1.本发明涉及催化加氢反应领域,具体涉及一种电催化乙炔加氢反应方法。
背景技术:
2.乙烯(c2h4)的工业生产依赖于石脑油或饱和碳2
‑
碳6烃的热解。然而,在热解衍生的乙烯产物物流中会残留0.5
‑
2.0%(体积分数)的乙炔(c2h2)。该杂质会严重毒化后续用于聚乙烯合成的齐格勒
‑
纳塔催化剂(a.borodzi
ń
ski,g.c.bond,catal.rev.48,91
‑
144(2006)),导致生产成本升高、产品质量下降。因此,如何有效地从富含乙烯的气流中选择性地去除乙炔,使乙烯纯度达到聚合物生产级别,具有重要的现实意义。经过近百年的研究,相继发展出了溶剂吸收及催化加氢等技术路线。
3.自1950年代以来,将c2h2选择性催化加氢为c2h4的方法已成为脱除乙炔的更有效方法(图1,路线1)(t.kenzi,bull.chem.soc.japan 23,180
‑
184(1950))。商用钯基催化剂在200℃下,以氢气为质子源,可实现大于90%的c2h2转化率和85%的乙烯选择性(m.armbruster et al.,nat.mater.11,690
‑
693(2012)),c2h2在钯基催化剂上的氢化反应机理也得到了系统地研究。现有很多研究期望开发良好的用于乙炔选择性加氢的催化剂,例如cn102247876a,其研究了一种用于乙炔选择性加氢的磷化钼催化剂,使得在常压以及200
‑
240℃下能够以高的乙炔转化率实现乙炔的选择性催化加氢。近年来,更多注意力转移到了如何进一步降低催化反应的温度(q.feng et al.,j.am.chem.soc.139,7294
‑
7301(2017);s.zhou et al.,adv.mater.31,e1900509(2019);q.feng et al.,adv.mater.31,e1901024(2019))。cn108147938公开了一种常压下乙炔选择性氢化到乙烯的方法,其采用特定的pd
x
m/sio2型催化剂,可以实现在50
‑
300℃下乙炔的选择性加氢。然而,在更低温度(例如室温)条件下实现c2h2的高转化率、高选择性氢化制c2h4,仍面临巨大挑战。
4.此外,目前开发的实验室和工业规模的c2h2加氢反应系统都需要在反应过程中引入过量的氢气以促进c2h2的加氢转化。但是这种方法会不可避免地导致c2h4过度加氢生成乙烷(c2h6),造成大量c2h4原料的浪费,增加了经济成本(a.s
á
rk
á
ny,a.horv
á
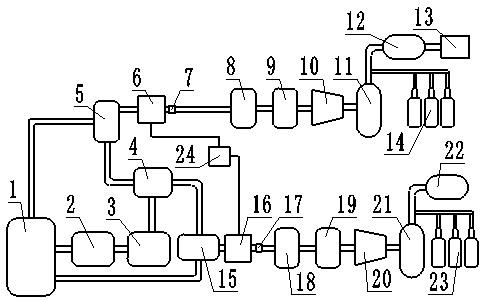
th,a.beck,appl.catal.a
‑
gen.229,117
‑
125(2002))。以50万吨/年乙烯生产线为例:根据乙烯市场价格7000元/吨,年产值约35亿;热催化加氢通常会造成2
‑
3%的乙烯损耗(过度加氢至乙烷),因此带来约1亿元损失。若采用最先进的进口催化剂(clariant260)可将损耗降至接近0%,但催化剂成本高。因此,无论从能量和原子利用的角度来看,都有必要创新乙炔加氢反应系统及方法,以在低温下实现乙炔向乙烯的高效转化。
5.电催化策略能够以水分子为氢源,在无需氢气输入的低温条件下将乙炔加氢还原为乙烯(图1,路线2)。理论上,每消除1吨乙烯原料气中的乙炔杂质,需工业用电费用约7元,仅占乙烯市场价格的千分之一,说明该策略具备大规模应用的成本优势。然而,由于现有电催化系统的局限性,如富乙烯气氛中乙炔气体的传质问题、反应分子/离子在催化界面的接触问题、电催化全反应系统的搭建问题等等,现有电催化c2h2加氢(ear)反应系统的转化率和产物选择性与热催化工艺相比仍有巨大差距。迄今为止,基于路线2的电催化乙炔加氢反
应系统及方法还存在诸多研究空白。
6.基于以上现有技术,本发明人期望建立一种电催化乙炔加氢反应的方法,以在富含乙烯的物流中将乙炔有效地转化为乙烯。
技术实现要素:
7.为提升乙炔反应分子在催化剂表面的浓度与扩散速率,实现低温条件下乙炔到乙烯的高活性、高选择性电催化转化,本发明提供了一种电催化乙炔加氢反应的方法。
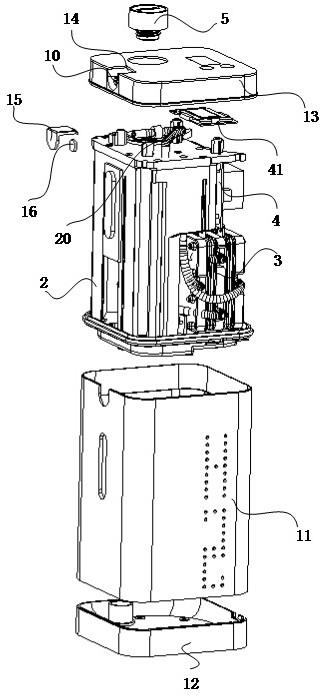
8.本发明的一个目的是提供一种电催化乙炔加氢反应的方法,所述方法包括:
9.注入原料气:将含乙炔的气体原料气通入通过工作电极与电化学装置分隔的乙炔气室中;
10.电催化乙炔加氢反应:接通电源,使电化学装置工作发生电解反应,以使扩散至工作电极的含乙炔的气体进行电催化乙炔加氢反应,使乙炔还原为乙烯。
11.根据本发明,所述电催化乙炔加氢反应在5
‑
30℃的温度下进行。
12.根据本发明,所述含乙炔的气体是乙炔浓度在0.5体积%
‑
5体积%之间的富乙烯气体。
13.根据本发明,反应空速为500
‑
6000h
‑1,优选1000
‑
3000h
‑1。
14.根据本发明,所述电化学装置的阴极电势可以为
‑
0.4v至
‑
0.6v(相对于可逆氢电极,rhe),优选
‑
0.4v至
‑
0.5v;或者电池电压为1.95v至2.00v
15.根据本发明,所述工作电极为气体扩散电极,所述气体扩散电极包括气体扩散层(gdl)和负载在所述gdl上的催化剂。
16.根据本发明,所述催化剂为通过涂覆在gdl上而形成的催化剂层。
17.根据本发明,所述工作电极上负载的催化剂为选自钯基催化剂、和铜基催化剂的一种或多种,例如为选自钯、铜及其合金、氧化物和氢氧化物的一种或多种;优选地,所述铜基催化剂为选自cual层状双金属氢氧化物(cual
‑
ldh)纳米片、cu纳米颗粒、cu2o纳米颗粒和cu/cu2o的混合物中的一种或多种。
18.根据本发明的一个实施方式,作为铜基催化剂的cual
‑
ldh纳米片可通过如下方法制备:
19.制备ph值在9
‑
10之间的弱碱性水溶液a;
20.将铜盐和铝盐溶于水中制备溶液b,其中cu/al摩尔比为1:1至3:1;
21.制备ph值在12
‑
14之间的强碱性水溶液c;
22.搅拌下将溶液b和c同时滴加到溶液a中,在整个滴加过程中,使混合溶液的ph值保持恒定在9
‑
10之间;
23.在完全加入溶液b和c之后,得天蓝色悬浮液,离心、水洗,得cual双层氢氧化物纳米片。
24.根据本发明的方法,在将负载有cual
‑
ldh纳米片的气体扩散电极连接到电化学装置电池后,进行电化学反应之前,原位电还原负载的cual
‑
ldh纳米片。
25.根据本发明的一个实施方式,所述电化学装置的工作电极为气体扩散电极,所述电化学装置还包括电解室、电源以及设于所述电解室内以将所述电解室分隔成阴极电解室和阳极电解室的离子交换膜,所述阴极电解室与所述乙炔气室邻接并由所述气体扩散电极
分隔;所述阳极电解室设有对电极,所述气体扩散电极连接所述电源的负极,所述对电极连接所述电源的正极。
