1.相关申请的交叉引用
2.本技术要求于2019年11月11日向韩国知识产权局提交的韩国专利申请第10
‑
2019
‑
0143630号的申请日和于2020年11月11日向韩国知识产权局提交的韩国专利申请第10
‑
2020
‑
0150222号的申请日的权益,其全部内容通过引用并入本文。
3.本公开内容涉及具有改善的驱动电压、效率和寿命的有机发光器件。
背景技术:
4.通常,有机发光现象是指通过利用有机材料将电能转化成光能的现象。利用有机发光现象的有机发光器件具有诸如宽的视角,优异的对比度,快速的响应时间,优异的亮度、驱动电压和响应速度的特性,因此已进行了许多研究。
5.有机发光器件通常具有包括阳极、阴极以及介于阳极与阴极之间的有机材料层的结构。有机材料层通常具有包含不同材料的多层结构以提高有机发光器件的效率和稳定性,例如,有机材料层可以由空穴注入层、空穴传输层、发光层、电子传输层、电子注入层等形成。在有机发光器件的结构中,如果在两个电极之间施加电压,则空穴从阳极注入至有机材料层中并且电子从阴极注入至有机材料层中,并且当注入的空穴和电子彼此相遇时形成激子,并且当激子再次落至基态时发光。
6.在如上所述的有机发光器件中,持续需要开发具有改善的驱动电压、效率和寿命的有机发光器件。
7.[现有技术文献]
[0008]
[专利文献]
[0009]
(专利文献0001)韩国未审查专利公开第10
‑
2000
‑
0051826号
技术实现要素:
[0010]
技术问题
[0011]
本公开内容涉及具有改善的驱动电压、效率和寿命的有机发光器件。
[0012]
技术方案
[0013]
本文提供了以下有机发光器件:
[0014]
一种有机发光器件,
[0015]
包括:
[0016]
阳极、阴极以及介于阳极与阴极之间的发光层。
[0017]
其中发光层包含由以下化学式1表示的化合物和由以下化学式2表示的化合物:
[0018]
[化学式1]
[0019][0020]
在化学式1中,
[0021]
x为o或s,
[0022]
每个y独立地为n或ch,条件是至少一个y为n,
[0023]
l1为单键、或者经取代或未经取代的c6‑
60
亚芳基,
[0024]
ar1和ar2各自独立地为经取代或未经取代的c6‑
60
芳基;或者经取代或未经取代的包含选自n、o和s中的任一者或更多者的c2‑
60
杂芳基,
[0025]
[化学式2]
[0026][0027]
在化学式2中,
[0028]
l2为经取代或未经取代的c6‑
60
亚芳基,
[0029]
l3和l4各自独立地为单键、或者经取代或未经取代的c6‑
60
亚芳基,
[0030]
ar3和ar4各自独立地为经取代或未经取代的c6‑
60
芳基;或者经取代或未经取代的包含选自n、o和s中的任一者或更多者的c2‑
60
杂芳基,
[0031]
r为氢、氘、或者经取代或未经取代的c6‑
60
芳基,以及
[0032]
n为0至9的整数。
[0033]
有益效果
[0034]
通过在发光层中包含由化学式1表示的化合物和由化学式2表示的化合物,上述有机发光器件具有优异的驱动电压、效率和寿命。
附图说明
[0035]
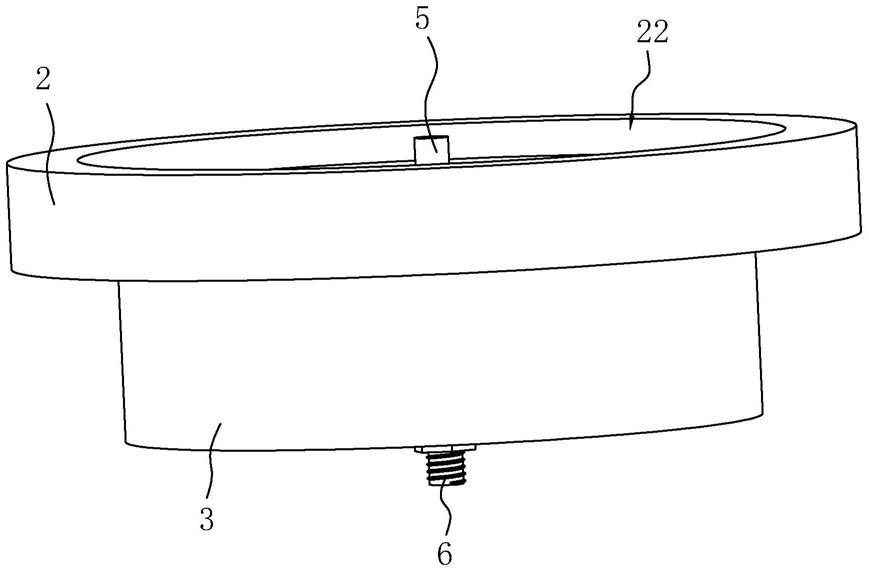
图1示出了包括基底1、阳极2、发光层3和阴极4的有机发光器件的一个实例。
[0036]
图2示出了包括基底1、阳极2、空穴传输层5、发光层3、电子传输层6和阴极4的有机发光器件的一个实例。
具体实施方式
[0037]
在下文中,将更详细地描述本公开内容的实施方案以促进理解本发明。
[0038]
如本文中所使用,符号或意指与另一个取代基连接的键。
[0039]
如本文中所使用,术语“经取代或未经取代的”意指未经取代或经选自以下中的一个或更多个取代基取代:氘;卤素基团;腈基;硝基;羟基;羰基;酯基;酰亚胺基;氨基;氧化膦基;烷氧基;芳氧基;烷基硫基;芳基硫基;烷基磺酰基;芳基磺酰基;甲硅烷基;硼基;烷基;环烷基;烯基;芳基;芳烷基;芳烯基;烷基芳基;烷基胺基;芳烷基胺基;杂芳基胺基;芳基胺基;芳基膦基;或者包含n、o和s原子中的至少一者的杂芳基,或者未经取代或经以上例示的取代基中的两个或更多个取代基连接的取代基取代。例如,“两个或更多个取代基连接的取代基”可以为联苯基。即,联苯基可以为芳基,或者其也可以被解释为两个苯基连接的取代基。
[0040]
在本公开内容中,羰基的碳数没有特别限制,但优选为1至40。具体地,羰基可以为具有以下结构式的化合物,但不限于此。
[0041][0042]
在本公开内容中,酯基可以具有其中酯基的氧可以经具有1至25个碳原子的直链、支链或环状烷基,或者具有6至25个碳原子的芳基取代的结构。具体地,酯基可以为具有以下结构式的化合物,但不限于此。
[0043][0044]
在本公开内容中,酰亚胺基的碳数没有特别限制,但优选为1至25。
[0045]
具体地,酰亚胺基可以为具有以下结构式的化合物,但不限于此。
[0046]
[0047]
在本公开内容中,甲硅烷基具体地包括三甲基甲硅烷基、三乙基甲硅烷基、叔丁基二甲基甲硅烷基、乙烯基二甲基甲硅烷基、丙基二甲基甲硅烷基、三苯基甲硅烷基、二苯基甲硅烷基、苯基甲硅烷基等,但不限于此。
[0048]
