1.本发明关于中药分析技术领域,尤其涉及一种银杏蜜环口服溶液多成分含量检测及其指纹图谱方法的构建。
背景技术:
2.银杏蜜环口服溶液是由银杏叶提取物和天麻蜜环菌粉运用现代制剂工艺制成的浅棕红色至棕红色澄清液体,味甜微苦,收载于国家药品西药标准(化学药品地标升国标第十六册,标准编号:ws1
‑
xg
‑
004
‑
2001)。该口服液具有降血脂、降血压、改善心脑缺血的作用,临床广泛应用于治疗冠心病、心绞痛和缺血性脑血管疾病。
3.银杏蜜环口服溶液临床疗效显著,但质量检测方法尚不完善。国家药品西药标准中含量检测方面仅规定总黄酮醇苷不得少于0.2mg/ml,现有技术中还记载了采用高效液相色谱法检测腺苷的含量作为银杏蜜环复方制剂的质量检测方法。(刘翠翠,银杏蜜环口服溶液中多糖的质量标准研究,成都大学,2021
‑
05
‑
01)该文章采用改良硫酸
–
苯酚法进行多糖的显色反应,采用酶标仪测定银杏蜜环口服溶液中多糖含量,为银杏蜜环口服溶液标准提高提供了参考。但目前现有标准缺乏对银杏蜜环口服溶液中多成分同时检测的研究,不能综合反映出该口服制剂的产品质量,难以满足生产中检测大量样品的需求。
4.但银杏蜜环口服溶液所含有化学成分的复杂性及上述现有技术检测方法的局限性,不能全面的评价银杏蜜环口服溶液整体内在质量。鉴于指纹图谱技术能够整体反映中药制剂多指标成分的化学特征,能提供丰富药物成分的鉴别信息,为了更好地对银杏蜜环口服溶液质量进行控制,保证临床疗效,建立其多指标成分检测及特征指纹图谱分析方法显得尤为必要。
技术实现要素:
5.针对以上技术问题,本发明提供一种银杏蜜环口服溶液多成分含量检测及其指纹图谱方法的构建,本发明建立的含量检测分析方法其精密度、重现性、稳定性、加样回收率良好。由此可知,该含量检测方法操作简便,准确可靠,可用于银杏蜜环口服溶液含量测定。
6.本发明指纹图谱检测方法,能同时检测银杏蜜环口服溶液中共有19个特征指纹峰,通过液质联用技术及对照品比对鉴定了其中15个指纹峰,分别为尿苷、腺苷、鸟苷、安赛蜜、5
‑
羟甲基糠醛、原儿茶酸、对羟基苯甲酸、苯甲酸钠、芦丁和异槲皮苷、槲皮素
‑3‑
o
‑
(2"
‑
β
‑
d
‑
葡萄糖基]
‑
α
‑
l
‑
鼠李糖苷、山柰酚
‑3‑
o
‑
芸香糖苷、异鼠李素
‑3‑
o
‑
芸香糖苷、槲皮素
‑3‑
o
‑
[2
”‑
(6”'
‑
p
‑
香豆酰基)
‑
β
‑
d
‑
葡萄糖基]
‑
α
‑
l
‑
鼠李糖苷、山柰酚
‑3‑
o
‑
(2"
‑
β
‑
d
‑
葡萄糖基)
‑
α
‑
l
‑
鼠李糖苷,山柰酚
‑3‑
o
‑
[2
”‑
(6”'
‑
p
‑
香豆酰基)
‑
β
‑
d
‑
葡萄糖基]
‑
α
‑
l
‑
鼠李糖苷。本发明所建立的检测方法具有全面、有效、高效特点,该检测方法可用于银杏蜜环口服溶液的内在质量检测。
[0007]
为达到上述发明目的,本发明提供了如下技术方案:
[0008]
一种银杏蜜环口服溶液多成分检测方法,所述检测方法包括如下步骤:
[0009]
⑴
、供试品溶液的制备:精密量取银杏蜜环口服溶液,置于量瓶中,用5~12%甲醇水溶液定容至刻度线,摇匀,取适量进行离心,其离心转速为10000~14000rpm,离心时间为8~12min,取上清液即得;
⑵
、混合对照品溶液的制备:分别取称定安赛蜜、5
‑
羟甲基糠醛、原儿茶酸、对羟基苯甲酸、苯甲酸钠,使用5~15%甲醇水溶液稀释成对照品储备液;再称定尿苷、腺苷、鸟苷、山柰酚
‑3‑
o
‑
芸香糖苷、异鼠李素
‑3‑
o
‑
芸香糖苷和槲皮素
‑3‑
o
‑
[2
”‑
(6”'
‑
p
‑
香豆酰基)
‑
β
‑
d
‑
葡萄糖基]
‑
α
‑
l
‑
鼠李糖苷,并使用30~50%甲醇水溶液及少量二甲基亚砜助溶稀释制备而成;
⑶