26.根据本发明的另一个实施方式,所述电化学装置的工作电极为气体扩散电极,所述电化学装置还包括电解室、对电极、阳极腔室以及电源,所述电解室与所述乙炔气室邻接并由所述气体扩散电极分隔;所述电解室与所述阳极腔室邻接并由所述对电极分隔;所述气体扩散电极连接所述电源的负极,所述对电极连接所述电源的正极。
27.根据本发明,所述电解室内容置有选自液体电解质和固体电解质的电解质;例如,所述阴极电解室内容置有碱性电解液;和/或所述阳极电解室内容置有碱性电解液。
28.根据本发明,所述乙炔气室的厚度可为0.5
‑
2.0mm。
29.根据本发明,乙炔催化加氢转化率可为99.95%以上,乙烯选择性可为90%以上。
30.根据本发明的方法,能够在低温下高活性、高转化率、高选择性地将乙炔电催化转化为乙烯,能够为电催化乙炔加氢的大规模应用创造基本条件,并有望替代现有热催化乙炔加氢化工过程,促进聚乙烯工业生产的绿色可持续发展。
附图说明
31.图1示出了现有热催化加氢系统将c2h2选择性氢化为c2h4的路线(路线1),以及设计的新型电催化加氢系统将c2h2选择性氢化为c2h4的路线(路线2)。
32.图2a和图2b分别为根据本发明实施方式的电催化乙炔加氢反应系统的示意图。
33.图3
‑
1和3
‑
2分别为根据本发明实施方式的电催化乙炔加氢反应系统的分解图。
34.图4
‑
1中a中示出了制备实施例制备的cual
‑
ldh的x射线衍射图,b示出了cual
‑
ldh的能量色散x射线光谱(edx)图,其中测得cu和al的原子比为2.5:1。
35.图4
‑
2中a和b分别示出了涂覆在碳基gdl上的cual
‑
ldh的透射电子显微成像(tem)(a)和扫描电子显微成像(sem)(b)。
36.图5
‑
1显示了cual
‑
ldh前体和在不同还原电势下制备的一系列ld
‑
cu催化剂的x射线光电子能谱(xps)。
37.图5
‑
2示出了在1m koh中于不同电势下电化学还原后的ld
‑
cu的扫描电子显微成像:(a,b)
‑
0.3v;(c,d)
‑
0.4v;(e,f)
‑
0.5v;(g,h)
‑
0.6v。
38.图5
‑
3示出了
‑
0.5v还原的ld
‑
cu的透射电子显微成像,其中a示出了高角环形暗场
‑
透射电子显微成像以及ld
‑
cu的cu、o、al元素分布图,比例尺:50nm;b示出了ld
‑
cu的透射电子显微成像,比例尺:20nm;c是从图b中的选定区域放大的ld
‑
cu的高分辨透射电子显微成像,比例尺:2nm。
39.图6显示了ld
‑
cu催化体系以及电催化乙炔加氢反应系统中的电化学乙炔还原反应评估,其中a示出了用于ear测试的阴极(工作电极)部分的示意图;b示出了在gdl上的ld
‑
cu的扫描电子显微成像,比例尺:200nm;c示出了在gdl上的ld
‑
cu的截面扫描电子显微成像和相应的元素分布图,比例尺:10μm;d示出了电催化生成c2h4和h2的分电流密度与阴极还原电势的关系,反应条件:反应气成分为5%c2h2,氩气(ar)作为平衡气,流速为10ml min
‑1,电解质为1m koh;e示出了ear产物的法拉第效率曲线;f比较ld
‑
cu和cu nps和先前报道(参考文献/现有文献)的电催化乙炔加氢制c2h4的分电流密度和法拉第效率。
40.图7示出了使用不同原料气在有或无ld
‑
cu催化剂的情况下的线性扫描伏安曲线。
41.图8示出了涂覆在碳基gdl上的cu nps在1m koh中电催化反应前(a,b)、后(c,d)的扫描电子显微成像。
42.图9显示了cu nps催化体系在电催化乙炔加氢反应系统中的电化学乙炔还原反应评估,其中a为使用5%
‑
c2h2和ar平衡气作为气体来源的cu nps的线性扫描伏安曲线;b为电催化生成c2h4和h2的分电流密度与施加电势的关系;c为不同ear产物的法拉第效率。
43.图10显示了在乙烯存在下关于ld
‑
cu的ear性能评估(实施例3),其中a显示了相对于阴极电势和流速绘制的乙炔转化率,b示出了在1ml min
‑1的流速下乙炔的转化率和ear产物的分布(选择性),c示出了在阴极电势为
‑
0.5v下乙炔还原产物的选择性随气体流速的变化曲线,d示出了对于0.5cm2和2.0cm2电极面积,乙炔转化率及氢气体积百分比随时间的变化曲线。
44.图11
‑
a示出了实施例4中采用的扩大的流动反应器的显示蛇形气道的乙炔气室的剖视图,图11
‑
b示出了a的左视图,图11
‑
c示出了a的俯视图。
45.图12为实施例4中示出的两电极全电池的示意结构图及其ear性能评估和比较。a为两电极ear电解池的示意图;b为组装好的流动反应器的照片;c
‑
e为全电池ear测试结果,其中c示出了乙炔转化率和氢气体积百分比随时间的变化曲线,d示出了产物选择性随时间的变化曲线,e示出了在1m koh中恒定电流为35ma时的电池电压;气体进料:0.5%c2h2、20%c2h4与ar平衡气。气体流速:50ml min
‑1;e图中的插入图显示了ear测试前后气体扩散电极背面水接触角的照片;f显示了本发明与已知的热催化乙炔加氢系统的性能比较;g显示了不同乙炔向乙烯转化路线的原子和能量经济性,每个路径均标明了反应吉布斯自由能(每摩尔乙炔分子)。
46.图13示出了nife
‑
ldh纳米片的结构图及阳极lsv曲线:a为nife
‑
ldh纳米片的电子显微成像;b为nife
‑
ldh纳米片的x射线衍射图;c为在1m koh中负载有nife
‑
ldh的对电极上的线性扫描伏安曲线。
47.图14示出了在实施例4的ear性能测试中原料气流和电催化加氢产物气流(10min)的气相色谱谱图。
48.图15示出了pd/c(商业钯碳)催化剂ear产物的分电流密度与阴极还原电势的关系图(5%c2h2,ar为平衡气)。
49.图16示出了ld
‑
cu分别在本发明系统与传统h型电催化系统的ear乙炔反应速率随温度变化曲线。
50.图17示出了富乙烯原料气在不同气室厚度时的乙炔转化率随空速的变化曲线。
51.附图标记列表
52.1,1’:进气口
53.2,2’:乙炔气室
54.3,3’:出气口
55.4,4’:气体扩散层
56.5,5’:催化剂层
57.6:离子交换膜
58.7,7’:对电极
59.8:阳极电解室
[0060]8’
:阳极腔室
[0061]
9:阴极电解室
[0062]9’
:电解室
[0063]
10,10’:外电路
[0064]
11,11’:电源
[0065]
12,12’:第一侧壁
[0066]
13,13’:第一开口
[0067]
14,14’:导体
[0068]
15,15’:气体扩散电极
[0069]
16,16’:气室壁
[0070]
17,17’:第三开口
[0071]
18,18’:垫圈
[0072]
h:乙炔气室的厚度
具体实施方式
[0073]
为了更清楚地说明本发明,下面结合根据本发明的示例性附图对本发明做进一步的说明。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