在本公开内容中,硼基具体包括三甲基硼基、三乙基硼基、叔丁基二甲基硼基、三苯基硼基和苯基硼基,但不限于此。
[0049]
在本公开内容中,卤素基团的实例包括氟、氯、溴、或碘。
[0050]
在本公开内容中,烷基可以为直链或支链,并且其碳数没有特别限制,但优选为1至40。根据一个实施方案,烷基的碳数为1至20。根据另一个实施方案,烷基的碳数为1至10。根据另一个实施方案,烷基的碳数为1至6。烷基的具体实例包括甲基、乙基、丙基、正丙基、异丙基、丁基、正丁基、异丁基、叔丁基、仲丁基、1
‑
甲基
‑
丁基、1
‑
乙基
‑
丁基、戊基、正戊基、异戊基、新戊基、叔戊基、己基、正己基、1
‑
甲基戊基、2
‑
甲基戊基、4
‑
甲基
‑2‑
戊基、3,3
‑
二甲基丁基、2
‑
乙基丁基、庚基、正庚基、1
‑
甲基己基、环戊基甲基、环己基甲基、辛基、正辛基、叔辛基、1
‑
甲基庚基、2
‑
乙基己基、2
‑
丙基戊基、正壬基、2,2
‑
二甲基庚基、1
‑
乙基
‑
丙基、1,1
‑
二甲基
‑
丙基、异己基、2
‑
甲基戊基、4
‑
甲基己基、5
‑
甲基己基等,但不限于此。
[0051]
在本公开内容中,烯基可以为直链或支链,并且其碳数没有特别限制,但优选为2至40。根据一个实施方案,烯基的碳数为2至20。根据另一个实施方案,烯基的碳数为2至10。根据又一个实施方案,烯基的碳数为2至6。其具体实例包括乙烯基、1
‑
丙烯基、异丙烯基、1
‑
丁烯基、2
‑
丁烯基、3
‑
丁烯基、1
‑
戊烯基、2
‑
戊烯基、3
‑
戊烯基、3
‑
甲基
‑1‑
丁烯基、1,3
‑
丁二烯基、烯丙基、1
‑
苯基乙烯基
‑1‑
基、2
‑
苯基乙烯基
‑1‑
基、2,2
‑
二苯基乙烯基
‑1‑
基、2
‑
苯基
‑2‑
(萘基
‑1‑
基)乙烯基
‑1‑
基、2,2
‑
双(二苯基
‑1‑
基)乙烯基
‑1‑
基、茋基、苯乙烯基等,但不限于此。
[0052]
在本公开内容中,环烷基没有特别限制,但其碳数优选为3至60。根据一个实施方案,环烷基的碳数为3至30。根据另一个实施方案,环烷基的碳数为3至20。根据又一个实施方案,环烷基的碳数为3至6。其具体实例包括环丙基、环丁基、环戊基、3
‑
甲基环戊基、2,3
‑
二甲基环戊基、环己基、3
‑
甲基环己基、4
‑
甲基环己基、2,3
‑
二甲基环己基、3,4,5
‑
三甲基环己基、4
‑
叔丁基环己基、环庚基、环辛基等,但不限于此。
[0053]
在本公开内容中,芳基没有特别限制,但其碳数优选为6至60,并且其可以为单环芳基或多环芳基。根据一个实施方案,芳基的碳数为6至30。根据一个实施方案,芳基的碳数为6至20。作为单环芳基,芳基可以为苯基、联苯基、三联苯基等,但不限于此。多环芳基包括萘基、蒽基、菲基、芘基、苝基、基、芴基等,但不限于此。
[0054]
在本公开内容中,芴基可以为经取代的,并且两个取代基可以彼此连接以形成螺环结构。在芴基为经取代的情况下,可以形成等。然而,该结构不限于此。
[0055]
在本公开内容中,杂环基为包含o、n、si和s中的一者或更多者作为杂原子的杂环基,并且其碳数没有特别限制,但优选为2至60。杂环基的实例包括噻吩基、呋喃基、吡咯基、
咪唑基、噻唑基、唑基、二唑基、三唑基、吡啶基、联吡啶基、嘧啶基、三嗪基、吖啶基、哒嗪基、吡嗪基、喹啉基、喹唑啉基、喹喔啉基、酞嗪基、吡啶并嘧啶基、吡啶并吡嗪基、吡嗪并吡嗪基、异喹啉基、吲哚基、咔唑基、苯并唑基、苯并咪唑基、苯并噻唑基、苯并咔唑基、苯并噻吩基、二苯并噻吩基、苯并呋喃基、菲咯啉基、异唑基、噻二唑基、吩噻嗪基、二苯并呋喃基等,但不限于此。
[0056]
在本公开内容中,芳烷基、芳烯基、烷基芳基和芳基胺基中的芳基与前述芳基的实例相同。在本公开内容中,芳烷基、烷基芳基和烷基胺基中的烷基与前述烷基的实例相同。在本公开内容中,杂芳基胺中的杂芳基可以应用前述杂环基的描述。在本公开内容中,芳烯基中的烯基与前述烯基的实例相同。在本公开内容中,除了亚芳基为二价基团之外,可以应用前述芳基的描述。在本公开内容中,除了亚杂芳基为二价基团之外,可以应用前述杂芳基的描述。在本公开内容中,除了烃环不是一价基团而是通过使两个取代基结合而形成的之外,可以应用前述芳基或环烷基的描述。在公开中,除了杂环不是一价基团而是通过使两个取代基结合而形成的之外,可以应用前述杂环基的描述。
[0057]
在下文中,将针对各配置详细地描述本公开内容。
[0058]
阳极和阴极
[0059]
本公开内容中使用的阳极和阴极意指有机发光器件中使用的电极。
[0060]
作为阳极材料,通常优选使用具有大功函数的材料,使得空穴可以顺利地注入到有机材料层中。阳极材料的具体实例包括:金属,例如钒、铬、铜、锌和金、或其合金;金属氧化物,例如锌氧化物、铟氧化物、铟锡氧化物(ito)和铟锌氧化物(izo);金属和氧化物的组合,例如zno:al或sno2:sb;导电聚合物,例如聚(3
‑
甲基噻吩)、聚[3,4
‑
(亚乙基
‑
1,2
‑
二氧)噻吩](pedot)、聚吡咯和聚苯胺;等等,但不限于此。
[0061]
作为阴极材料,通常优选使用具有小功函数的材料,使得电子可以容易地注入到有机材料层中。阴极材料的具体实例包括:金属,例如镁、钙、钠、钾、钛、铟、钇、锂、钆、铝、银、锡和铅、或其合金;多层结构材料,例如lif/al或lio2/al;等等,但不限于此。
