本发明涉及功能化材料技术领域,具体涉及一种氮和磷共掺杂碳层包裹磷化二钴催化剂及其制备方法和应用。
背景技术:
通过水分解生产氢气是一种有前途的解决方案,无二氧化碳排放,可以满足迅速增长的全球能源需求和环境问题。水分解过程由两电子转移反应(氢析出反应,her)和四电子-质子耦合反应(氧析出反应,oer)组成,需要的热力学吉布斯自由能(δg)约为237.13kj·mol-1,对应于可逆氢电极(rhe)的标准电势(δe)为1.23v。太阳能驱动的水分解总是在恶劣的条件下进行,例如间歇性与位置有关的太阳辐射,以及实验室规模的当前太阳能制氢(sth)值仍然非常小。
电化学水分解被认为是在温和条件下运行并由可再生能源发电生产氢气的重要策略,然而常规的贵金属电催化剂,例如铂和钌/铱基催化剂(及其合金和金属氧化物),具有高成本和地球稀缺性的特点,限制了其应用。
技术实现要素:
本发明的目的在于提供一种氮和磷共掺杂碳层包裹磷化二钴催化剂及其制备方法和应用,本发明提供的催化剂具有优异的双功能电催化活性,且成本较低,对环境友好。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了一种氮和磷共掺杂碳层包裹磷化二钴催化剂,包括碳基材料以及分布在所述碳基材料上的氮和磷共掺杂碳层包裹磷化二钴纳米颗粒;
所述氮和磷共掺杂碳层包裹磷化二钴纳米颗粒包括磷化二钴纳米颗粒和包覆在所述磷化二钴纳米颗粒表面的碳层,所述碳层中掺杂有氮元素和磷元素。
优选地,所述氮和磷共掺杂碳层包裹磷化二钴纳米颗粒中氮元素的含量为0.5~15wt%,磷元素的含量为0.5~15wt%。
优选地,所述磷化二钴纳米颗粒的粒径为100~200nm。
本发明提供了上述技术方案所述氮和磷共掺杂碳层包裹磷化二钴催化剂的制备方法,包括以下步骤:
将二价锌盐、二价钴盐、尿素、氟化铵和水混合,得到混合溶液;
将碳纤维布置于所述混合溶液中,进行水热反应,得到znco/cc前驱体;
将所述znco/cc前驱体置于2-甲基咪唑水溶液中,进行浸渍,得到znco-mof/cc;
将所述znco-mof/cc进行煅烧,得到co/nc;
在所述co/nc中掺杂磷元素和氮元素,得到氮和磷共掺杂碳层包裹磷化二钴催化剂。
优选地,所述二价锌盐、二价钴盐、尿素和氟化铵的摩尔比为0.5~1:1~2:6~10:2~4。
优选地,所述水热反应的温度为100~160℃;所述水热反应的时间为6~10h。
优选地,所述煅烧的温度为800~900℃;所述煅烧的时间为2~3h。
优选地,在所述co/nc中掺杂磷元素的方法包括:将所述co/nc和磷源分别置于两个瓷舟中,在管式炉的上游放置盛有磷源的瓷舟,下游放置盛有co/nc的瓷舟,在管式炉中充入流动的保护性气体,进行磷化反应。
本发明提供了上述技术方案所述氮和磷共掺杂碳层包裹磷化二钴催化剂或上述技术方案所述制备方法制备得到的氮和磷共掺杂碳层包裹磷化二钴催化剂在电催化分解水中的应用。
优选地,所述氮和磷共掺杂碳层包裹磷化二钴催化剂作为阳极和/或阴极。