[0074]
根据本发明的方法可通过图2a、图2b以及图3
‑
1或3
‑
2所示的电催化乙炔加氢反应系统来实施。
[0075]
具体地,结合图2a,示例说明根据本发明一个实施方式的电催化乙炔加氢反应系统,所述系统包括乙炔气室2和电化学装置,所述电化学装置的工作电极分隔所述乙炔气室2和所述电化学装置,以形成所述乙炔气室2和所述电化学装置共用的分隔壁,通入所述乙炔气室2内的含乙炔的气体能够在所述工作电极完成电化学催化加氢反应,反应生成的乙炔加氢产物气体能够导出乙炔气室2。
[0076]
可以理解的是,乙炔气室2具有进气口1和出气口3,通过进气口1向乙炔气室2内通入有含乙炔的气体,含乙炔的气体在工作电极完成电化学催化加氢反应后生成乙炔加氢产物气体,出气口3用于将乙炔加氢产物气体导出乙炔气室2。
[0077]
根据本发明的实施方式,进气口1和出气口3的设置方式可以根据实际需要选择,例如可以是上下、左右、前后或对角相对设置。
[0078]
本发明实施例的电催化乙炔加氢反应系统结构简单高效,能够实现流动相的含乙炔的气体的大电流密度催化转化,降低电催化乙炔加氢生产成本;而且该反应系统可以在低温(例如5
‑
30℃)条件下进行,填补低温条件下乙炔催化转化的空白,实现在低温条件下乙炔到乙烯的高活性、高选择性电催化转化。
[0079]
根据本发明,所述电化学装置用于发生电解反应,以产生乙炔电化学催化加氢反应所需的质子。即,电化学装置的对电极发生电化学氧化反应产生质子,质子与乙炔在工作电极发生电化学还原反应生成乙烯。
[0080]
根据本发明的一个实施方式,继续结合图2a,电化学装置的工作电极为气体扩散电极,所述电化学装置还包括电解室、电源11以及设于所述电解室内以将所述电解室分隔
成阴极电解室9和阳极电解室8的离子交换膜6,所述阴极电解室9与所述乙炔气室2邻接并由所述气体扩散电极分隔;所述阳极电解室8设有对电极7,所述气体扩散电极通过外电路10连接所述电源11的负极,所述对电极7通过外电路10连接所述电源11的正极。该电化学装置结构简单紧凑,可以缩小体积。另外,对电极7和气体扩散电极分别与电源11的正、负极连接,能够保证在打开电源11时,工作电极处于偏负压条件,给出电子到乙炔分子(工作电极处的乙炔分子),并克服电催化乙炔加氢反应过电势,从而促进乙炔加氢反应的进行。
[0081]
根据本发明的一个实施方式,所述气体扩散电极可以包括气体扩散层4和固定于所述气体扩散层4位于所述阴极电解室9一侧的催化剂层5。
[0082]
根据本发明的实施方式,催化剂层5例如可通过粘接、喷涂、电沉积、煅烧等方式固定在气体扩散层4一侧。
[0083]
根据本发明的实施方式,气体扩散层4用以作为导电载体和乙炔气体扩散通道,催化剂层5与阴极电解室9中的电解质相接触,催化剂层5提供乙炔加氢反应活性中心;所述气体扩散电极固定在乙炔气室2与阴极电解室9之间,其中气体扩散层4背离催化剂层5的一侧位于乙炔气室2内,以使乙炔气室2中的含乙炔的气体可以直接接触气体扩散层4,并通过其扩散至催化剂层5并与催化剂相接触而驱动加氢反应。
[0084]
根据本发明的实施方式,所述气体扩散层4例如可以为多孔碳、泡沫镍、泡沫铜、不锈钢网等。
[0085]
根据本发明,负载在气体扩散电极上的催化剂与阴极电解质溶液相接触,该设置方式可增强乙炔以及质子在催化剂层5上的相互作用,同时保证气体扩散电极的导电性,降低催化剂与电极、阴极电解质间的接触内阻。
[0086]
根据本发明,所述离子交换膜6固定在阴极电解室9与阳极电解室8之间,该设置方式可保证离子(例如质子)的定向迁移,同时保持较高的电导率,并阻止乙炔分子由气体扩散电极扩散至对电极7发生氧化副反应。
[0087]
根据本发明的一个实施方式,所述电化学装置还可包括参比电极,所述参比电极设于所述阴极电解室9。
[0088]
根据本发明,所述阴极电解室9中的阴极电解质分别与离子交换膜6和气体扩散电极或其上的催化剂层5相接触。
[0089]
根据本发明的实施方式,所述阴极电解室和阳极电解室内容置有电解质;其中,电解质可以为水溶液、离子液体等液态电解质,或聚合物、金属氧化物等固态电解质,例如1m koh溶液、0.5m h2so4溶液,emim
‑
bf4离子液体、阳离子聚合物、钙钛矿陶瓷等。
[0090]
根据本发明的实施方式,若阴极电解质为水溶液、离子液体等液态电解质,则所述气体扩散层4为疏液结构,以确保含乙炔的气体通过其接触催化剂,同时防止阴极电解室9内的电解质进入乙炔气室2。
[0091]
根据本发明的一个实施方式,所述阴极电解室内的电解质为碱性电解液;和/或所述阳极电解室内的电解质为碱性电解液。
[0092]
根据本发明的一个实施方式,乙炔气室2和电化学装置可以是一个腔室的两个部分,气体扩散电极将两部分密封地(例如流体密封地)分开,以防止电化学装置的电解质进入乙炔气室2。气体扩散电极4可以附在一支撑基体上,该支撑基体可以具有足够的强度以承载所分隔腔室的重量,且支撑基体不妨碍乙炔在气体扩散电极上的扩散及电化学反应。
[0093]
根据本发明的一个实施方式,乙炔气室2和电化学装置可以是分开的单独的部分,气体扩散电极设置在两者之间,其中,乙炔气室2和电化学装置彼此连接而将气体扩散电极夹持在其间。例如乙炔气室2、气体扩散电极、阴极电解室9、离子交换膜6、阳极电解室8、对电极7彼此之间密封连接。
[0094]
根据本发明的一个实施方式,如图3
‑
1所示,电催化乙炔加氢反应系统包括乙炔气室2和电化学装置,所述电化学装置包括气体扩散电极15、阴极电解室9、阳极电解室8、离子交换膜6、对电极7以及电源(未示出),所述阴极电解室9的第一侧壁12上具有与所述阴极电解室9内部连通的第一开口13,所述阳极电解室8的第二侧壁上具有与所述阳极电解室8内部连通的第二开口(未示出);所述阴极电解室9的所述第一侧壁12与所述阳极电解室8的所述第二侧壁贴合,且所述第一开口13与所述第二开口密封对接,并将所述离子交换膜6夹持于所述第一开口13和所述第二开口处以分隔所述阴极电解室9和所述阳极电解室8;所述对电极7设于所述阳极电解室8,所述气体扩散电极15连接所述电源的负极,所述对电极7连接所述电源的正极。
[0095]
根据本发明的实施方式,所述乙炔气室2的气室壁16上设有第三开口17,所述阴极电解室的与所述第一侧壁相对的第三侧壁上设有第四开口(未示出),所述气室壁16与所述第三侧壁贴合,且所述第三开口17与所述第四开口密封对接,并将所述气体扩散电极15夹持于所述第三开口17和所述第四开口处,以分隔所述乙炔气室和所述阴极电解室。
[0096]
根据本发明的实施方式,乙炔气室2的内部形状可以是矩形、圆形或方形等。另外,为了增加乙炔气室2内含乙炔的气体与气体扩散电极15的接触面积,如图11
‑
a至11
‑