[0062]
空穴注入层
[0063]
如有必要,根据本公开内容的有机发光器件还可以包括在阳极上的空穴注入层。
[0064]
空穴注入层是注入来自电极的空穴的层,并且空穴注入材料优选为这样的化合物:其具有传输空穴的能力、注入阳极中的空穴的效应和优异的对发光层或发光材料的空穴注入效应,防止发光层中产生的激子移动至电子注入层或电子注入材料,并且具有优异的薄膜形成能力。优选的是,空穴注入材料的homo(最高占据分子轨道)在阳极材料的功函数与周围有机材料层的homo之间。
[0065]
空穴注入材料的具体实例包括金属卟啉,低聚噻吩,基于芳基胺的有机材料,基于六腈六氮杂苯并菲的有机材料,基于喹吖啶酮的有机材料,基于苝的有机材料,基于蒽醌、聚苯胺和聚噻吩的导电聚合物等,但不限于此。
[0066]
空穴传输层
[0067]
如果必要,根据本公开内容的有机发光器件可以包括在阳极上(或者在存在空穴注入层时,在空穴注入层上)的空穴传输层。
[0068]
空穴传输层为接收来自阳极或空穴注入层的空穴并将空穴传输至发光层的层。空穴传输材料适当地为具有大的空穴迁移率的材料,其可以接收来自阳极或空穴注入层的空
穴并将空穴传输至发光层。
[0069]
空穴传输材料的具体实例包括基于芳基胺的有机材料、导电聚合物、同时存在共轭部分和非共轭部分的嵌段共聚物等,但不限于此。
[0070]
发光层
[0071]
本公开内容中使用的发光层意指可以通过使从阳极和阴极传输的空穴和电子结合而发出可见光区域中的光的层。通常,发光层包含主体材料和掺杂剂材料,并且在本公开内容中,包含由化学式1表示的化合物和由化学式2表示的化合物作为主体。
[0072]
在化学式1中,优选地,所有的y均为n。
[0073]
优选地,l1为单键、亚苯基或亚萘基。更优选地,l1为单键、或
[0074]
优选地,ar1和ar2各自独立地为苯基、联苯基、三联苯基、萘基、菲基、(苯基)萘基、(萘基)苯基、二甲基芴基、二苯基芴基、二苯并呋喃基、二苯并噻吩基、咔唑
‑9‑
基、9
‑
苯基
‑
9h
‑
咔唑基,ar1和ar2各自独立地未经取代或经至少一个氘取代。当ar1或ar2经至少一个氘取代时,优选地,它们各自为选自以下中的任一者:
[0075][0076]
优选地,ar1为苯基、联苯基或萘基,ar1未经取代或经至少一个氘取代;以及ar2为苯基、联苯基、三联苯基、萘基、菲基、(苯基)萘基、(萘基)苯基、二甲基芴基、二苯基芴基、二苯并呋喃基、二苯并噻吩基、咔唑
‑9‑
基、9
‑
苯基
‑
9h
‑
咔唑基,ar2未经取代或经至少一个氘取代。
[0077]
由化学式1表示的化合物的代表性实例如下:
[0078]
[0079]
[0080]
[0081]
[0082]
[0083]
[0084]
[0085]
[0086]
[0087]
[0088]
[0089]
[0090]
[0091]
[0092]
[0093]
[0094]
[0095]
[0096]
[0097]
[0098]
[0099]
[0100]
[0101]
[0102]
[0103]
[0104][0105]
本文还提供了如以下反应方案1所示的用于制备由化学式1表示的化合物的方法。
[0106]
[反应方案1]
[0107]
[0108]
在反应方案1中,除x'以外的剩余取代基的限定与以上限定的相同,并且x'为卤素,优选为溴或氯。以上反应为suzuki偶联反应,其优选在钯催化剂和碱的存在下进行,并且用于suzuki偶联反应的反应性基团可以如本领域已知地进行改变。以上制备方法可以进一步呈现在下文所述的制备例中。
[0109]
在化学式2中,优选地,化学式2由以下化学式2
‑
1表示:
[0110]
[化学式2
‑
1]
[0111][0112]
其中在化学式2
‑
1中,
[0113]
r1为氢、氘或苯基,
[0114]
n1为0至8的整数,
[0115]
l2、l3、l4、ar3、ar4和r如以上所限定。
[0116]
优选地,l2为亚苯基、或经至少一个氘取代的亚苯基。经至少一个氘取代的亚苯基优选为选自以下中的任一者:
[0117][0118]
优选地,l3和l4各自独立地为单键、亚苯基、联苯二基或亚萘基,l3和l4各自独立地未经取代或经至少一个氘取代。当l3或l4经至少一个氘取代时,优选地,它们各自为选自以下中的任一者:
[0119][0120]
优选地,ar3和ar4各自独立地为苯基、联苯基、三联苯基、萘基、菲基、(苯基)菲基、三亚苯基、苯基萘基、萘基苯基、二甲基芴基、二苯基芴基、二苯并呋喃基、(苯基)二苯并呋喃基、二苯并噻吩基、(苯基)二苯并噻吩基、咔唑
‑9‑
基或9
‑
苯基
‑
9h
‑
咔唑基,ar3和ar4各自独立地未经取代或经至少一个氘取代。当ar3或ar4经至少一个氘取代时,它们各自优选为选自以下中的任一者:
[0121][0122]
由化学式2表示的化合物的代表性实例如下:
[0123]
[0124]
[0125]
[0126]
[0127]
[0128]
[0129]
[0130]
[0131]
[0132]
[0133]
[0134]
[0135]
[0136]
[0137]
[0138]
[0139]
[0140]
[0141]
[0142]
[0143]
[0144]
[0145]
