一种利用ddpcr检测小耳畸形患者ecr区域的拷贝数的引物及试剂盒
技术领域
1.本发明涉及一种利用ddpcr检测小耳畸形患者ecr区域的拷贝数的引物及试剂盒,属于分子遗传学检测技术领域。
背景技术:
2.小耳畸形(omim 600674)是一种外耳发育异常,临床表现为小的形状异常的耳廓,为常见的面部先天性畸形。表型严重程度不等,可表现为轻微的结构异常,也可表现为外耳完全消失。小耳畸形可以作为一种独立的出生缺陷,也可以作为发育异常或综合征的一部分,合并外耳道、中耳等的畸形。其中,耳甲腔型小耳畸形伴有残余耳垂、耳甲腔、耳道、耳屏和耳屏间切迹,是一种轻度的小耳畸形。
3.目前关于小耳畸形产生的遗传和细胞机制研究有限。位于4p16.1的h6家族同源盒1转录因子基因(hmx1)在人类胚胎发育的颅面部分化过程中起着重要作用,hmx1的纯合功能缺失突变可导致眼耳综合征(ocacs,omim 142992),该综合征以外耳和眼睛的发育异常为特征。关于小鼠的研究表明,hmx1下游的一段进化保守区域(evolutionarily conserved region,ecr)是促进耳发育的重要调节因素,hoxa2、meis和pbx可以协同作用于ecr区域内32bp的核心序列,调节hmx1的表达。累及hmx1
‑
ecr区域的重复和缺失存在于多种外耳畸形的动物中,如小鼠、大鼠和高原牛等。
4.目前拷贝数检测方法主要包括定量pcr、多重连接探针扩增(multiplex ligation
‑
dependent probe amplification,简称mlpa)、染色体芯片(如比较基因组杂交芯片、单核苷酸多态性微阵列芯片等)、下一代测序(next generation sequencing,简称ngs)等。其中,定量pcr利用pcr体系中的荧光信号积累检测pcr进程以及对目的片段进行定量,可用于快速筛查验证已知cnv变异的存在情况,也是实验室检测的常规操作,但是定量pcr依赖扩增效率计算结果,可能受到pcr抑制剂的作用从而影响检测结果的准确性,因此对检测样本的质量要求较高。
5.数字pcr是一种基于pcr的全新核酸检测技术,又被称为第三代pcr技术,是一种单分子水平的大规模分区扩增核酸定量技术。基于反应体系有限分割原理,将一个样本分成上万份,以微腔室/微孔或微滴作为独立反应器,dna样品在独立的反应单元中进行扩增,含有或不含待检目标分子。在扩增结束后,对各分区以终点荧光的“有”或“无”作为判断标准采集每个独立反应体系的信号,根据泊松分布(poisson distribution)原理进行数据分析,实现对核酸分子的绝对定量。数字pcr对样品需求量低,并且具有良好的灵敏度和耐受性,是目前pcr中最先进的技术。
6.根据样品分散方式,目前市场上常用的数字pcr仪器可以分为基于微流控芯片的数字pcr以及基于油包水技术的微滴式数字pcr(droplet digital pcr,简称ddpcr)。其中,基于微流控芯片的数字pcr制造芯片的成本较高,而ddpcr通过液滴发生器将乳化的液滴分散成>20000个均一的反应单元。因此,ddpcr比基于微流控芯片的数字pcr更为经济适用,且
与传统的定量pcr相比具有更好的精确性、灵敏度和特异性。
7.但是目前,尚未有利用ddpcr检测小耳畸形患者ecr区域的拷贝数的引物及试剂盒的报道。鉴于此,有必要提供一种利用ddpcr检测小耳畸形患者ecr区域的拷贝数的引物及试剂盒,以解决现有技术的不足。
技术实现要素:
8.本发明的目的之一,是提供一种利用ddpcr检测小耳畸形患者ecr区域的拷贝数的引物。本发明首次发现了利用ddpcr检测小耳畸形患者ecr区域的拷贝数的引物,可以特异性扩增并检测小耳畸形患者ecr区域的拷贝数,特异性强,检测结果可靠。
9.本发明解决上述技术问题的方案如下:一种利用ddpcr检测小耳畸形患者ecr区域的拷贝数的引物,包括如seq id no.1所示的上游引物,以及如seq id no.2所示的下游引物。
10.seq id no.1:5'
‑