c所示,乙炔气室2内可以形成蛇形气道,蛇形气道内含乙炔的气体的流动方向与气体扩散层4所在的平面平行,以使含乙炔的气体能够在气体扩散层4上呈蛇形迂回流动,使得含乙炔的气体与气体扩散层4形成巨大化的接触面积。
[0097]
根据本发明的实施方式,所述乙炔气室的厚度(乙炔气室的厚度是指乙炔气室朝向气体扩散层的内壁至气体扩散层的垂直距离)为0.5
‑
2.0mm,优选0.5
‑
1.5mm,例如1.0mm。例如,所述乙炔气室的厚度在图11
‑
b中以h示出。
[0098]
根据本发明的实施方式,当乙炔气室2内形成蛇形气道时,气道的深度与乙炔气室的厚度相同,例如当气道由隔板围成时,隔板的高度(垂直于气体扩散层4所在的平面的方向的尺寸)可与乙炔气室的厚度相当。
[0099]
当厚度处于以上范围时,能够使乙炔的电催化加氢转化率保持在较高的水平,例如在500
‑
3000h
‑1空速下乙炔转化率保持在98%以上。
[0100]
在图3
‑
1中,气体扩散电极15上连接有导体14,气体扩散电极15通过导体14连接电源的阴极,导体14例如可为铝箔、铜箔、不锈钢片等;气体扩散电极15的gdl例如可为碳基gdl(如h14c9,德国科德宝),离子交换膜例如可为质子交换膜(如n
‑
117,美国杜邦)、对电极例如可为pt。根据本发明的实施方式,还可在图3
‑
1所述系统的阴极电解室中设置参比电极,参比电极可以为ag/agcl。
[0101]
在图3
‑
1中,乙炔气室2、气体扩散电极15、阴极电解室9、离子交换膜6、阳极电解室8、对电极7彼此之间可以通过螺栓连接。
[0102]
在图3
‑
1中,乙炔气室2、阴极电解室9和阳极电解室8的外壳分别形成矩形壳体结构;乙炔气室内部结构为圆柱形。
[0103]
如图3
‑
1所示,在本实施例中,进气口1设置在乙炔气室2的前部,通过进气口1向乙炔气室2内通入含乙炔的气体,而出气口3设置在乙炔气室2的后部,通过出气口3排出乙炔加氢产物气体。
[0104]
根据本发明的另一个实施方式,如图2b所示,所述电化学装置的工作电极为气体扩散电极,所述电化学装置还包括电解室9’、对电极7’、阳极腔室8’以及电源11’,所述电解室9’与所述乙炔气室2’邻接并由所述气体扩散电极分隔;所述电解室9’与所述阳极腔室8’邻接并由所述对电极7’分隔;所述气体扩散电极通过外电路10’连接所述电源11’的负极,所述对电极7’通过外电路10’连接所述电源11’的正极。
[0105]
根据本发明的一个实施方式,如图3
‑
2所示,所述电解室9’的第一侧壁12’上具有与所述电解室9’内部连通的第一开口13’,所述阳极腔室8’的第二侧壁上具有与所述阳极腔室8’内部连通的第二开口(未示出);所述电解室9’的所述第一侧壁12’与阳极腔室8’的所述第二侧壁贴合,且所述第一开口13’与所述第二开口密封对接,并将所述对电极7’夹持于所述第一开口13’和所述第二开口处。
[0106]
根据本发明的实施方式,所述乙炔气室2’的气室壁16’上设有第三开口17’,所述电解室9’的与所述第一侧壁相对的第三侧壁上设有第四开口(未示出),所述气室壁16’与所述第三侧壁贴合,且所述第三开口17’与所述第四开口密封对接,并将所述气体扩散电极15’夹持于所述第三开口17’和所述第四开口处,以分隔所述乙炔气室2’和所述电解室9’。
[0107]
根据本发明的实施方式,所述阳极腔室8’内可以容置有阳极反应物和阳极产物,例如,所述阳极腔室8’中可以容置有电解质,可以容置有水,还可以容置有气体,如,所述阳极腔室8’内容置有水溶液、氧气产物等。
[0108]
根据本发明的实施方式,所述对电极7’可以是多孔电极。
[0109]
根据本发明,如本领域技术人员可以理解,各电解室和阳极腔室可以设置进口和出口,以供电解质的注入或产物的导出。
[0110]
根据本发明,所述连接可以是本领域熟知的连接方式,例如螺栓连接、卡接、铆接、材料连接(如粘接、焊接)等。
[0111]
根据本发明,如有必要,各部件之间可以通过本领域常规的方式密封,以保证其中的气、液流通而确保电化学反应而不泄露,例如可以采用密封圈、密封垫(橡胶、硅胶、氟硅胶、聚四氟乙烯等材质)等方式密封,如在图3
‑
1和3
‑
2中以附图标记18示出的垫圈。例如,可以在用螺栓连接各部件时,将密封圈夹在各部件之间,用螺丝拧紧固定实现密封。
[0112]
如有必要,各电极和隔膜与各自对应的壳体或槽体的接触或连接方式可以通过本领域如电化学领域常规的方式绝缘和密封,比如可以绝缘套管、密封垫圈等方式绝缘和密封,以避免其中的气、液外泄以及短路而确保电化学反应顺利进行。
[0113]
根据本发明的一个实施方式,所述催化剂涂覆在气体扩散电极面向电解室或阴极电解质的一侧,形成催化剂层,如图2a和图2b中的催化剂层5,5’。
[0114]
根据本发明的一个实施方式,所述催化剂可通过浸渍、煅烧、物理或化学沉积而负载在gdl上从而形成合二为一的气体扩散电极。
[0115]
根据本发明,所述催化剂可为选自钯基催化剂和铜基催化剂的一种或多种,例如为选自钯、铜及其合金、氧化物和氢氧化物的一种或多种。
[0116]
根据本发明的实施方式,所述铜基催化剂可为选自cual
‑
ldh纳米片、cu纳米颗粒、
cu2o纳米颗粒和cu/cu2o的混合物中的一种或多种。
[0117]
根据本发明的一个实施方式,作为铜基催化剂的cual
‑
ldh纳米片可通过如下方法制备:
[0118]
制备ph值在9
‑
10之间的弱碱性水溶液a;
[0119]
将铜盐和铝盐溶于水中制备溶液b,其中cu/al摩尔比为1:1至3:1;
[0120]
制备ph值在12
‑
14之间的强碱性水溶液c;
[0121]
搅拌下将溶液b和c同时滴加到溶液a中,在整个滴加过程中,使混合溶液的ph值保持恒定在9
‑
10之间;
[0122]
在完全加入溶液b和c之后,得天蓝色悬浮液,离心、水洗,得cual
‑
ldh纳米片。
[0123]
根据本发明的实施方式,所制备的cual
‑
ldh纳米片可重新分散在去离子水中,并在2℃下保存备用,cual
‑
ldh浓度不限定,例如可以约为6.0mg ml
‑1;另外,也可以将所制备的cual
‑
ldh纳米片干燥为粉末保存。
[0124]
根据本发明的实施方式,所述弱碱性水溶液a可以通过将na2co3或k2co3溶于去离子水中而制备。
[0125]
根据本发明的实施方式,所述强碱性水溶液c可以通过将naoh或koh溶于去离子水中而制备。
[0126]
根据本发明的实施方式,所述铜盐可以是硝酸铜,氯化铜,硫酸铜等。根据本发明的实施方式,所述铝盐可以是硝酸铝,例如al(no3)3·
9h2o。
[0127]
根据本发明的一个实施方式,可以如下方式制备气体扩散电极:
[0128]
将催化剂水分散体用乙二醇和正丙醇稀释以形成水/乙二醇/正丙醇混合浆料(例如溶剂体积比可以为(1
‑
3):(1
‑
3):(1
‑
3),如1:1:1),其中催化剂的浓度为0.5mg ml