[0146]
[0147]
[0148]
[0149][0150]
本文还提供了如以下反应方案2所示的用于制备由化学式2表示的化合物的方法。
[0151]
[反应方案2]
[0152][0153]
其中在反应方案2中,除x'以外的剩余取代基的限定与以上限定的相同,并且x'为卤素,优选为溴或氯。以上反应为胺取代反应,其优选在钯催化剂和碱的存在下进行,并且用于胺取代反应的反应性基团可以如本领域已知地进行改变。以上制备方法可以进一步呈现在下文所述的制备例中。
[0154]
优选地,在发光层中,由化学式1表示的化合物与由化学式2表示的化合物的重量比为10:90至90:10,更优选地20:80至80:20、30:70至70:30或40:60至60:40。
[0155]
同时,除了主体之外,发光层还可以包含掺杂剂。掺杂剂材料没有特别限制,只要其是用于有机发光器件的材料即可。作为实例,可以提及芳族胺衍生物、苯乙烯胺化合物、硼配合物、荧蒽化合物、金属配合物等。芳族胺衍生物的具体实例包括经取代或未经取代的具有芳基氨基的稠合芳族环衍生物,其实例包括具有芳基氨基的芘、蒽、和二茚并芘等。苯乙烯胺化合物为其中经取代或未经取代的芳基胺中取代有至少一个芳基乙烯基的化合物,其中选自芳基、甲硅烷基、烷基、环烷基和芳基氨基的一个或两个或更多个取代基是经
取代或未经取代的。其具体实例包括苯乙烯胺、苯乙烯二胺、苯乙烯三胺、苯乙烯四胺等,但不限于此。此外,金属配合物的实例包括铱配合物、铂配合物等,但不限于此。
[0156]
电子传输层
[0157]
如有必要,根据本公开内容的有机发光器件可以包括在发光层上的电子传输层。
[0158]
电子传输层是接收来自阴极或形成在阴极上的电子注入层的电子并将电子传输至发光层,并且抑制空穴从发光层转移的层,并且电子传输材料适当地为这样的材料:其可以良好地接收来自阴极的电子并将电子转移至发光层,并且具有大的电子迁移率。
[0159]
电子传输材料的具体实例包括:8
‑
羟基喹啉的al配合物;包含alq3的配合物;有机自由基化合物;羟基黄酮
‑
金属配合物;等等,但不限于此。电子传输层可以与如根据常规技术使用的任何期望的阴极材料一起使用。特别地,阴极材料的合适实例为具有低功函数的典型材料,后接铝层或银层。其具体实例包括铯、钡、钙、镱和钐,在每种情况下都后接铝层或银层。
[0160]
电子注入层
[0161]
如果必要,根据本公开内容的有机发光器件还可以包括在发光层上(或者在存在电子传输层时,在电子传输层上)的电子注入层。
[0162]
电子注入层是注入来自电极的电子的层,并且优选为这样的化合物:其具有传输电子的能力,具有注入来自阴极的电子的效应和优异的将电子注入到发光层或发光材料中的效应,防止由发光层产生的激子移动至空穴注入层,并且在形成薄膜的能力方面也优异。
[0163]
可以用作电子注入层的材料的具体实例包括芴酮、蒽醌二甲烷、联苯醌、噻喃二氧化物、唑、二唑、三唑、咪唑、四羧酸、亚芴基甲烷、蒽酮等、及其衍生物,金属配合物化合物,含氮5元环衍生物,等等,但不限于此。
[0164]
金属配合物化合物的实例包括8
‑
羟基喹啉锂、双(8
‑
羟基喹啉)锌、双(8
‑
羟基喹啉)铜、双(8
‑
羟基喹啉)锰、三(8
‑
羟基喹啉)铝、三(2
‑
甲基
‑8‑
羟基喹啉)铝、三(8
‑
羟基喹啉)镓、双(10
‑
羟基苯并[h]喹啉)铍、双(10
‑
羟基苯并[h]喹啉)锌、双(2
‑
甲基
‑8‑
喹啉)氯镓、双(2
‑
甲基
‑8‑
喹啉)(邻甲酚)镓、双(2
‑
甲基
‑8‑
喹啉)(1
‑
萘酚)铝、双(2
‑
甲基
‑8‑
喹啉)(2
‑
萘酚)镓等,但不限于此。
[0165]
有机发光器件
[0166]
图1和图2示出了根据本公开内容的有机发光器件的结构。图1示出了包括基底1、阳极2、发光层3和阴极4的有机发光器件的一个实例。图2示出了包括基底1、阳极2、空穴传输层5、发光层3、电子传输层6和阴极4的有机发光器件的一个实例。
[0167]
根据本公开内容的有机发光器件可以通过顺序地堆叠上述结构来制造。在这种情况下,有机发光器件可以通过如下来制造:通过使用pvd(物理气相沉积)法例如溅射法或电子束蒸镀法在基底上沉积金属、具有导电性的金属氧化物、或其合金以形成阳极,在阳极上形成上述各层,然后在其上沉积可以用作阴极的材料。除了这样的方法之外,有机发光器件还可以通过在基底上以上述配置的相反顺序从阴极材料顺序地沉积至阳极材料来制造(wo 2003/012890)。此外,发光层可以通过使主体和掺杂剂经受真空沉积法和溶液涂覆法来形成。在本文中,溶液涂覆法意指旋涂、浸涂、刮刀涂覆、喷墨印刷、丝网印刷、喷洒法、辊涂等,但不限于此。
[0168]
另一方面,根据使用的材料,根据本公开内容的有机发光器件可以为前侧发射型、
[0196][0197]
在氮气气氛下将化合物sub9(15g,40.8mmol)和化合物a(11.8g,44.9mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(16.9g,122.3mmol)溶解在水(51ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.4mmol)。在反应12小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物9(14.1g)。(产率:63%,ms:[m h]