本发明提供了一种氮和磷共掺杂碳层包裹磷化二钴催化剂,包括碳基材料以及分布在所述碳基材料上的氮和磷共掺杂碳层包裹磷化二钴纳米颗粒;所述氮和磷共掺杂碳层包裹磷化二钴纳米颗粒包括磷化二钴纳米颗粒和包覆在所述磷化二钴纳米颗粒表面的碳层,所述碳层中掺杂有氮元素和磷元素。在本发明中,磷化二钴纳米颗粒包裹在n、p掺杂的碳层中,由于n的掺杂效应,调控了co2p的电子结构,使得本发明提供的催化剂不仅提供了更多的活性位点,而且在碱性电解质中,n、p掺杂的碳层具有较高的稳定性,可以有效的保护催化剂免于降解和团聚。本发明提供的氮和磷共掺杂碳层包裹磷化二钴催化剂具有优异的双功能电催化活性,远远优于市售pt/c和ruo2以及大多数先前报道的催化剂,而且本发明提供的催化剂成本较低,适宜大规模推广应用。
附图说明
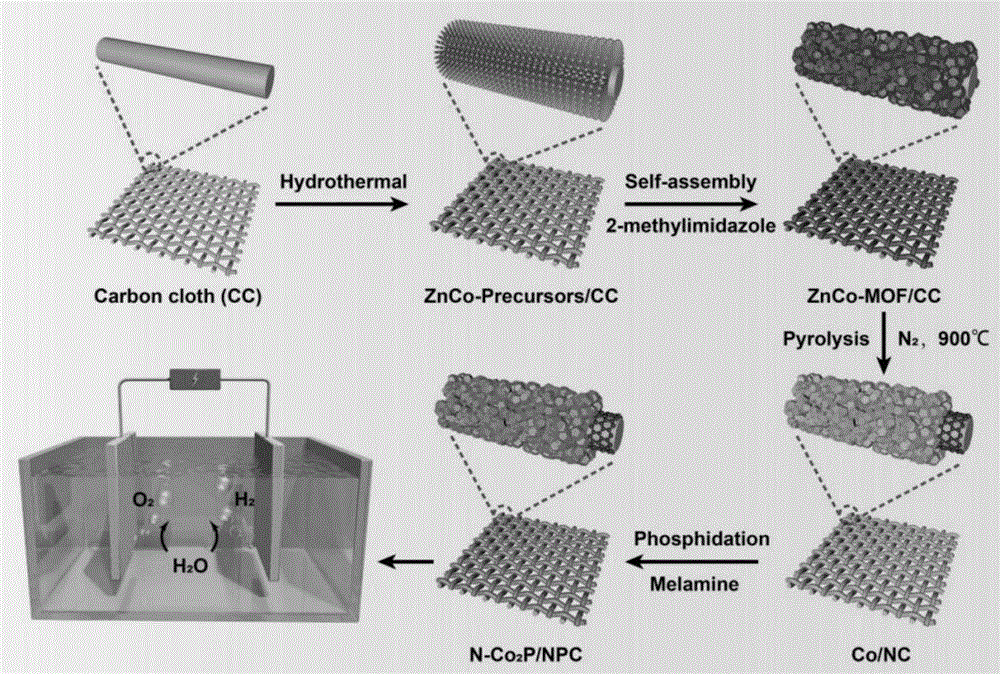
图1为本发明制备氮和磷共掺杂碳层包裹磷化二钴催化剂的原理示意图;
图2为对比例1~2和实施例1制备的催化剂的x射线衍射图;
图3为对比例2和实施例1制备的催化剂的sem图和tem图;
图4为对比例1~2和实施例1制备的催化剂的电催化析氢线性扫描曲线图;
图5为对比例1~2和实施例1制备的催化剂的电催化析氧线性扫描曲线图;
图6为实施例1制备的氮和磷共掺杂碳层包裹磷化二钴催化剂的电催化水分解线性扫描曲线图。
具体实施方式
本发明提供了一种氮和磷共掺杂碳层包裹磷化二钴催化剂,包括碳基材料以及分布在所述碳基材料上的氮和磷共掺杂碳层包裹磷化二钴纳米颗粒;
所述氮和磷共掺杂碳层包裹磷化二钴纳米颗粒包括磷化二钴纳米颗粒和包覆在所述磷化二钴纳米颗粒表面的碳层,所述碳层中掺杂有氮元素和磷元素。
本发明提供的催化剂包括碳基材料。在本发明中,所述碳基材料优选包括碳纤维布。在本发明中,所述碳基材料优选为多孔结构,孔径优选为0.05~0.1mm,孔隙率优选为20~40%。
在本发明中,所述碳基材料的厚度优选为0.1~0.2mm,更优选为0.15mm。
在本发明中,所述碳基材料作为导电载体,负载氮和磷共掺杂碳层包裹磷化二钴纳米颗粒。
本发明提供的催化剂包括分布在所述碳基材料上的氮和磷共掺杂碳层包裹磷化二钴纳米颗粒。在本发明中,所述氮和磷共掺杂碳层包裹磷化二钴纳米颗粒优选包裹在所述碳基材料的碳纤维表面。在本发明中,基于碳基材料,所述氮和磷共掺杂碳层包裹磷化二钴纳米颗粒在碳基材料上的负载量优选为1~3mg/cm2,更优选为2.5mg/cm2。
在本发明中,所述氮和磷共掺杂碳层包裹磷化二钴纳米颗粒包括磷化二钴纳米颗粒和包覆在所述磷化二钴纳米颗粒表面的碳层,所述碳层中掺杂有氮元素和磷元素。在本发明中,所述氮和磷共掺杂碳层包裹磷化二钴纳米颗粒中氮元素的含量优选为0.5~15wt%,更优选为5~10wt%;磷元素的含量优选为0.5~15wt%,更优选为5~10wt%;所述磷元素的含量包括磷化二钴纳米颗粒中的磷元素和碳层中的磷元素的总含量。在本发明中,所述磷化二钴纳米颗粒中磷元素的含量优选为15wt%。
在本发明中,所述磷化二钴纳米颗粒的粒径优选为100~200nm。在本发明中,所述磷化二钴纳米颗粒表面的碳层的厚度优选为2~10nm,更优选为5nm。在本发明中,所述磷化二钴纳米颗粒是双功能催化剂的主要活性体;所述氮磷掺杂的碳层能够抑制磷化二钴的表面氧化并提供催化位点。