、色谱条件:色谱柱为十八烷基键合硅胶色谱柱;所述检测波长:252~256nm,检测流速:0.25~0.35ml/min,检测柱温:48~52℃,流动相:流动相a为0.1%~0.3甲酸水溶液,流动相b为甲醇;采用下述梯度洗脱程序为:
[0010]
0~3min,流动相a的体积百分比由94%匀速降至86%,流动相b的体积百分比由6%匀速上升至14%;3~6min流动相a的体积百分比由86%匀速降至77%,流动相b的体积百分比由14%匀速上升至23%;6~8.3min,流动相a的体积百分比由77%匀速降至69.5%,流动相b的体积百分比由23%匀速上升至30.5%;8.3~10.3min流动相a的体积百分比由69.5%匀速降至66.5%,流动相b的体积百分比由30.5%匀速上升至33.5%;10.3~12.5min,流动相a的体积百分比由66.5%匀速降至58.5%,流动相b的体积百分比由33.5%匀速上升至41.5%;12.5~19min流动相a的体积百分比由58.5%匀速降至26%;流动相b的体积百分比由41.5%匀速上升至74%;
[0011]
⑷
、色谱峰的检测:对上述步骤
⑴
供试品溶液和步骤
⑵
对照品溶液,按照上述步骤
⑶
色谱条件,采用超高效液相色谱法检测色谱峰面积,采用外标法计算本发明药物中对应的多成分的含量。优选的,所述检测方法步骤
⑴
甲醇水溶液的浓度为10%。
[0012]
优选的,所述检测方法步骤
⑴
离心转速为12700rpm,离心时间为10min。
[0013]
优选的,所述步骤
⑵
混合对照品中尿苷浓度为0.7688~49.20μg/ml、腺苷浓度为0.3712~23.76μg/ml、鸟苷浓度为0.2944~18.84μg/ml、安赛蜜浓度为2.400~153.6μg/ml、5
‑
羟甲基糠醛浓度为0.1024~6.552μg/ml、原儿茶酸浓度为0.2680~17.15μg/ml、对羟基苯甲酸浓度为0.09656~6.180μg/ml、苯甲酸钠浓度为28.30~1811μg/ml、山柰酚
‑3‑
o
‑
芸香糖苷浓度为0.3717~23.79μg/ml、异鼠李素
‑3‑
o
‑
芸香糖苷浓度为0.3698~23.67μg/ml、槲皮素
‑3‑
o
‑
[2
”‑
(6”'
‑
p
‑
香豆酰基)
‑
β
‑
d
‑
葡萄糖基]
‑
α
‑
l
‑
鼠李糖苷浓度为0.6102~39.05μg/ml。
[0014]
优选的,所述步骤
⑵
混合对照品中尿苷浓度为12.30μg/ml、腺苷浓度为5.940μg/ml、鸟苷浓度为4.710μg/ml、安赛蜜浓度为38.40μg/ml、5
‑
羟甲基糠醛浓度为1.638μg/ml、原儿茶酸浓度为4.288μg/ml、对羟基苯甲酸浓度为1.545μg/ml、苯甲酸钠浓度为452.8μg/ml、山柰酚
‑3‑
o
‑
芸香糖苷浓度为5.948μg/ml、异鼠李素
‑3‑
o
‑
芸香糖苷浓度为5.918μg/ml、槲皮素
‑3‑
o
‑
[2
”‑
(6”'
‑
p
‑
香豆酰基)
‑
β
‑
d
‑
葡萄糖基]
‑
α
‑
l
‑
鼠李糖苷浓度为9.763μg/ml。
[0015]
优选的,所述检测方法步骤
⑶
检测波长:254nm,检测流速:0.3ml/min,检测柱温:50℃。
[0016]
优选的,所述色谱柱型号为:acquityhss t3。
[0017]
优选的,所述检测方法为指纹图谱检测方法,其包括以下步骤:
[0018]
⑴
、采用超高效液相色谱法对多批银杏蜜环口服溶液供试品溶液和对照品溶液进
行检测,并记录指纹图谱;
[0019]
⑵
、再将所述步骤
⑴
记录的指纹图谱全部导入中药色谱指纹图谱相似度评价系统软件,并选择不同批次银杏蜜环口服溶液的指纹图谱中的共有特征色谱峰作为特征指纹峰,生成标准对照指纹图谱,计算各共有特征指纹峰的相对保留时间、相对峰面积。