=550)
[0198]
制备例1
‑
10
[0199][0200]
在氮气气氛下将化合物sub10(15g,38.1mmol)和化合物a(11g,41.9mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(15.8g,114.3mmol)溶解在水(47ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.4mmol)。在反应8小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物10(15.8g)。(产率:72%,ms:[m h]
=576)
[0201]
制备例1
‑
11
[0202][0203]
在氮气气氛下将化合物sub11(15g,38.1mmol)和化合物a(11g,41.9mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(15.8g,114.3mmol)溶解在水(47ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.4mmol)。在反应8小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物11(16.6g)。(产率:76%,ms:[m h]
=576)
[0204]
制备例1
‑
12
[0205][0206]
在氮气气氛下将化合物sub12(15g,41.9mmol)和化合物a(12.1g,46.1mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(17.4g,125.8mmol)溶解在水(52ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.4mmol)。在反应12小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物12(13.8g)。(产率:61%,ms:[m h]
=540)
[0207]
制备例1
‑
13
[0208]
[0209]
在氮气气氛下将化合物sub13(15g,41.9mmol)和化合物a(12.1g,46.1mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(17.4g,125.8mmol)溶解在水(52ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.4mmol)。在反应9小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物13(15.4g)。(产率:68%,ms:[m h]
=540)
[0210]
制备例1
‑
14
[0211][0212]
在氮气气氛下将化合物sub14(15g,36.8mmol)和化合物a(10.6g,40.5mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(15.2g,110.3mmol)溶解在水(46ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.4mmol)。在反应11小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物14(16.3g)。(产率:75%,ms:[m h]
=590)
[0213]
制备例1
‑
15
[0214][0215]
在氮气气氛下将化合物sub15(15g,36.8mmol)和化合物a(10.6g,40.5mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(15.2g,110.3mmol)溶解在水(46ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.4mmol)。在反应12小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物15(15.2g)。(产率:70%,ms:[m h]
=590)
[0216]
制备例1
‑
16
[0217][0218]
在氮气气氛下将化合物sub16(15g,40.1mmol)和化合物a(11.6g,44.1mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(16.6g,120.4mmol)溶解在水(50ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.4mmol)。在反应12小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物16(13.8g)。(产率:62%,ms:[m h]
=556)
[0219]
制备例1
‑
17
[0220][0221]