本发明还提供了上述技术方案所述氮和磷共掺杂碳层包裹磷化二钴催化剂的制备方法,如图1所示,包括以下步骤:
将二价锌盐、二价钴盐、尿素、氟化铵和水混合,得到混合溶液;
将碳纤维布置于所述混合溶液中,进行水热反应,得到znco/cc前驱体;
将所述znco/cc前驱体置于2-甲基咪唑水溶液中,进行浸渍,得到znco-mof/cc;
将所述znco-mof/cc进行煅烧,得到co/nc;
在所述co/nc中掺杂磷元素和氮元素,得到氮和磷共掺杂碳层包裹磷化二钴催化剂。
在本发明中,若没有特殊说明,所采用的制备原料均为本领域技术人员所熟知的市售商品。
本发明采用改进的湿化学合成策略制备氮和磷共掺杂碳层包裹磷化二钴催化剂,制备方法简单易操作,催化剂的催化活性高,成本低,对环境友好无污染。
本发明将二价锌盐、二价钴盐、尿素、氟化铵和水混合,得到混合溶液。在本发明中,所述二价锌盐、二价钴盐、尿素和氟化铵的摩尔比优选为0.5~1:1~2:6~10:2~4,更优选为0.5~0.6:1~1.5:6:2.5。在本发明中,所述二价锌盐优选包括硝酸锌、氯化锌、硫酸锌和醋酸锌中的一种,更优选为zn(no3)2·6h2o;所述二价钴盐优选包括硝酸钴、氯化钴、醋酸钴和硫酸钴中的一种,更优选为co(no3)2·6h2o。
在本发明中,所述混合溶液的浓度优选为9~12mmol/l,更优选为10mmol/l。
本发明对所述二价锌盐、二价钴盐、尿素、氟化铵和水混合的具体方法没有特殊要求,采用本领域技术人员所熟知的混合方式即可,具体优选为磁力搅拌。在本发明中,所述混合的时间优选为30min。
得到混合溶液后,本发明将碳纤维布置于所述混合溶液中,进行水热反应,得到znco/cc前驱体。在本发明中,所述碳纤维布的尺寸优选为2cm×3cm,厚度优选为0.1~0.2mm,更优选为0.15mm。在本发明中,所述碳纤维布优选为亲水性碳纤维布。在本发明中,所述碳纤维布优选先进行预处理。在本发明中,所述预处理的方法优选包括:将碳纤维布依次进行水洗、醇洗和酸洗。在本发明中,所述水洗、醇洗和酸洗优选独立地在超声条件下进行;所述醇洗采用的洗液优选为乙醇;所述酸洗采用的酸洗液优选包括硝酸和硫酸;所述硝酸的浓度优选为5mol/l,所述硫酸的浓度优选为3mol/l。本发明对碳纤维布进行预处理能够增加其亲水性。
本发明对所述碳纤维布和混合溶液的用量没有特殊要求,以将所述碳纤维布浸泡在所述混合溶液中为宜。在本发明中,所述水热反应的温度优选为100~160℃,更优选为120~140℃;所述水热反应的时间优选为6~10h,更优选为6~8h。本发明在所述水热反应过程中,尿素和氟化铵与锌离子、钴离子结合,形成锌钴金属氢氧化物前驱体。
本发明优选在所述水热反应后,将所得碳布依次进行洗涤和干燥,得到znco/cc前驱体。在本发明中,所述洗涤优选包括采用去离子水和无水乙醇分别清洗三次。在本发明中,所述干燥的温度优选为60~80℃;所述干燥的时间优选为4~8h;所述干燥优选为真空干燥。
在本发明中,所述znco/cc前驱体(zn-coprecursor/cc)优选为纳米针阵列结构,该结构有利于催化反应气体的释放以及有利于电荷转移。