[0020]
优选的,根据所述指纹图谱检测方法生成指纹图谱包括1个特征指纹峰,通过液质联用技术及对照品比对鉴定了其中15个指纹峰,分别为:1号峰为尿苷、2号峰为腺苷、3号峰为鸟苷、4号峰为安赛蜜、5号峰为5
‑
羟甲基糠醛、7号峰为原儿茶酸、10号峰为对羟基苯甲酸、12号峰为苯甲酸钠、13号峰为芦丁和异槲皮苷、14号峰为槲皮素
‑3‑
o
‑
(2"
‑
β
‑
d
‑
葡萄糖基]
‑
α
‑
l
‑
鼠李糖苷、15号峰为山柰酚
‑3‑
o
‑
芸香糖苷、16号峰为异鼠李素
‑3‑
o
‑
芸香糖苷、17号峰为槲皮素
‑3‑
o
‑
[2
”‑
(6”'
‑
p
‑
香豆酰基)
‑
β
‑
d
‑
葡萄糖基]
‑
α
‑
l
‑
鼠李糖苷、18号峰为山柰酚
‑3‑
o
‑
(2"
‑
β
‑
d
‑
葡萄糖基)
‑
α
‑
l
‑
鼠李糖苷,19号峰为山柰酚
‑3‑
o
‑
[2
”‑
(6”'
‑
p
‑
香豆酰基)
‑
β
‑
d
‑
葡萄糖基]
‑
α
‑
l
‑
鼠李糖苷。
[0021]
本发明采用的技术方案有益效果如下:
[0022]
⑴
、本发明建立一种银杏蜜环口服溶液中多成分含量检测方法,该检测方法各化合物日内精密度试验rsd值均小于2.5%,日间精密度试验rsd值均小于2.6%,表明该定量分析方法精密度良好;重现性试验结果表明,各化合物含量检测的rsd值均小于1.%;稳定性试验结果表明,各化合物的rsd值均小于2.6%;加样回收率试验结果表明,6份样品中被测化合物的加样回收率在96.07%
–
104.8%,rsd值均小于2.%,表明各化合物的加样回收率良好。由此可知,本发明构建的银杏蜜环口服溶液多成分定量检测分析方法其精密度、重现性、稳定性、加样回收率良好。由此,该含量检测方法操作简便,准确可靠,可用于银杏蜜环口服溶液含量的测定。
[0023]
⑵
、本发明还建立银杏蜜环口服溶液的指纹图谱检测方法,同时检测银杏蜜环口服溶液中的共有19个特征指纹峰,通过液质联用技术及对照品比对鉴定了其中15个指纹峰,分别为:尿苷(1号峰)、腺苷(2号峰)、鸟苷(3号峰)、安赛蜜(4号峰)、5
‑
羟甲基糠醛(5号峰)、原儿茶酸(7号峰)、对羟基苯甲酸(10号峰)、苯甲酸钠(12号峰)、芦丁 异槲皮苷(13号峰)、槲皮素
‑3‑
o
‑
(2"
‑
β
‑
d
‑
葡萄糖基]
‑
α
‑
l
‑
鼠李糖苷(14号峰)、山柰酚
‑3‑
o
‑
芸香糖苷(15号峰)、异鼠李素
‑3‑
o
‑
芸香糖苷(16号峰)、槲皮素
‑3‑
o
‑
[2
”‑
(6”'
‑
p
‑
香豆酰基)
‑
β
‑
d
‑
葡萄糖基]
‑
α
‑
l
‑
鼠李糖苷(17号峰)、山柰酚
‑3‑
o
‑
(2"
‑
β
‑
d
‑
葡萄糖基)
‑
α
‑
l
‑
鼠李糖苷(18号峰)和山柰酚
‑3‑
o
‑
[2
”‑
(6”'
‑
p
‑
香豆酰基)
‑
β
‑
d
‑
葡萄糖基]
‑
α
‑
l
‑
鼠李糖苷(19号峰)。其中13号峰为芦丁与异槲皮苷的混合峰。本发明所建立22批银杏蜜环口服溶液的指纹图谱与对照指纹图谱的相似度值均大于0.99。
[0024]
综上所述,本发明所建立的检测方法具有全面、有效、高效特点,该检测方法可用于银杏蜜环口服溶液的内在质量检测。
附图说明
[0025]
此处所说明的附图用来提供对本发明的进一步理解,构成本发明的一部分,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。下面将结合附图及实施例对本发明作进一步说明,附图中:
[0026]
图1
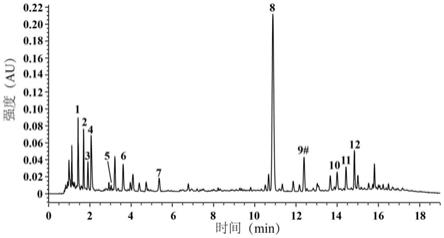
‑
本发明银杏蜜环口服溶液供试品溶液的uplc色谱图(其中色谱图中具体检测