‑1;将混合浆料滴涂在gdl(例如碳基gdl,h14c9,德国科德宝)上,干燥得气体扩散电极,即负载催化剂的气体扩散电极。
[0129]
其中,所述催化剂可以为如上所述制备的cual
‑
ldh纳米片、铜纳米粒子(金属含量为99.9%,10
‑
30nm,上海麦克林生物化学有限公司)等。
[0130]
根据本发明的一个实施方式,在将涂有cual
‑
ldh纳米片的气体扩散电极连接到电化学电池上后,进行电化学反应之前,原位电还原负载的cual
‑
ldh纳米片。在一个实施方式中,在进一步进行电化学反应之前,立即在1m koh中于
‑
0.4v下进行cual
‑
ldh电还原一定时间,例如10
‑
15分钟。
[0131]
根据本发明的一个实施方式,催化剂层的平均厚度可以为1.0
±
0.2μm。
[0132]
本领域技术人员可根据实验需要对上述组件材料以及形状进行选择。例如,所述乙炔气室(气体腔室)的尺寸和内部结构可以根据电催化乙炔加氢的实际需要进行调整;气体腔室和电解质容器的材质可采用可承受一定压力的铝合金、石墨或不锈钢材质;可根据实际需要对催化剂的组分、形貌、结构以及负载量等参数进行调控;电解质材质可根据需要采用水溶液、离子液体等液态电解质或聚合物、金属氧化物等固态电解质;所述离子交换膜可采用质子交换膜或阴离子交换膜等商品化离子交换膜,本发明对此并不加以限制;所述对电极材料可采用铂电极、碳棒电极、氧化铱电极等商品化电极,本发明对此并不加以限制;所述电源可以是电化学工作站、恒电流、恒电压电源,本发明对此并不加以限制。
[0133]
根据本发明的另一方面,提供一种电催化乙炔加氢反应的方法,所述方法包括:
oxidation.adv.energy mater.9,1900881(2019))。简要地,将20ml的含有ni(no3)2·
6h2o(2.181g,北京化工厂)和fe(no3)3·
9h2o(1.010g,北京化工厂)的水溶液逐滴加入到在80℃磁力搅拌下的23体积%甲酰胺水溶液(20ml,北京化工厂)中。同时,逐滴添加2.5m naoh溶液以保持ph=10。反应在10分钟内完成。冷却至室温后,通过离心收集产物,用去离子水和乙醇洗涤五次,然后再分散在水中以备后用。
[0152]
3.气体扩散电极的准备
[0153]
负载cual
‑
ldh纳米片的气体扩散电极
[0154]
将制备的cual
‑
ldh纳米片水分散体用乙二醇和正丙醇稀释以形成水/乙二醇/正丙醇混合溶剂(体积比为1:1:1)浆料,其中cual
‑
ldh浓度为0.5mg ml
‑1。将浆料滴涂在碳基gdl(h14c9,德国科德宝)上,并在红外灯下干燥得到负载cual
‑
ldh的气体扩散电极前体(例如,对于使用几何面积(接触电解质的工作电极面积)为0.5cm2的电化学电池进行的实验,将70μl浆料滴涂在gdl(h14c9,德国科德宝)上,并在红外灯下干燥得气体扩散电极;对于使用几何面积为2.0cm2的电化学电池进行的实验,将280μl的浆料滴涂在碳基gdl(h14c9,德国科德宝)。
[0155]
然后将准备好的气体扩散电极前体连接到电催化乙炔加氢反应系统上。通常在进一步进行电化学测量之前,立即在1m koh中于
‑
0.4v下进行cual
‑
ldh电还原10分钟,以制备负载ldh衍生的铜催化剂(ld
‑
cu)的气体扩散电极。催化剂层的平均厚度为1.0
±
0.2μm。
[0156]
对ld
‑
cu进行结构、组成和形态分析,其中:图5
‑
1显示了cual
‑
ldh前体和在不同电还原电势下制备的一系列ld
‑
cu催化剂的x射线光电子能谱(xps)数据。对于cual
‑
ldh纳米片,仅检测到cu(ii)。对于在
‑
0.3v至
‑
0.6v电位范围内还原的ld
‑
cu样品,它们的铜价态均降低,且具有相似的纳米粒子形态(图5
‑
2)。透射电子显微成像(tem)结果显示,
‑
0.5v还原的ld
‑
cu由cu(0)和cu(i)物种的混合物组成(图5
‑
3)。元素分析进一步显示平均cu/o原子比为2.7
±
0.3。在ld
‑
cu中仅发现痕量的al(电感耦合等离子体
‑
原子发射光谱测试结果为0.008wt.%),表明它仅作为牺牲模板,对乙炔的电还原几乎没有贡献(表s1)。
[0157]
图6示出了ld
‑
cu/gdl催化体系以及流动系统中的电化学乙炔还原反应。在电化学还原系统中,气相乙炔通过定义明确的气体
‑
电解质
‑
催化剂界面(图6中的a图)扩散到催化剂层的表面,从而克服了碱性电解质中乙炔的界面传质限制。如图6中的a、b和c图所示,ld
‑
cu催化剂固定在gdl上,催化剂层的平均厚度为1.0
±
0.2μm。
[0158]
表s1.从碳基气体扩算层上剥离的ld
‑
cu的edx元素分析
[0159][0160][0161]
负载cu nps催化剂的气体扩散电极
[0162]
首先将市售的铜纳米粒子(金属含量为99.9%,10
‑
30nm,上海麦克林生物化学有
限公司)分散在水
‑
乙二醇
‑
正丙醇混合溶液(体积比为1:1:1)中,以形成cu纳米粒子浓度为0.5mg ml
‑1的浆料。然后将这些浆料施加到碳基gdl(h14c9)上,所有其他步骤均与上述cual
‑
ldh纳米片负载过程相同,以制备负载有金属铜纳米粒子催化剂(cu nps)的气体扩散电极。
[0163]
负载nife
‑
ldh纳米片的气体扩散电极
[0164]
以与上述cual
‑
ldh纳米片负载过程相同的方式,将如上制备的nife
‑
ldh纳米片分散负载在gdl(h14c9,德国科德宝)上,以制备负载有nife
‑
ldh纳米片的气体扩散电极。
[0165]
负载pd/c纳米颗粒的气体扩散电极
[0166]
首先将市售的pd/c纳米颗粒(金属含量为10%,10
‑
30nm,上海麦克林生物化学有限公司)分散在水
‑
乙二醇
‑
正丙醇混合溶液(体积比为1:1:1)中,以形成pd/c纳米颗粒浓度为0.5mg ml
‑1的浆料。然后将这些浆料施加到gdl(h14c9,德国科德宝)上,所有其他步骤均与上述cual
‑
ldh纳米片负载过程相同,以制备负载有pd/c纳米颗粒催化剂(pd/c)的气体扩散电极。
[0167]
测试设备和方法
[0168]
1.显微表征
[0169]
cual
‑
ldh的透射电子显微镜(tem)图像是在ht7700显微镜(日立,日本,以100kv的加速电压运行)上获得的;ld
‑
cu的tem图像在jeol
‑
2100f显微镜(日本jeol,以200kv的加速电压运行)上采集的,该仪器还配备了高角环形暗场扫描tem(haadf
‑
stem)和能量色散x射线光谱仪(edx),用于元素映射和催化剂样品的定量分析;将所有用于tem和edx分析的样品分散在镍网支撑的碳膜上;在hitachi s
‑
4800仪器上以5kv和10kv的加速电压获得场发射扫描电子显微镜(fesem)图像,该仪器还配备了edx光谱仪,用于gdl负载的ld