得到znco/cc前驱体后,本发明将所述znco/cc前驱体置于2-甲基咪唑水溶液中,进行浸渍,得到znco-mof/cc。在本发明中,所述2-甲基咪唑水溶液的浓度优选为50~200mg/ml,更优选为100~150mg/ml。在本发明中,所述浸渍优选在室温条件下进行,所述浸渍的时间优选为6~12h,更优选为8~10h。本发明在所述浸渍过程中,2-甲基咪唑和锌离子、钴离子耦合为金属有机框架结构。
本发明优选将浸渍后的碳布依次进行洗涤和干燥,得到znco-mof/cc。在本发明中,所述洗涤优选包括采用去离子水和无水乙醇分别清洗三次。在本发明中,所述干燥的温度优选为60~80℃;所述干燥的时间优选为4~8h,更优选为6h;所述干燥优选为真空干燥。
得到znco-mof/cc后,本发明将所述znco-mof/cc进行煅烧,得到co/nc。在本发明中,所述煅烧优选在保护性气氛条件下进行,更优选为氩气气氛或氮气气氛。在本发明中,所述煅烧的温度优选为800~900℃,更优选为900℃;所述煅烧的时间优选为2~3h,更优选为3h。在本发明中,由室温升至所述煅烧的温度的升温速率优选为2~5℃/min,更优选为3~4℃/min。本发明在所述煅烧过程中,发生高温碳化反应,znco-mof在煅烧过程中,与金属络合的有机配体(包含碳和氮配体)发生碳化,形成氮掺杂的碳层结构,锌原子煅烧之后气化,只剩下钴纳米颗粒包裹于氮掺杂碳层中;碳纤维布作为催化剂的载体,无变化。
本发明优选在所述煅烧后,将所得碳布自然冷却至室温,得到co/nc。
得到co/nc后,本发明在所述co/nc中掺杂磷元素和氮元素,得到氮和磷共掺杂碳层包裹磷化二钴催化剂。本发明中在所述co/nc中掺杂磷元素的方法优选包括:将所述co/nc和磷源分别置于两个瓷舟中,在管式炉的上游放置盛有磷源的瓷舟,下游放置盛有co/nc的瓷舟,在管式炉中充入流动的保护性气体,进行磷化反应。在本发明中,所述磷源优选包括次磷酸钠和红磷。在本发明中,所述保护性气体优选包括氩气或氮气。在本发明中,所述磷化反应的温度优选为300~400℃;所述磷化反应的时间优选为1~3h,更优选为1~2h;由室温升至所述磷化反应的温度的升温速率优选为2~5℃/min,更优选为2~3℃/min。在所述磷化反应过程中,磷源(次磷酸钠)高温气化产生磷化氢,磷化氢和钴结合,生成co2p。
本发明在所述co/nc中掺杂氮元素的方法优选包括:将所述co/nc和氮源分别置于两个瓷舟中,在管式炉的上游放置盛有氮源的瓷舟,下游放置盛有co/nc的瓷舟,在管式炉中充入流动的保护性气体,进行氮化反应。在本发明中,所述氮源优选包括三聚氰胺或尿素。在本发明中,所述保护性气体优选包括氩气或氮气。在本发明中,所述氮化反应的温度优选为500~700℃,更优选为600℃;所述氮化反应的时间优选为0.5~2h,更优选为1h;由室温升至所述氮化反应的温度的升温速率优选为2~5℃/min,更优选为2~3℃/min。在所述氮化反应过程中,氮源高温气化产生氮化碳气体,氮化碳与磷化二钴结合,完成氮掺杂。