成分为:1、尿苷;2、腺苷;3、鸟苷;4、安赛蜜;5、5
‑
羟甲基糠醛;6、原儿茶酸;7、对羟基苯甲酸;8、苯甲酸钠;9、芦丁 异槲皮苷的混合峰;10、山柰酚
‑3‑
o
‑
芸香糖苷;11、异鼠李素
‑3‑
o
‑
芸香糖苷;12、槲皮素
‑3‑
o
‑
[2
”‑
(6”'
‑
p
‑
香豆酰基)
‑
β
‑
d
‑
葡萄糖基]
‑
α
‑
l
‑
鼠李糖苷);
[0027]
图2
‑
是本发明对照品溶液的uplc色谱图(其中色谱图中具体检测成分为:1、尿苷;2、腺苷;3、鸟苷;4、安赛蜜;5、5
‑
羟甲基糠醛;6、原儿茶酸;7、对羟基苯甲酸;8、苯甲酸钠;9、芦丁/异槲皮苷;10、山柰酚
‑3‑
o
‑
芸香糖苷;11、异鼠李素
‑3‑
o
‑
芸香糖苷;12、槲皮素
‑3‑
o
‑
[2
”‑
(6”'
‑
p
‑
香豆酰基)
‑
β
‑
d
‑
葡萄糖基]
‑
α
‑
l
‑
鼠李糖苷);
[0028]
图3
‑
本发明银杏蜜环口服溶液uplc对照指纹图谱;
[0029]
图4
‑
本发明银杏蜜环口服溶液uplc指纹图谱叠加图。
具体实施方式
[0030]
为了更加充分理解本发明的实施,下面通过典型的实施例对本发明做进一步的说明。除非另作定义,本发明专利申请说明书以及权利要求书中使用的技术术语或者科学术语应当为本发明所属领域内具有一般技能的人士所理解的通常意义。
[0031]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
[0032]
以下实施例中所使用的银杏蜜环口服溶液为邛崃天银制药有限公司产品。
[0033]
实施例1
[0034]
本发明实施例1提供了一种银杏蜜环口服溶液多成分的含量检测方法。
[0035]
仪器:waters acquity uplc h class plus,pda检测器;
[0036]
色谱条件如下:色谱柱:acquityhss t3(2.1
×
100mm,1.8μm)、
[0037]
柱温:50
°
、进样量:2μl、检测波长:254nm、流速:0.3ml/min;
[0038]
流动相:a相为0.2%甲酸水溶液,b相为甲醇;梯度洗脱,洗脱程序如下:
[0039]
表1流动相梯度洗脱程序如下
[0040][0041]
具体包括如下步骤:
[0042]
步骤a、供试品溶液的制备
[0043]
精密量取银杏蜜环口服溶液1ml,置于5ml容量瓶中,用10%甲醇水溶液(v/v)定容至刻度线,摇匀,取适量于12700rpm离心10min,取上清液即得银杏蜜环口服溶液供试品溶
液。
[0044]
步骤b、对照品溶液的制备
[0045]
分别取11种对照品适量,精密称定,使用10%甲醇水溶液(v/v)制备安赛蜜、5
‑
羟甲基糠醛、原儿茶酸、对羟基苯甲酸、苯甲酸钠对照品储备液;使用40%甲醇水溶液(v/v)及少量二甲基亚砜助溶制备尿苷、腺苷、鸟苷、山柰酚
‑3‑
o
‑
芸香糖苷、异鼠李素
‑3‑
o
‑
芸香糖苷和槲皮素
‑3‑
o
‑
[2
”‑
(6”'
‑
p
‑
香豆酰基)
‑
β
‑
d
‑
葡萄糖基]
‑
α
‑
l
‑
鼠李糖苷对照品储备液。取各储备液适量,加入10%甲醇水溶液(v/v)配制成浓度分别为尿苷12.30μg/ml、腺苷5.940μg/ml、鸟苷4.710μg/ml、安赛蜜38.40μg/ml、5
‑
羟甲基糠醛1.638μg/ml、原儿茶酸4.288μg/ml、对羟基苯甲酸1.545μg/ml、苯甲酸钠452.8μg/ml、山柰酚
‑3‑
o
‑
芸香糖苷5.948μg/ml、异鼠李素
‑3‑
o
‑
芸香糖苷5.918μg/ml、槲皮素
‑3‑
o
‑
[2
”‑
(6”'
‑
p
‑
香豆酰基)
‑
β
‑
d
‑
葡萄糖基]
‑
α
‑
l
‑
鼠李糖苷9.763μg/ml的混合对照品溶液。
