,在200~800nm波段进行紫外吸收光谱扫描,测量bodipy-ak在四氢呋喃溶液中的紫外-可见吸收光谱,测试结果如图1所示。由图1可知bodipy-ak在大约250~375nm和500~600nm处出现强的吸收峰。荧光发射光谱测试:在室温条件下,使用spexfluorolog-3fluorometer/phosphorometer荧光仪,在333nm波长下进行激发,获得荧光发射光谱。结果如图2所示。由图2可知,bodipy-ak在大约550~700nm处出现了较大的发射峰。溶剂化效应与量子产率测试:在室温条件下,使用spexfluorolog-3fluorometer/phosphorometer荧光仪,分别以正己烷、甲苯、四氢呋喃、二氯甲烷和乙腈为溶剂在530nm波长进行激发,获得不同溶剂下荧光发射光谱。结果如图3所示。以罗丹明6g为标准样品,获得在不同溶剂中bodipy-ak的相对量子产率(λem/nm(фema),其中λem/nm为发射波长,фema为量子产量),结果如表1所示。由图3可知,bodipy-ak在不同溶剂中都具有较强的荧光发射,没有观察到明显的溶剂化显色效应,说明其激发态主要可归属为π-π*跃迁。由表1可知,本发明所提供的bodipy-ak在不同溶剂中均有较高的量子产率。表1不同溶剂中bodipy-ak的相对量子产率溶剂正己烷甲苯四氢呋喃二氯甲烷乙腈λem/nm(фema)580(0.92)587(0.64)584(0.85)584(0.53)581(0.69)以上所述仅是本发明的优选实施方式,应当指出,对于本
技术领域:
的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。当前第1页12
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。