在氮气气氛下将化合物sub17(15g,40.1mmol)和化合物a(11.6g,44.1mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(16.6g,120.4mmol)溶解在水(50ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.4mmol)。在反应9小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物17(15.1g)。(产率:68%,ms:[m h]
=556)
[0222]
制备例1
‑
18
[0223]
[0224]
在氮气气氛下将化合物sub18(15g,40.1mmol)和化合物a(11.6g,44.1mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(16.6g,120.4mmol)溶解在水(50ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.4mmol)。在反应9小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物18(17.8g)。(产率:80%,ms:[m h]
=556)
[0225]
制备例1
‑
19
[0226][0227]
在氮气气氛下将化合物sub19(15g,34.6mmol)和化合物a(10g,38.1mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(14.4g,103.9mmol)溶解在水(43ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。在反应10小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物19(15.5g)。(产率:73%,ms:[m h]
=615)
[0228]
制备例1
‑
20
[0229][0230]
在氮气气氛下将化合物sub20(15g,34.6mmol)和化合物a(10g,38.1mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(14.4g,103.9mmol)溶解在水(43ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。在反应11小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物20(17g)。(产率:80%,ms:[m h]
=61)
[0231]
制备例1
‑
21
[0232][0233]
在氮气气氛下将化合物sub21(15g,42mmol)和化合物a(12.1g,46.2mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(17.4g,126.1mmol)溶解在水(52ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.4mmol)。在反应9小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物21(14.5g)。(产率:64%,ms:[m h]
=539)
[0234]
制备例1
‑
22
[0235][0236]
在氮气气氛下将化合物sub22(15g,31.1mmol)和化合物a(9g,34.2mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(12.9g,93.2mmol)溶解在水(39ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。在反应11小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物22(12.4g)。(产率:60%,ms:[m h]
=665)
[0237]
制备例1
‑
23
[0238][0239]
在氮气气氛下将化合物sub2(15g,47.2mmol)和化合物b(7.4g,47.2mmol)添加至
thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(19.6g,141.6mmol)溶解在水(59ml)中,添加至混合物中,并将混合物充分搅拌,然后添加四(三苯基膦)钯(0)(0.5g,0.5mmol)。在反应10小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物subb
‑
1(13.9g)。(产率:75%,ms:[m h]
=394)
[0240][0241]
在氮气气氛下将化合物subb
‑
1(15g,38.1mmol)和化合物a(11g,41.9mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(15.8g,114.3mmol)溶解在水(47ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.4mmol)。在反应12小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物23(15.3g)。(产率:70%,ms:[m h]