[0046]
步骤c、按照上述色谱条件,采用超高效液相色谱法对上述对照品溶液和供试品溶液进行检测。
[0047]
供试品溶液的uplc色谱图参照附图1所示;
[0048]
对照品溶液的uplc色谱图参照附图2所示。其中9号色谱峰经质谱鉴定、对照品比对,为芦丁和异槲皮苷色谱峰重叠,因此9号混合峰不作为含量检测质控的指标成分。
[0049]
采用外标法计算样品中对应的多成分的含量,公式如下:
[0050][0051]
其中,c为待测银杏蜜环口服溶液中多成分的含量,单位为μg/ml;
[0052]
c
s
为相应的对照品溶液浓度,单位为μg/ml;
[0053]
a
n
为供试品溶液中相应成分的峰面积;
[0054]
a
s
为对照品溶液中相应成分的峰面积。
[0055]
按照上述检测色谱条件对银杏蜜环口服溶液多成分含量检测方法的方法学考察:
[0056]
(1)线性关系和检测限研究
[0057]
按照实施例1中步骤b的方法制备银杏蜜环口服溶液混合对照品溶液,逐级稀释成系列不同浓度混合对照品溶液,采用超高效液相色谱法检测各化合物的峰面积,色谱条件同实施例1。平行进样2次,以各化合物对照品浓度为横坐标x(μg/ml),相应的对照品峰面积为纵坐标y,绘制回归方程;以信噪比s/n=3为检测限(lod)、s/n=10为定量限(loq)检测各化合物的检测限和定量限。结果见表2。
[0058]
表2银杏蜜环口服溶液11个对照品化合物线性关系、检测限和定量限
[0059]
[0060][0061]
由表2可知,11个化合物在各自的浓度范围内线性关系良好(r2≥0.9997),检测限的浓度范围为0.011
‑
0.35μg/ml,定量限的浓度范围为0.032
‑
1.0μg/ml。
[0062]
(2)精密度试验
[0063]
(a)日内精密度试验
[0064]
按照实施例1中步骤a的方法制备银杏蜜环口服溶液供试品溶液,精密吸取同一份上述供试品溶液,采用超高效液相色谱法检测各化合物的峰面积,重复进样6次,色谱条件同实施例1。以各成分峰面积计算rsd值,结果见表3。
[0065]
表3银杏蜜环口服溶液多成分含量检测的日内精密度试验结果(n=6)
[0066][0067]
由表3可知,各化合物的rsd值均小于2.5%,表明该定量分析方法日内精密度良好。
[0068]
(b)日间精密度试验
[0069]
按照实施例1中步骤a的方法制备银杏蜜环口服溶液供试品溶液,连续3天,每天精密吸取同一份上述供试品溶液,采用超高效液相色谱法检测各化合物的峰面积,每天连续重复进样6次,色谱条件同实施例1。以各成分每天进样的平均峰面积计算rsd值,结果见表4。
[0070]
表4银杏蜜环口服溶液多成分含量检测日间精密度试验结果(n=3)
[0071][0072][0073]
由表4可知,各化合物的rsd值均小于2.6%,表明该定量分析方法日间精密度良
好。
[0074]
由表3和表4可知,本发明提供的银杏蜜环口服溶液多成分含量检测方法精密度良好。
[0075]
(3)重现性试验
[0076]
按照实施例1中步骤a的方法平行制备6份银杏蜜环口服溶液供试品溶液,采用超高效液相色谱法检测各化合物的峰面积,每份样品重复进样2次,色谱条件同实施例1。以实施例1步骤c中的外标法计算各化合物的含量并计算rsd值,结果见表5。
[0077]
表5银杏蜜环口服溶液多成分含量检测的重现性试验结果(n=6,μg/ml)
[0078][0079]
由表5可知,各化合物含量检测的rsd值均小于1.0%,表明本发明构建的银杏蜜环口服溶液多成分定量分析方法重现性良好。
[0080]
(4)稳定性试验
[0081]
按照实施例1中步骤a的方法制备银杏蜜环口服溶液供试品溶液,在10℃下保存,分别于0、2、4、6、8、10、12h精密吸取上述供试品溶液,采用超高效液相色谱法检测各化合物的峰面积,色谱条件同实施例1。以各化合物的峰面积计算rsd值,结果见表6。