‑
cu催化剂的元素映射。
[0170]
使用配备有cu kαx射线源的bruker d8 focus衍射仪收集xrd图。xps数据是在escalab 250xi(thermo fisher scientific,美国)上使用单色al
‑
kα辐射(hν=1486.6ev)作为激发源获得的,结合能通过284.8ev处的c1s峰进行校准。对于x射线表征研究,将样品干燥,然后在测试之前在ar气氛下保存。所有x射线表征研究均在干燥后24小时内进行,以最大程度地减少样品的氧化。
[0171]
2.电化学测量
[0172]
电化学还原乙炔试验系统
[0173]
实验中采用的电化学还原乙炔试验系统视实验需要可选择图3
‑
1和图3
‑
2所示的系统,其可以是三电极体系,也可以是二电极体系,三电极体系只需在例如图3
‑
1所示系统阴极电解室中增设一参比电极,负载有ld
‑
cu、cu nps或pd/c的气体扩散电极作为工作电极(阴极);负载nife
‑
ldh纳米片的气体扩散电极或铂电极作为对电极;ag/agcl(饱和kcl)电极用作参比电极;其中电化学实验是使用chi660e电化学工作站(中国上海辰华);使用1m koh作为阴极电解质和阳极电解质(除非另有说明)。
[0174]
两种类型的乙炔气室用于电化学测量,一种乙炔气室为圆形腔室,如图3
‑
1和3
‑
2所示,几何电极面积为0.5cm2,2.0cm2或2.5cm2,另一种乙炔气室为矩形腔室,其中含有蛇形气道,几何面积为25cm2,如图11
‑
a所示。
[0175]
使用以下公式工作电极电势与rhe校正:
[0176]
e
rhe
=e
ag/agcl
0.1976 0.0591
×
ph
ꢀꢀ
[1]
[0177]
电化学还原乙炔过程
[0178]
在测量之前,首先将气体扩散电极(ld
‑
cu或cu nps)放置在乙炔气室和阴极电解室之间的界面处,然后以30ml min
‑1的流速用反应气体(5.0%c2h2 ar平衡气)吹扫气室5分钟,气体流速由质量流量计(sevenstar,d07
‑
19b),中国)控制。然后,将5ml 1m koh充满电化学电池的阴极腔室(或阴极电解室)和阳极腔室(或阳极电解室)。随后,将电极连接到电化学工作站。在每次原位电还原处理或ear测量之前,都要测试系统的电阻,以使干扰最小化(<5ohm)。在测量期间未应用ir校正。
[0179]
对于在没有乙烯的情况下的ear测试,以5.0%c2h2和ar平衡气的混合气为原料气。对于在过量乙烯存在下的ear测试,使用0.5%c2h2、20.0%c2h4和79.5%ar的混合气为原料气。除稳定性测试外,所有电化学测量均进行30分钟,并通过注射器从出口物流中抽取气体样品进行分析。使用配有三个通道的gc
‑
2014色谱仪(日本岛津制作所)分析气体样品。第一个通道使用hp plot al2o3色谱柱进行分离,使用he作为载气和氢火焰离子化检测器(fid)进行烃类分析。第二个通道使用微填充柱haysep q,h
‑
n色谱柱和molsieve 13x色谱柱进行分离,使用he作为载气和热导检测器(tcd)分析co2,n2,ar,o2,ch4和co。第三通道使用微填充柱hayesep q和molsieven2作为载气和tcd检测器分析h2。
[0180]
计算公式
[0181]
乙炔的转化率(conversion)和乙烯的选择性(selectivity)计算如下:
[0182][0183][0184]
其中c
feed
代表进料中乙炔的浓度。c
x
,c2h6和c4h
x
是产物中乙炔,乙烷和碳4烯烃的浓度。为了计算选择性,假定乙炔仅被氢化为乙烯,其又可以被氢化为乙烷。在此电化学实验中仅检测到碳2和碳4烃,碳平衡(检测到的产物中的总碳原子/进料气中的总碳原子)在97
‑
99%之间。
[0185]
出口物流中的h2体积(h
2 volume)计算如下:
[0186][0187]
其中,c2h2(feed)表示进料中乙炔的体积,c2h4(feed)表示进料中乙烯的体积,h2(generation)表示产生的氢气的体积,balance表示平衡气。
[0188]
在本研究中,公式[4]用于ear测试。
[0189]
乙炔100%转化所需的理论总电流(i)计算如下:
[0190][0191]
其中z是电子转移数(对于将乙炔还原为乙烯而言,等于2),f是法拉第常数(96485c mol
‑1),p是环境压力(101.325kpa),v是乙炔的速度,r为气体摩尔常数(8.314j mol
‑1k
‑1),t为温度(293.15k)。对于流量为1ml min
‑1的富乙烯气源(0.5%c2h2、20%c2h4与
ar平衡气),v为8.3
×
10
‑8l s
‑1,计算得出的理论总电流i为0.67ma。
[0192]
实施例
[0193]
使用如图3
‑
1或3
‑
2所示的电催化乙炔加氢反应系统作为反应系统。
[0194]
照图3
‑
1或3
‑
2中所示,将制备实施例所制备的负载有cual
‑
ldh纳米片、cu nps或pd/c的气体扩散电极(包括gdl和催化剂层)用作工作电极,固定在乙炔气室和阴极电解室之间,并按照图3
‑
1或3
‑
2搭建反应系统,其中,采用1m koh溶液为阴极电解质和阳极电解质,采用n
‑
117作为离子交换膜,碳布、pt或负载有nife
‑
ldh纳米片的碳基气体扩散电极被用作对电极,chi660e电化学工作站(中国上海辰华)为电源。根据需要,乙炔气室分别为几何截面积为0.5cm2,2.0cm2和2.5cm2,厚度为1.0mm的平面气室,或几何截面积为25cm2,厚度为1.0mm的矩形腔室,其中包含一个蛇形气道。
[0195]
除特殊说明外,本说明书中所采用的设备和方法均为本领域常规的设备和方法。
[0196]
实施例1 ld
‑
cu的ear性能
[0197]
工作电极为制备实施例所制备的负载有cual
‑
ldh纳米片的气体扩散电极,碳布作为阳极,气室为0.5cm2的圆形平面气室,如上所述搭建反应系统(图3
‑
1)后,接通电源,立即在1m koh中于
‑
0.4v下进行cual
‑
ldh电还原10分钟,以制备负载ldh衍生的铜催化剂(ld
‑
cu)(表s1)的气体扩散电极,之后由进气口向乙炔气室中注入原料气;持续接通电源,使电化学装置工作发生电解反应,以使扩散至工作电极的含乙炔的气体进行电催化乙炔加氢反应,使乙炔还原为乙烯,产物由出气口排出。实验过程中,控制气体流速为20ml min
‑1进行电催化乙炔加氢线性伏安极化曲线测试,测试结果如图6和图7所示。
[0198]
室温(20℃)下研究了在不存在乙烯的情况下ld
‑
cu的ear性能。在5%
‑
c2h2(ar为平衡气)下进行的线性扫描伏安法(lsv)测量显示,在
‑
0.3v时电流密度为2.5ma cm
‑2,随着电势降至