在本发明的具体实施例中,在所述co/nc中掺杂磷元素和氮元素方法优选包括:将所述co/nc置于管式炉的下游,将磷源和氮源的混合物置于管式炉的上游,在管式炉中充入流动的保护性气体,依次进行磷化反应和氮化反应。在本发明中,所述磷源优选包括次磷酸钠和红磷;所述氮源优选包括三聚氰胺或尿素;所述保护性气体优选包括氩气或氮气。在本发明中,所述磷化反应的温度优选为300~400℃;所述磷化反应的时间优选为2~3h;所述氮化反应的温度优选为500~700℃,更优选为600℃;所述氮化反应的时间优选为0.5~2h,更优选为1h;由磷化反应的温度升至所述氮化反应的温度的升温速率优选为2~3℃/min。
在本发明的具体实施例中,当所述碳纤维布的尺寸为2cm×3cm时,所述磷源的添加量优选为5~8mmol,更优选为5mmol;所述氮源的添加量优选为0.5~2mmol,更优选为1mmol。
本发明优选在所述氮化反应后,将所得碳布自然冷却至室温,得到氮和磷共掺杂碳层包裹磷化二钴催化剂(n-co2p/npc)。
本发明还提供了上述技术方案所述氮和磷共掺杂碳层包裹磷化二钴催化剂或上述技术方案所述制备方法制备得到的氮和磷共掺杂碳层包裹磷化二钴催化剂在电催化分解水中的应用。
在本发明中,所述氮和磷共掺杂碳层包裹磷化二钴催化剂优选作为阳极和/或阴极。本发明提供的氮和磷共掺杂碳层包裹磷化二钴催化剂为双功能水分解电催化剂,既可以作为阳极也可以作为阴极,用于整体水分解。
在本发明中,所述在电催化分解水的电解质优选为碱性电解质,更优选为1mol/l的氢氧化钠溶液。
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
将0.5mmolzn(no3)2·6h2o、1mmolco(no3)2·6h2o、6mmol尿素和2.5mmol氟化铵溶于35ml水中,并剧烈搅拌30分钟后,得到混合溶液;
将尺寸为2cm×3cm,厚度为0.15mm的碳纤维布依次进行超声波水洗、超声波乙醇洗和超声波酸洗,得到预处理后的碳纤维布;所述超声波酸洗用酸洗液由硝酸和硫酸组成,硝酸的浓度为5mol/l,硫酸的浓度为3mol/l;
将所述混合溶液转移到有teflon内衬的不锈钢高压釜中,并将所述预处理后的碳纤维布浸入混合溶液中,在120℃进行水热反应8小时,当温度冷却到室温时,用去离子水和无水乙醇分别清洗三次,并在真空干燥箱中于60℃干燥6h,在碳布上合成zn-co纳米针阵列前体(zn-coprecursor/cc);
将所述zn-coprecursor/cc浸入100mg/ml的2-甲基咪唑水溶液中12小时,碳布从粉红色变为紫色,用去离子水和无水乙醇分别清洗三次,然后在60℃条件下真空干燥24小时,得到znco-mof/cc;
将所述znco-mof/cc在流动的氩气气氛下加热至900℃保持3小时,加热速率为3℃/min,然后自然冷却至室温,znco-mof纳米阵列变成氮掺杂碳层包裹钴纳米颗粒,得到co/nc;
将所述co/nc置于管式炉的下游,将500mg次磷酸铵和100mg三聚氰胺的混合粉末置于管式炉的上游,在流动的氩气气氛中,将管式炉以2℃/min的加热速率加热至300℃并保持1小时;接着以3℃/min的加热速率加热至600℃保温1小时,然后自然冷却至室温,得到n-co2p/npc。