[0082]
表6银杏蜜环口服溶液多成分含量检测的稳定性试验结果(n=7)
[0083]
[0084][0085]
由表6可知,各化合物的rsd值均小于2.6%,表明供试品溶液在10℃条件下放置12h稳定。
[0086]
(5)加样回收率试验
[0087]
精密量取0.5ml的银杏蜜环口服溶液6份,准确加入与样品中各成分质量相当的混合对照品溶液ml(尿苷11.07μg/ml、腺苷6.336μg/ml、鸟苷5.024μg/ml、安赛蜜30.72μg/ml、5
‑
羟甲基糠醛1.179μg/ml、原儿茶酸3.920μg/ml、对羟基苯甲酸1.442μg/ml、苯甲酸钠523.3μg/ml、山柰酚
‑3‑
o
‑
芸香糖苷7.137μg/ml、异鼠李素
‑3‑
o
‑
芸香糖苷6.312μg/ml、槲皮素
‑3‑
o
‑
[2
”‑
(6”'
‑
p
‑
香豆酰基)
‑
β
‑
d
‑
葡萄糖基]
‑
α
‑
l
‑
鼠李糖苷14.20μg/ml),按照实施例1中步骤a的方法制备6份银杏蜜环口服溶液供试品溶液,每份样品平行检测2次,采用超高效液相色谱法检测各化合物的峰面积,色谱条件同实施例1。根据实施例1步骤c中的外标法和加样回收率公式计算各化合物的加样回收率及rsd值,考察该方法的准确性。
[0088]
加样回收率公式如下:
[0089]
结果见表7。
[0090]
表7银杏蜜环口服溶液多成分加样回收率试验结果(n=6)
[0091]
[0092]
[0093][0094]
由表7可知,6份样品中被测化合物的加样回收率在96.07%
‑
104.8%,rsd值均小于2.%,表明各化合物的加样回收率良好,本发明构建的银杏蜜环口服溶液多成分定量分析方法准确可行。
[0095]
综上所述,在方法学考察中,被测化合物在各自的浓度范围内线性关系良好,相关系数r2>0.9997;检测限的浓度范围为0.011
‑
0.35μg/ml,定量限的浓度范围为0.032
‑
1.0μ
g/ml;日内精密度rsd值均小于2.5%,日间精密度rsd值均小于2.6%;稳定性rsd值均小于2.6%;重现性rsd值均小于1.%;加样回收率在96.07%
‑
104.8%,rsd值均小于2.1%。因此,本发明中所构建的银杏蜜环口服溶液多成分定量分析方法合理可行。
[0096]
实施例2
[0097]
本发明实施例2提供了一种银杏蜜环口服溶液多成分定量分析方法在检测不同批次银杏蜜环口服溶液含量检测中的应用。
[0098]
(1)对照品溶液的制备
[0099]
按照同实施例1中的“对照品溶液的制备”方法。
[0100]
(2)供试品溶液的制备
[0101]
取2020年生产的22批次有效期内银杏蜜环口服溶液产品(b1
‑
b22),按照实施例1中的“供试品溶液的制备”方法制备供试品溶液。
[0102]
(3)采用超高效液相色谱法对上述对照品溶液和供试品溶液进行检测,并利用实施例1步骤c中的外标法计算供试品溶液中目标成分的含量,色谱条件同实施例1。
[0103]
不同批次银杏蜜环口服溶液多成分含量检测结果如表7所示。
[0104]
由表8可知,通过本发明建立的银杏蜜环口服溶液多成分的定量分析方法研究不同批次银杏蜜环口服溶液中多成分的含量,其中22批有效期内银杏蜜环口服溶液样品中,不同成分含量波动的rsd值为0.8%
‑
20.6%,表明银杏蜜环口服溶液中各批次成分含量存在一定波动。从整体上看,被测化学成分在不同批次间含量较稳定,有较好的批间一致性,表明制剂生产过程中原料的添加及工艺检测良好。
[0105]
本方法对银杏蜜环口服溶液中的安赛蜜及苯甲酸钠两种添加剂含量进行了检测。中国药典2020版在合剂(第四部,通则0181)中规定苯甲酸钠的用量不超过0.3%(以苯甲酸计);世界卫生组织(who)规定安赛蜜的日最大摄取量(adi)为0