‑
0.6v逐渐增加至70ma cm
‑2以上(图7)。相比之下,使用5%
‑
c2h4(ar为平衡气)或纯ar进行的实验显示在ld
‑
cu上电化学响应可以忽略不计,这表明ld
‑
cu在所检查的电位范围内不催化c2h4的电还原。还测试了不添加催化剂的gdl的lsv性能。同样,没有检测到电流密度。
[0199]
如图6中的d图所示,在ld
‑
cu上,乙烯生产的起始电势(在10ma cm
‑2时)为
‑
0.39v,比析氢竞争反应的起始电位大210mv,证明了无氢ear的可能性流程(即不需要外部氢气进料即可生产乙烯的流程)。
[0200]
图6中的e图进一步表明,在ld
‑
cu上,乙烯是ear的主要产物,而c2h6的产生可以忽略不计(法拉第效率小于1%)。还检测到一些碳4低聚物(主要为c4h6),在负阴极电位更高时c4h6的法拉第效率降低(从
‑
0.28v的11.9%降至
‑
0.60v的2.7%)。与乙炔还原反应相比(c2h2 2h
2e
‑
→
c2h4,e0=0.73v),可以基于乙炔偶联反应的正极还原电位(2c2h2 2h
2e
‑
→
c4h6,e0=1.39v)理解产品选择性随施加的电位的变化。乙烯的法拉第效率从
‑
0.28v时的39.2
±
4.2%增加到
‑
0.52v时的79.5
±
5.0%。在
‑
0.6v时,法拉第效率略微下降至74.9
±
0.8%。因此,对阴极电位的明智选择是实现对乙烯的选择性ear的重要要求。
[0201]
如图6中的f图所示,使用ld
‑
cu作为催化剂的流动电化学系统提供的c2h4产品部分电流密度和法拉第效率分别比以前报道(现有文献1,davitt,h.j.&albright,l.f.electrochemical hydrogenation of ethylene,acetylene,and ethylene
‑
acetylene mixtures.j.electrochem.soc.118,236
‑
242(1971);现有文献2huang,b.,
durante,c.,isse,a.a.&gennaro,a.highly selective electrochemical hydrogenation of acetylene to ethylene at ag and cu cathodes.electrochem.commun.34,90
‑
93(2013))的ear结果高10倍和40%,数据如下表s2中所示。
[0202]
表s2.本发明ld
‑
cu and cu nps上的ear c2h4的分电流密度(j)和法拉第效率(fe)与现有技术的比较
[0203][0204]
实施例2 cu nps的ear性能
[0205]
除了使用以上制备实施例所制备的负载有商用cu nps(金属含量为99.9%,10
‑
30nm,上海麦克林生物化学有限公司)的气体扩散电极外,以与实施例1相似的方式进行电催化加氢反应过程:由进气口向乙炔气室中注入原料气;接通电源,使电化学装置工作发生电解反应,以使扩散至工作电极的含乙炔的气体进行电催化乙炔加氢反应,使乙炔还原为乙烯,产物由出气口排出。由此,测试了商用金属铜纳米颗粒催化剂(cu nps)的lsv曲线和电势相关的ear性能以及电化学反应前后gdl上的cu nps的sem图,如图8和9所示。cu nps上生成乙烯和氢气的起始电位之差为140mv。乙烯法拉第效率在较高的部分电流密度下急剧下降,这是由于乙烯生产起始电位较负(
‑
0.48v),her竞争反应严重。
[0206]
如图6中的f图所示,使用cu nps作为催化剂的流动电化学系统提供的c2h4产品部分电流密度和法拉第效率亦分别比以前报道的ear结果高,数据如表s2中所示。
[0207]
实施例3在乙烯存在下关于ld
‑
cu的ear性能
[0208]
除了使用富含乙烯的气体源(0.5%c2h2、20%c2h4和ar平衡气)以及根据需要更改
流速和气室结构之外,以与实施例1相似的方式在室温(20℃)下测试。还监测出口物流中的氢气量,以证明ear工艺的无氢气特性。由于进料气中存在大量的乙烯,因此除h2以外,以碳为基础计算乙烯的生产选择性。
[0209]
对于0.5cm2的平面气室,图10中的a图显示了在不同进料气流速下的乙炔转化率。通过阴极电势调节,以1ml min
‑1的流速可以转化超过98%的乙炔,在
‑
0.4v至
‑
0.5v的电势下观察到最佳的转化。较高的流速导致乙炔的转化率降低(流速分别为5和20ml min
‑1时分别达到88.9%和33.4%)。然后在1ml min
‑1的固定流速下测试随时间变化的ear性能。在这些条件下,将乙炔100%转化所需的理论总电流经计算为0.67ma(请参阅以上计算公式和方法)。从图10中的b图可以看出,乙炔转化率保持恒定在99.1
±
0.4%。重要的是,乙烯是乙炔电还原的主要产物,在5小时的操作中,平均乙烯选择性达到93.2
±
0.6%(碳4烯烃和乙烷分别为6.3%和0.5%)。出口物流中的h2体积百分比保持为0.07
±
0.02%,表明竞争性氢的释放得到了有效抑制。与乙炔转化率不同,产物的选择性主要取决于电势,并且对气体流速的敏感性低得多(图10中的c图)。即使当流速增加到20ml min
‑1时,乙烯的选择性仍保持在90%以上。此外,所产生的任何h2均以较高的流速稀释。重要的是,发现乙炔转化高度依赖于气室面积(或电极面积),如图10中的d图所示。在将流速提高到10ml min
‑1时,在0.5cm2电极上的乙炔转化率降低到小于50%,而对于2.0cm2电极面积,乙炔转化率超过99%。由于阴极电位从
‑
0.63v增加到
‑
0.46v,h2体积百分比也降低了。
[0210]
实施例4扩大乙炔气室中在乙烯存在下关于ld
‑
cu的ear性能
[0211]
为了进一步提高在较高流速下的乙炔转化性能并评估ear的应用前景,本实施例采用了一种扩大的类似图3
‑
2的两电极流动反应器,该反应器的乙炔气室为矩形腔室,其中包含一个蛇形气道(如图11
‑
a至11
‑
c所示),其几何电极面积为5.0cm
×
5.0cm=25.0cm2,高度为1.0mm(图12中的a,b图)。制备实施例中制备的负载ld
‑
cu铜催化剂的气体扩散电极被用于工作电极(阴极),沉积有nife
‑
ldh纳米片的碳基气体扩散电极被用于析氧反应(oer)的阳极(图13示出了nife
‑
ldh纳米片的结构及阳极性能:a为nife
‑
ldh纳米片的透射电子显微成像;b为nife
‑
ldh纳米片的x射线衍射图谱;c为在1m koh中nife
‑
ldh负载阳极上的线性扫描伏安曲线)。反应温度为室温(20℃),反应原料气为0.5%c2h2、20%c2h4和ar平衡气的混合气。
[0212]
测试了该扩大流量反应器上的ear,流速为50ml min
‑1(空速为1200h