=576)
[0242]
制备例1
‑
24
[0243][0244]
在氮气气氛下将化合物sub23(15g,35.7mmol)和化合物b(5.6g,35.7mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(14.8g,107.2mmol)溶解在水(44ml)中,添加至混合物中,并将混合物充分搅拌,然后添加四(三苯基膦)钯(0)(0.4g,0.4mmol)。在反应12小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物subb
‑
2(12g)。(产率:68%,ms:[m h]
=496)
[0245][0246]
在氮气气氛下将化合物subb
‑
2(15g,30.2mmol)和化合物a(8.7g,33.3mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(12.5g,90.7mmol)溶解在水(38ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。在反应8小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物24(13.1g)。(产率:64%,ms:[m h]
=678)
[0247]
制备例1
‑
25
[0248][0249]
在氮气气氛下将化合物sub12(15g,41.9mmol)和化合物b(6.6g,41.9mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(17.4g,125.8mmol)溶解在水(52ml)中,添加至混合物中,并将混合物充分搅拌,然后添加四(三苯基膦)钯(0)(0.5g,0.4mmol)。在反应10小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物subb
‑
3(12.9g)。(产率:71%,ms:[m h]
=434)
[0250][0251]
在氮气气氛下将化合物subb
‑
3(15g,34.6mmol)和化合物a(10g,38mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(14.3g,103.7mmol)溶解在水(43ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。在反应12小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物25(17g)。(产率:80%,ms:[m h]
=616)
[0252]
制备例1
‑
26
[0253][0254]
在氮气气氛下将化合物sub17(15g,40.1mmol)和化合物b(6.3g,40.1mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(16.6g,120.4mmol)溶解在水(50ml)中,添加至混合物中,并将混合物充分搅拌,然后添加四(三苯基膦)钯(0)(0.5g,0.4mmol)。在反应11小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物subb
‑
4(12.1g)。(产率:67%,ms:[m h]
=450)
[0255][0256]
在氮气气氛下将化合物subb
‑
4(15g,33.3mmol)和化合物a(9.6g,36.7mmol)添加
至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(13.8g,100mmol)溶解在水(41ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。在反应11小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物26(15.8g)。(产率:75%,ms:[m h]
=632)
[0257]
制备例1
‑
27
[0258][0259]
在氮气气氛下将化合物sub3(15g,38.1mmol)和化合物b(10g,38.1mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(15.8g,114.3mmol)溶解在水(47ml)中,添加至混合物中,并将混合物充分搅拌,然后添加四(三苯基膦)钯(0)(0.4g,0.4mmol)。在反应8小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物subb
‑
5(14.1g)。(产率:79%,ms:[m h]
=470)
[0260][0261]
在氮气气氛下将化合物subb
‑