实施例2
将0.6mmolzn(no3)2·6h2o、1.5mmolco(no3)2·6h2o、6mmol尿素和2.5mmol氟化铵溶于35ml水中,并剧烈搅拌30分钟后,得到混合溶液;
将尺寸为2cm×3cm,厚度为0.15mm的碳纤维布依次进行超声波水洗、超声波乙醇洗和超声波酸洗,得到预处理后的碳纤维布;所述超声波酸洗用酸洗液由硝酸和硫酸组成,硝酸的浓度为5mol/l,硫酸的浓度为3mol/l;
将所述混合溶液转移到有teflon内衬的不锈钢高压釜中,并将所述预处理后的碳纤维布浸入混合溶液中,在120℃进行水热反应6小时,当温度冷却到室温时,用去离子水和无水乙醇分别清洗三次,并在真空干燥箱中于60℃干燥6h,在碳布上合成zn-co纳米针阵列前体(zn-coprecursor/cc);
将所述zn-coprecursor/cc浸入100mg/ml的2-甲基咪唑水溶液中12小时,碳布从粉红色变为紫色,用去离子水和无水乙醇分别清洗三次,然后在60℃条件下真空干燥24小时,得到znco-mof/cc;
将所述znco-mof/cc在流动的氩气气氛下加热至800℃保持3小时,加热速率为2℃/min,然后自然冷却至室温,znco-mof纳米阵列变成氮掺杂碳层包裹钴纳米颗粒,得到co/nc;
将所述co/nc置于管式炉的下游,将600mg次磷酸铵和150mg三聚氰胺的混合粉末置于管式炉的上游,在流动的氩气气氛中,将管式炉以2℃/min的加热速率加热至300℃并保持1小时;接着以2℃/min的加热速率加热至600℃保温1小时,然后自然冷却至室温,得到n-co2p/npc。
对比例1
将0.6mmolzn(no3)2·6h2o、1.5mmolco(no3)2·6h2o、6mmol尿素和2.5mmol氟化铵溶于35ml水中,并剧烈搅拌30分钟后,得到混合溶液;
将尺寸为2cm×3cm,厚度为0.15mm的碳纤维布依次进行超声波水洗、超声波乙醇洗和超声波酸洗,得到预处理后的碳纤维布;所述超声波酸洗用酸洗液由硝酸和硫酸组成,硝酸的浓度为5mol/l,硫酸的浓度为3mol/l;
将所述混合溶液转移到有teflon内衬的不锈钢高压釜中,并将所述预处理后的碳纤维布浸入混合溶液中,在120℃进行水热反应6小时,当温度冷却到室温时,用去离子水和无水乙醇分别清洗三次,并在真空干燥箱中于60℃干燥6h,在碳布上合成zn-co纳米针阵列前体(zn-coprecursor/cc);
将所述zn-coprecursor/cc浸入100mg/ml的2-甲基咪唑水溶液中12小时,碳布从粉红色变为紫色,用去离子水和无水乙醇分别清洗三次,然后在60℃条件下真空干燥24小时,得到znco-mof/cc;
将所述znco-mof/cc在流动的氩气气氛下加热至800℃保持3小时,加热速率为2℃/min,然后自然冷却至室温,znco-mof纳米阵列变成氮掺杂碳层包裹钴纳米颗粒,得到co/nc;
将所述co/nc和次磷酸钠分别放在两个瓷舟中,并在管式炉的上游放置500mg次磷酸钠,下游放置co/nc;随后,将管式炉以2℃/min的加热速率加热至300℃,并在流动的氩气气氛中保持2小时,然后自然冷却至室温,得到co2p/npc。