–
15mg/kg,苯甲酸钠的adi为05mg/kg。在被测的22批有效期内银杏蜜环口服溶液中,苯甲酸钠的含量范围在2.6
‑
2.7mg/ml,以苯甲酸计约为0.23%,符合中国药典2020版在合剂(第四部,通则0181)中关于苯甲酸钠的限量规定;按成年人体重60kg计算,每日苯甲酸钠最大摄取量约为81mg,符合who关于日最大摄入苯甲酸钠的规定。安赛蜜的浓度范围在0.1682
–
0.1809mg/ml,且每日最大摄取量约为5.4mg,符合who关于日最大摄入安赛蜜的规定。
[0106]
表8 22批银杏蜜环口服溶液多成分含量检测结果(n=2,μg/ml)
[0107][0108]
实施例3
[0109]
本发明实施例3提供了一种银杏蜜环口服溶液uplc指纹图谱的构建方法。
[0110]
仪器:waters acquity uplc h class plus,pda检测器;
[0111]
uplc色谱检测条件如下:色谱柱:acquityhss t3(2.1
×
100mm,1.8μm)、柱温:50℃、进样量:2μl、检测波长:254nm、检测流速:0.3ml/min;流动相:a相为0.2%甲酸水溶液,b相为甲醇;梯度洗脱,洗脱程序如下:0~3min,流动相a的体积百分比由94%匀速降至86%,流动相b的体积百分比由6%匀速上升至14%;3~6min流动相a的体积百分比由86%匀速降至77%,流动相b的体积百分比由14%匀速上升至23%;6~8.3min,流动相a的体积百分比由77%匀速降至69.5%,流动相b的体积百分比由23%匀速上升至30.5%;8.3~10.3min流动相a的体积百分比由69.5%匀速降至66.5%,流动相b的体积百分比由30.5%匀速上升至33.5%;10.3~12.5min,流动相a的体积百分比由66.5%匀速降至58.5%,流动相b的体积百分比由33.5%匀速上升至41.5%;12.5~19min流动相a的体积百
分比由58.5%匀速降至26%;流动相b的体积百分比由41.5%匀速上升至74%。
[0112]
具体包括如下步骤:
[0113]
步骤a、供试品溶液的制备
[0114]
精密量取银杏蜜环口服溶液1ml,置于5ml容量瓶中,用10%甲醇水溶液(v/v)定容至刻度线,摇匀,取适量于12700rpm离心10min,取上清液,即得银杏蜜环口服溶液供试品溶液。
[0115]
步骤b、按上述色谱条件,采用超高效液相色谱法对上述供试品溶液进行检测分析,获得相应色谱图。
[0116]
步骤c、指纹图谱的建立
[0117]
将所得22批银杏蜜环口服溶液色谱图导入国家药典委员会推荐的《中药色谱指纹图谱相似度评价系统2012版》软件分析,以1号样品的指纹图谱为参照指纹图谱,时间窗口设为0.min,利用中位数法,经过多点校正自动匹配,得到银杏蜜环口服溶液uplc对照指纹图谱,标定出19个特征指纹峰。
[0118]
步骤d、指纹图谱相似度评价分析
[0119]
使用《中药色谱指纹图谱相似度评价系统2012版》软件分析22批银杏蜜环口服溶液指纹图谱的相似度。
[0120]
银杏蜜环口服溶液uplc对照指纹图谱如图3所示;
[0121]
银杏蜜环口服溶液uplc指纹图谱叠加图如图4所示。
[0122]
通过与对照品比对、质谱鉴定,对其中15个特征指纹峰进行指认,分别为尿苷(1号峰)、腺苷(2号峰)、鸟苷(3号峰)、安赛蜜(4号峰)、5
‑
羟甲基糠醛(5号峰)、原儿茶酸(7号峰)、对羟基苯甲酸(10号峰)、苯甲酸钠(12号峰)、芦丁 异槲皮苷(13号峰)、槲皮素
‑3‑
o
‑
(2"
‑
β
‑
d
‑
葡萄糖基]
‑
α
‑
l
‑
鼠李糖苷(14号峰)、山柰酚
‑3‑