‑1)。如图12中的c
‑
e图所示,在4小时的操作中保持了99.9
±
0.1%的平均乙炔转化率,显示出高的c2h2加氢比活性834mmol g
‑1h
‑1(24.8μmol cm
‑2h
‑1)。产物中乙烯/乙炔的体积比超过1
×
105,满足了聚乙烯级乙烯原料的纯度要求(图14和表s3)。此外,该方法在出口物流中保持非常低的氢气体积百分比,为0.08
±
0.01%。如图12中的d图所示,在扩大的流动反应器中ld
‑
cu上的产物分布与图10中的b图所示的慢流量下的小型反应器相似,平均乙烯选择性为90.1
±
0.8%(对于碳4烯烃和乙烷,分别为9.2
±
0.9%和0.68
±
0.12%)。在测试过程中,电池电压从1.95v略微增加到1.98v(图12中的e图),这可能是由于ld
‑
cu催化剂的不稳定性或电解质渗透作用所致,因为测试后gdl的疏水性降低了(水接触角从149
°
减小至112
°
)。如图12中的f图和表4所示,ld
‑
cu上的ear工艺(就乙炔转化率和乙烯选择性而言)与最先进的热氢化催化剂参考文献3(armbruster,m.et al.al
13
fe
4 as a low
‑
cost alternative for palladium in heterogeneous hydrogenation.nat.mater.11,690
‑
693(2012))、4(feng,
q.et al.isolated single
‑
atom pd sites in intermetallic nanostructures:high catalytic selectivity for semihydrogenation of alkynes.j.am.chem.soc.139,7294
‑
7301(2017))、5(hu,m.et al.mof
‑
confined sub
‑
2nm atomically ordered intermetallic pdzn nanoparticles as high
‑
performance catalysts for selective hydrogenation of acetylene.adv.mater.1801878(2018))、6(pei,g.x.et al.performance of cu
‑
alloyed pd single
‑
atom catalyst for semihydrogenation of acetylene under simulated front
‑
end conditions.acs catal.7,1491
‑
1500(2017))7(huang,f.et al.anchoring cu
1 species over nanodiamond
‑
graphene for semi
‑
hydrogenation of acetylene.nat.commun.10,4431(2019))相比具有优势,同时展示了残留氢气可忽略不计的室温操作和高纯度乙烯生产的特殊优势。
[0213]
表s3.在图12中的从全电池ear性能测试中获得的c2h4和c2h2的fid通道积分面积
[0214][0215]
表s4.ld
‑
cu与先前报道的热氢化催化剂(现有文献3
‑
7)的乙炔向乙烯转化的性能比较
[0216][0217]
考虑到聚乙烯工业的大规模,乙炔制乙烯的原子经济性和能源经济性是需要考虑的重要评估标准。如图12中的g图所示,对于乙炔经加氢路线转化为乙烯,h2的生产会消耗
该过程所需的大部分总能量,因为随后的乙炔加氢反应是自发的(标准吉布斯自由能,δg=
‑
141kj mol
‑1)。蒸汽甲烷重整和水分解是两种典型的h2生产技术。甲烷的蒸汽重整具有相对较低的δg,为33kj mol
‑1,但每个氢化的c2h2分子产生0.25当量的co2。用于生产h2的水分解过程不含co2,但在热力学上不利(δg=237kj mol
‑1),这意味着串联水分解
‑
乙炔加氢路线在能源上是浪费的。相反,ear过程代表了一种一步策略,该策略既具有较低的理论能量输入要求(δg=96kj mol
‑1),又具有原子效率(即,只要用于驱动反应的电力来自可再生能源,就不会有温室气体的排放)。考虑到原子和能量的经济性以及性能,相对于传统的热加氢路线,在富含乙烯的物流中室温电化学乙炔还原看来是一种绿色有效的方法,应引起塑料行业的关注。
[0218]
实施例5 pd/c(商业钯碳)催化剂的电催化乙炔加氢性能(5%c2h2)
[0219]
采用制备实施例制备的负载pd/c(商业钯碳)催化剂的气体扩散电极,以与实施例1相同的反应系统,乙炔气室为0.5cm2的平面气室,5%
‑
c2h2(ar为平衡气)作为气体来源,流速为20ml min
‑1,测试了pd/c(商业钯碳)催化剂的电催化乙炔加氢性能,结果如图15所示,示出了电催化乙炔加氢产物的电流密度与施加电势的关系,显示了良好的乙烯选择性。
[0220]
实施例6 ld
‑
cu的温度相关ear性能
[0221]
以与实施例1相同的反应系统,工作电极面积为0.5cm2,阴极电势为
‑
0.4v,流速为20ml min
‑1,在5℃至25℃的温度范围内研究了ld
‑
cu的温度相关ear性能。如图16所示,对于乙炔气室中5%
‑
c2h2(ar为平衡气),观察到乙炔转化率与反应温度之间的阿累尼乌斯(arrhenius)线性关系,在
‑
0.4v提供了21.4kj mol
‑1的表观活化能。
[0222]
还测试了h型电解池(传统h电解池结构)中ld
‑
cu随温度变化的ear性能,在h电池中,将电极浸入5%
‑
c2h2(ar为平衡气)饱和的1m koh中,gdl被硅脂封闭,以消除气体通过gdl扩散。与本发明反应系统乙炔气室中5%
‑
c2h2在ld
‑
cu上的反应相比,乙炔转化率降低了约70倍。另外,发现低温促进反应动力学(呈异常的非线性关系),表明溶解度控制的液相扩散过程是h电池中的速率决定步骤。
[0223]
实施例7不同乙炔气室厚度对乙炔转化率的影响
[0224]
以与实施例1相似的反应系统,实验对比了两种乙炔气室厚度(1.0mm和10.0mm)在不同空速下的乙炔转化率。电极面积均为0.5cm2,原料气为0.5%c2h2 20%c2h4 79.5%ar平衡气,阴极电压为
‑
0.5v vs.rhe,电解时长1小时。厚度为1.0mm时,500
‑
3000h
‑1空速下乙炔转化率保持在98%以上,6000h
‑1下略有下降,为94.4%。厚度为10.0mm时,转化率随空速升高迅速下降,由500h
‑1下的58.7%降至6000h
‑1下的13.0%,具体如图17所示。该实验证明降低乙炔气室厚度能够大幅提高乙炔的电催化加氢转化率。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。