对比例2
将0.5mmolzn(no3)2·6h2o、1mmolco(no3)2·6h2o、6mmol尿素和2.5mmol氟化铵溶于35ml水中,并剧烈搅拌30分钟后,得到混合溶液;
将尺寸为2cm×3cm,厚度为0.15mm的碳纤维布依次进行超声波水洗、超声波乙醇洗和超声波酸洗,得到预处理后的碳纤维布;所述超声波酸洗用酸洗液由硝酸和硫酸组成,硝酸的浓度为5mol/l,硫酸的浓度为3mol/l;
将所述混合溶液转移到有teflon内衬的不锈钢高压釜中,并将所述预处理后的碳纤维布浸入混合溶液中,在120℃进行水热反应8小时,当温度冷却到室温时,用去离子水和无水乙醇分别清洗三次,并在真空干燥箱中于60℃干燥6h,在碳布上合成zn-co纳米针阵列前体(zn-coprecursor/cc);
将所述zn-coprecursor/cc浸入100mg/ml的2-甲基咪唑水溶液中12小时,碳布从粉红色变为紫色,用去离子水和无水乙醇分别清洗三次,然后在60℃条件下真空干燥24小时,得到znco-mof/cc;
将所述znco-mof/cc在流动的氩气气氛下加热至900℃保持3小时,加热速率为3℃/min,然后自然冷却至室温,znco-mof纳米阵列变成氮掺杂碳层包裹钴纳米颗粒,得到co/nc;
将所述co/nc和次磷酸钠分别放在两个瓷舟中,并在管式炉的上游放置600mg次磷酸钠,下游放置co/nc;随后,将管式炉以3℃/min的加热速率加热至300℃,并在流动的氩气气氛中保持2小时,然后自然冷却至室温,得到co2p/npc。
测试例1
对比例1~2和实施例1制备的催化剂的x射线衍射图如图2所示。其中co/nc是对比例1中制备的;co2p/npc是对比例2中制备的;n-co2p/npc是实施例1制备的。由图2可以看出,co2p/npc和n-co2p/npc在42.5°、43.3°、44.1°、50.4°、52.0°和56.2°的衍射峰对应co2p(jcpds#32-0306)的(220)、(211)、(130)、(310)、(002)和(320)晶面,证明co2p/npc和n-co2p/npc的成功制备。其中,位于36.2°和61.3°处有两个衍射峰,可以联系到con(jcpds#16-0116)的(111)和(220)晶面;大约26.4°的宽衍射峰,对应碳布的石墨2h相(jcpds#141-1487)的(002)晶面。
测试例2
对比例2和实施例1制备的催化剂的sem图和tem图如图3所示。其中图3中的a是对比例2制备的co2p/npc的sem图;图3中的b是对比例2制备的co2p/npc的放大sem图;图3中的c是对比例2制备的co2p/npc的tem图;图3中的d是实施例1制备的n-co2p/npc的sem图;图3中的e是实施例1制备的n-co2p/npc的放大的sem图;图3中的f是实施例1制备的n-co2p/npc的tem图。由图3中的d和e可以看出,n-co2p/npc是由碳层、n-co2p纳米粒子和碳纤维均匀排列组成的分层结构,碳层包裹n-co2p纳米粒子,并生长在碳纤维上,形成了独特的异质结构纤维的内部基本单元,并提供了较大的表面积,相对于原始碳纤维产生更多的活性位,这有利于整个电极的离子和电子传输。图3中的f进一步表明,尺寸为100~200nm的co2p纳米粒子紧密嵌入n、p掺杂的碳层中。