o
‑
芸香糖苷(15号峰)、异鼠李素
‑3‑
o
‑
芸香糖苷(16号峰)、槲皮素
‑3‑
o
‑
[2
”‑
(6”'
‑
p
‑
香豆酰基)
‑
β
‑
d
‑
葡萄糖基]
‑
α
‑
l
‑
鼠李糖苷(17号峰)、山柰酚
‑3‑
o
‑
(2"
‑
β
‑
d
‑
葡萄糖基)
‑
α
‑
l
‑
鼠李糖苷(18号峰)、山柰酚
‑3‑
o
‑
[2
”‑
(6”'
‑
p
‑
香豆酰基)
‑
β
‑
d
‑
葡萄糖基]
‑
α
‑
l
‑
鼠李糖苷(19号峰)。
[0123]
本发明银杏蜜环口服溶液uplc指纹图谱检测方法的方法学考察:
[0124]
(1)精密度试验
[0125]
按照实施例3中步骤a的方法制备银杏蜜环口服溶液供试品溶液,精密吸取上述供试品溶液,采用超高效液相色谱法进行检测各化合物的保留时间和峰面积,重复进样6次,色谱条件同实施例3。以7号色谱峰为参照峰s,计算各成分的相对保留时间与相对峰面积的rsd值,结果见表9和表10。
[0126]
由表9和表10可知,各化合物的相对保留时间与相对峰面积rsd值均小于1.2%,表明该指纹图谱检测方法精密度良好。
[0127]
表9银杏蜜环口服溶液指纹图谱精密度试验结果(相对保留时间)(n=6)
[0128][0129][0130]
表10银杏蜜环口服溶液指纹图谱精密度试验结果(相对峰面积)(n=6)
[0131][0132][0133]
(2)稳定性试验
[0134]
按照实施例3中步骤a的方法制备银杏蜜环口服溶液供试品溶液,在10℃下保存,分别于0、2、4、6、8、10、12h精密吸取上述供试品溶液,采用超高效液相色谱法进行检测各化合物的保留时间和峰面积,色谱条件同实施例3。以7号色谱峰为参照峰s,计算各成分的相对保留时间与相对峰面积的rsd值,结果见表10和表11。
[0135]
由表10和表11可知,各化合物的相对保留时间与相对峰面积rsd值均小于3.0%,表明该供试品溶液在10℃条件下放置12h稳定。
[0136]
表11银杏蜜环口服溶液指纹图谱稳定性试验结果(相对保留时间,n=7)
[0137][0138]
表12银杏蜜环口服溶液指纹图谱稳定性试验结果(相对峰面积,n=7)
[0139][0140]
(3)重现性试验
[0141]
按照实施例3中步骤a的方法平行制备6份银杏蜜环口服溶液供试品溶液,采用超高效液相色谱法进行检测各化合物的保留时间和峰面积,色谱条件同实施例3。以7号色谱峰为参照峰s,计算各成分的相对保留时间与相对峰面积的rsd值,结果见表13和表14。
[0142]
由表13和表14可知,各化合物的相对保留时间与相对峰面积rsd值均小于2.5%,表明该指纹图谱检测检测方法重现性良好。
[0143]
表13银杏蜜环口服溶液指纹图谱重现性试验结果(相对保留时间)(n=6)
[0144][0145]
表14银杏蜜环口服溶液指纹图谱重现性试验结果(相对峰面积)(n=6)
[0146]
[0147][0148]
综上所述,通过系统的方法学考察,精密度、稳定性、重现性的相对保留时间和相对峰面积的rsd值均小于3.0%,本发明中所构建的银杏蜜环口服溶液指纹图谱检测方法合理可行。
[0149]
(4)指纹图谱相似度评价分析
[0150]
《中药色谱指纹图谱相似度评价系统2012版》软件计算22批银杏蜜环口服溶液指纹图谱的相似度。22批银杏蜜环口服溶液指纹图谱相似度评价结果见表14。
[0151]
表15 22批银杏蜜环口服溶液相似度评价结果
[0152][0153]
[0154]
由表15可知,22批银杏蜜环口服溶液的指纹图谱与对照指纹图谱的相似度值均大于0.99,各批次银杏蜜环口服溶液样品质量稳定。
[0155]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。