cacttccttcccactgatcc
‑
3';
11.seq id no.2:5'
‑
ctttatctaccctgtcccgg
‑
3'。
12.本发明的利用ddpcr检测小耳畸形患者ecr区域的拷贝数的引物的原理介绍:
13.本技术的发明人在人类单纯双侧耳甲腔型小耳畸形家系研究中首次发现累及hmx1
‑
ecr的拷贝数(cnv)重复与小耳畸形发生有关。上述用于检测小耳畸形患者ecr区域的拷贝数的引物,是本技术的发明人根据ucsc genome browser中已公布的hmx1基因下游进化保守区域(ecr)的部分序列(hg19,chr4:8701924
‑
8702539)而设计的。使用primer3软件搜索适合设计引物的区域,扩增产物片段避开所选用的内切酶酶切位点,参数设置如下:引物溶解温度(tm)=60℃
±
5℃,扩增产物长度60
‑
200bp,引物长度20bp
±
5bp,引物gc含量为40%
‑
60%,引物3
′
端最大自我互补值为3,引物间3
′
端最大互补值为3。在搜索获得的引物对中,选择最优引物对即上述第一引物和第二引物。使用ucsc在线工具blat,将第一引物和第二引物与数据库中的核苷酸序列进行比对,确认该引物对只能配对ecr序列,且不包含常见单核苷酸多态(snps)。
14.本发明的用于检测小耳畸形患者ecr区域的拷贝数的引物的有益效果:
15.本发明首次发现了用于检测小耳畸形患者ecr区域的拷贝数的引物,可以特异性扩增并检测小耳畸形患者ecr区域的拷贝数,特异性强,检测结果可靠。
16.本发明的目的之二,是提供一种利用ddpcr检测小耳畸形患者ecr区域的拷贝数的试剂盒。本发明的试剂盒利用上述ddpcr检测小耳畸形患者ecr区域的拷贝数的引物,可以快速筛查小耳畸形患者的ecr区域的拷贝数,可以为后续进行更精确的变异检测、变异机制研究以及疾病发生机制探索提供基础,加快变异筛选过程。
17.本发明解决上述技术问题的方案如下:一种利用ddpcr检测小耳畸形患者ecr区域的拷贝数的试剂盒,至少包括上述利用ddpcr检测小耳畸形患者ecr区域的拷贝数的引物。
18.本发明的利用ddpcr检测小耳畸形患者ecr区域的拷贝数的试剂盒的有益效果是:
19.本发明的试剂盒利用上述ddpcr检测小耳畸形患者ecr区域的拷贝数的引物,可以快速筛查小耳畸形患者的ecr区域的拷贝数,可以为后续进行更精确的变异检测、变异机制研究以及疾病发生机制探索提供基础,加快变异筛选过程。
20.在上述技术方案的基础上,本发明还可以做如下改进。
21.进一步,所述试剂盒还包括内参基因,其包括如seq id no.3所示的上游引物,以及如seq id no.4所示的下游引物。
22.seq id no 3:5'
‑
aggaacttggccaggatctc
‑
3';
23.seq id no 4:5'
‑
tgatctggatgtggcatgtc
‑
3'。
24.采用上述进一步的有益效果是:以内参基因为参照,可以得到ecr序列相应的拷贝数改变。
25.更进一步,所述试剂盒还包括ecr区域拷贝数正常的对照dna和ecr区域拷贝数增加的阳性对照dna。
26.采用上述更进一步的有益效果是:正常对照和阳性对照的存在可以保证实验过程的顺利进行,避免实验出现假阴性与假阳性。
27.更进一步,所述ecr区域拷贝数正常的对照dna为来自于表型正常个体的基因组dna,所述ecr区域拷贝数增加的阳性对照dna为来自于小耳畸形患者的基因组dna。
28.更进一步,所述试剂盒还包括限制性内切酶、酶切缓冲液以及pcr反应液。
29.采用上述更进一步的有益效果是:酶切基因组dna可使dna片段分散为小片段,得到更为精确的实验结果。
30.更进一步,所述限制性内切酶为hindⅲ。
31.采用上述更进一步的有益效果是:作为六碱基识别位点内切酶,hindⅲ可识别基因组中的aagctt序列,且对甲基化不敏感,能在目的片段外切割dna样本,使实验结果更准确。
附图说明
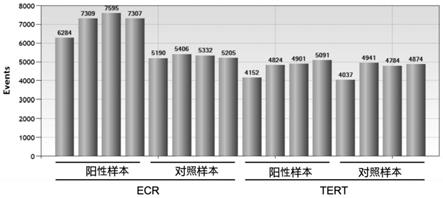