图3中的a、b以及c显示,co2p/npc是由碳层、co2p纳米粒子和碳纤维均匀排列组成的分层结构;与n-co2p/npc相比,颗粒大小不均匀。
测试例3
对比例1~2和实施例1制备的催化剂的电催化析氢线性扫描曲线图如图4所示。其中,pt/c为市售催化剂,将pt/c以2.5mg/cm2的负载量涂到碳布上进行检测,co/nc是对比例1中制备的;co2p/npc是对比例2制备的;n-co2p/npc是实施例1制备的。
检测方法为:在1.0mol/l的koh溶液中使用标准三电极系统,将催化剂作为工作电极,碳棒为对电极,汞/氧化汞(hg/hgo)电极作为参比电极,在电化学工作站(chi760e)上进行测试。根据以下公式校正所测得的电势:e(rhe)=e(hg/hgo) 0.098v 0.059×ph,并通过几何表面积将电流密度(j)归一化,所得结果如图4所示。
由图4可以看出,采用本发明制备的n-co2p/npc作为电极具有出色的催化活性,电流密度为10ma·cm-2和100ma·cm-2时所需的过电位分别为68mv和155mv,远小于co2p/npc(140mv和242mv),co/nc(158mv和291mv),同时,当电流密度超过100ma·cm-2时,催化活性优于商业20%wtpt/c催化剂。
测试例4
对比例1~2和实施例1制备的催化剂的电催化析氧线性扫描曲线图如图5所示。其中,ruo2为市售催化剂,co/nc是对比例1中制备的;co2p/npc是对比例2中制备的;n-co2p/npc是实施例1制备的。
检测方法为:在1.0mol/l的koh溶液中使用标准三电极系统,将催化剂作为工作电极,碳棒为对电极,汞/氧化汞(hg/hgo)电极作为参比电极,在电化学工作站(chi760e)上进行测试。根据以下公式校正所测得的电势:e(rhe)=e(hg/hgo) 0.098v 0.059×ph,并通过几何表面积将电流密度(j)归一化,所得结果如图5所示。
由图5可以看出,采用本发明制备的n-co2p/npc作为电极,在电流密度为10ma·cm-2和100ma·cm-2时所需的过电位分别为230mv和296mv,这远小于co2p/npc(270mv和347mv)、co/nc(295mv和390mv)和商业ruo2(290mv和360mv)。
测试例5
实施例1制备的氮和磷共掺杂碳层包裹磷化二钴催化剂的电催化水分解线性扫描曲线图如图6所示。检测方法为:在1.0mol/l的koh溶液中使用两电极测试,n-co2p/npc分别作为阴极和阳极测试。
由图6可以看出,n-co2p/npc催化剂在1.53v、1.69v和1.78v的电压下可分别提供10ma·cm-2、100ma·cm-2和200ma·cm-2的电流密度,这比商业pt/c//ruo2电极电催化剂更优越。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
本文用于企业家、创业者技术爱好者查询,结果仅供参考。