32.图1为quantasoft软件所读取的各反应体系阳性液滴数目。
33.图2为不同反应体系中阳性样本与对照样本的目的片段浓度(显示部分数据)。
34.图3为定量pcr方法得到的对照样本与阳性样本ecr拷贝数情况。
具体实施方式
35.以下结合具体附图对本发明的原理和特征进行描述,所举实例只用于解释本发明,并非用于限定本发明的范围。
36.实施例1:基因组dna制备
37.被检测个体的dna样本可来源于各种组织细胞,以外周血为例。外周血dna提取可选择经典的酚氯仿提取方法以及利用市场上各种外周血dna提取试剂盒,具体步骤可参见相应公司说明书。酚氯仿提取方法如下:
38.(1)抽取edta抗凝外周血。
39.(2)外周血样本平衡至室温,颠倒混匀,取400μl
‑
500μl加入2ml收集管中,加入去离子水补齐至2ml,颠倒混匀。
40.(3)低温高速14,000g离心10min,弃上清。
41.(4)再次加入2ml去离子水,指尖弹匀,重复上一步。
42.(5)加入50μl qiagen蛋白酶(蛋白酶k)和450μl消化液。
43.(6)混匀,56℃孵育,过夜。
44.(7)根据上一步得到的产物体积,按照1:1:1加入等体积的tris平衡酚和氯仿,颠倒混匀。
45.(8)4℃高速14,000g离心10min,吸取上清转移到新的离心管中。
46.(9)重复(7)以及(8),转移上清至新的离心管,注意不要吸取到下层。
47.(10)根据上一步得到的产物体积,加入2倍体积的冷无水乙醇,轻柔颠倒混匀,可见丝状dna出现。
48.(11)4℃高速14,000g离心10min,弃去上清。
49.(12)1ml 75%乙醇洗涤沉淀,轻弹管底,轻柔颠倒摇晃。
50.(13)4℃高速14,000g离心10min,弃上清。
51.(14)再次4℃高速14,000g离心2min,用10μl小枪头吸弃多余液体,室温开盖晾干。
52.(15)加入100μl去离子水溶解dna,采用nanodrop 2000微量分光光度计检测浓度以及质量,多余样本可储存在
‑
20℃备用。
53.实施例2:酶切dna
54.限制性内切酶选用hindⅲ,购自neb公司,该酶对哺乳动物cpg甲基化不敏感。
55.相应的酶切缓冲液为nebuffer 2.1,反应条件37℃,50μl酶切体系如下:10
×
nebuffer 2.1,5μl(1
×
);gdna,1μg;hindⅲ,10units;以及去离子水补齐至50μl。
56.hindⅲ酶从
‑
20℃冰箱取出后置于冰上保存,冰上配制体系,最后加酶,移液枪上下吹吸混匀或轻弹管壁,快速离心。该酶为省时酶,37℃反应只需10min,也可以过夜酶切而没有星号活性。反应结束后不必进行热失活,如果反应产物计划长期保存,可选择进行热失活,注意温度不要超过65℃,反应结束后不需要进行dna纯化,酶切产物需保存在
‑
20℃或更低的温度中。
57.实施例3:配制pcr反应体系
58.选用bio
‑
rad evagreen预混液,低温配制,可选择在冰上操作,提前准备evagreen预混液冰上融化,轻轻混匀,20μl反应体系如下:
[0059]2×
evagreen预混液,10μl;dna digestion mixture,1μl;10μm的seq id no.1或seq id no.3所示的上游引物,0.2μl;10μm的seq id no.2或seq id no.4所示的下游引物,0.2μl;以及去离子水补齐至20μl。
[0060]
实施例4:微滴生成
[0061]
采用bio
‑
rad qx100或qx200 droplet generator,利用其特定的试剂(droplet generation oil for evagreen)和配套耗材以及微流控系统,将每个样本分散成20000个nl级别的微滴,在分散过程中,目的dna片段以及其它背景dna片段被随机分散到液滴中。该过程能够生成大小以及体积均一的微滴,保证定量的精确性。此过程结束后,每个体系的体积约为40μl,与起始pcr反应液的体积相比加倍。
[0062]
实施例5:pcr扩增
[0063]
pcr扩增选择bio
‑
rad c1000 touch
tm thermal cycler with 96
‑
deep well reaction module for pcr,或选择其他温控较好的pcr仪器,市场上常用的pcr仪器均可尝试。初次反应,为确定最优的退火温度,可设计温度梯度,然后比较扩增结果以及特异性,进而选择最合适的温度,以便结果分析。液滴生成后,上机扩增之前对96孔板进行密封,可选择使用px1 pcr plate sealer,对孔板进行快速精确密封,最大程度上减少pcr扩增反应中
的样品挥发。
[0064]
反应程序为:95℃,预变性5min;95℃,变性30s,60℃,退火/延伸1min,共进行40个循环;4℃和90℃,分别信号稳定5min;4℃,结束;温度变化速率均设置为2.5℃/s。
[0065]
仪器顶盖温度设置为105℃,样品体积设置为40μl,这是因为微滴生成后,体系中增加了droplet generation oil,体积加倍。
[0066]
实施例6:结果读取以及分析
[0067]
pcr反应结束后,直接上机进行结果读取。选用bio
‑
rad qx200 droplet reader,打开quantasoft
tm
软件,根据实验安排设置孔板布局,具体步骤可参照qx200 droplet reader和quantasoft软件使用手册。双击孔板上的每一个孔均可以进行编辑,可以命名样本名称以及实验类型(本实验选择abs),选择pcr所用的预混液,如qx200 ddpcr evagreen supermix,并对目的基因进行命名(ecr和tert等)。设置完毕后,确定,点击run进行液滴读取,染料选择evagreen,读取结束后进行结果分析。
[0068]
结果的浓度表示为在每微升的1x的ddpcr反应液中,检测到的拷贝数,目的片段的拷贝数计算公式为:
[0069][0070]
其中,内参基因在基因组中的拷贝数为2。
[0071]
据此可快速计算出待测样本的ecr区域的拷贝数。同时,实验中加入正常对照以及阳性对照可以更好的对结果进行分析对比。以正常对照个体的dna以及已验证ecr拷贝数增加的小耳畸形患者的dna为模板,进行ddpcr,结果详见图1和图2。可知正常样本ecr拷贝数为2,阳性对照样本的ecr拷贝数约为3。
[0072]
对比例
[0073]
采用传统的定量pcr方法检测ecr拷贝数,根据定量pcr要求,设计如seq id no.5所示的上游引物和如seq id no.6所示的下游引物:
[0074]
ecr
‑
f:5'
‑
ttccaacccagcgaaattag
‑
3'(seq id no.5);
[0075]
ecr
‑
r:5'
‑
gggatcagtgggaaggaagt
‑
3'(seq id no.6)。
[0076]
调整dna模板浓度,选取阳性与对照样本,调整dna浓度至约5ng/μl备用;涡旋混匀dna样本与引物,瞬离;上下轻轻颠倒以及用枪头吹吸混匀hieffpower qpcr sybr green master mix,注意不要产生气泡,瞬离。
[0077]
qpcr反应体系配制,每个样本每对引物做4个重复孔,表1为单孔内所加体系,配制体系时同一对引物配成大的mix,分别加入每个孔内,最后再分别加入样本dna。
[0078]
表1 qpcr反应体系
[0079][0080]
反应条件,根据rotor
‑
gene 6000操作手册以及sybr green说明书,pcr扩增程序为:95℃,30s;95℃,10s,60℃,15s,72℃,20s,共40个循环。
[0081]
溶解曲线采用仪器默认程序,90s pre
‑
melting,72℃
‑
95℃逐渐升温,每步增加1℃,5s。
[0082]
实验结果,如图3所示。
[0083]
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。