5(15g,31.9mmol)和化合物a(9.2g,35.1mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(13.2g,95.8mmol)溶解在水(40ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。在反应10小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物27(12.5g)。(产率:60%,ms:[m h]
=652)
[0262]
制备例1
‑
28
[0263][0264]
在氮气气氛下将化合物sub24(15g,35.4mmol)和化合物b(5.5g,35.4mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(14.7g,106.2mmol)溶解在水(44ml)中,添加至混合物中,并将混合物充分搅拌,然后添加四(三苯基膦)钯(0)(0.4g,0.4mmol)。在反应9小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物subb
‑
6(12.5g)。(产率:71%,ms:[m h]
=500)
[0265][0266]
在氮气气氛下将化合物subb
‑
6(15g,30mmol)和化合物a(8.6g,33mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(12.4g,90mmol)溶解在水(37ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。在反应12小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物23(14.9g)。(产率:73%,ms:[m h]
=682)
[0267]
制备例1
‑
29
[0268][0269]
在氮气气氛下将化合物sub25(15g,56mmol)和化合物c(11.6g,56mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(23.2g,168.1mmol)溶解在水(70ml)中,添加至混合物中,并将混合物充分搅拌,然后添加四(三苯基膦)钯(0)(0.6g,0.6mmol)。在反应8小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物subc
‑
1(16.7g)。(产率:76%,ms:[m h]
=394)
[0270][0271]
在氮气气氛下将化合物subc
‑
1(15g,38.1mmol)和化合物a(10g,38.1mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(15.8g,114.3mmol)溶解在水(47ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.4mmol)。在反应11小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物29(16g)。(产率:73%,ms:[m h]
=576)
[0272]
制备例1
‑
30
[0273][0274]
在氮气气氛下将化合物sub2(15g,47.2mmol)和化合物c(9.7g,47.2mmol)添加至
thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(19.6g,141.6mmol)溶解在水(59ml)中,添加至混合物中,并将混合物充分搅拌,然后添加四(三苯基膦)钯(0)(0.5g,0.5mmol)。在反应8小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物subc
‑
2(14g)。(产率:67%,ms:[m h]
=444)
[0275][0276]
在氮气气氛下将化合物subc
‑
2(15g,33.8mmol)和化合物a(8.9g,33.8mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(14g,101.4mmol)溶解在水(42ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。在反应9小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物30(13.1g)。(产率:62%,ms:[m h]
=626)
[0277]
制备例1
‑
31
[0278][0279]
在氮气气氛下将化合物sub26(15g,40.8mmol)和化合物c(8.4g,40.8mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(16.9g,122.3mmol)溶解在水(51ml)中,添加至混合物中,并将混合物充分搅拌,然后添加四(三苯基膦)钯(0)(0.5g,0.4mmol)。在反应11小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物subc
‑
3(13.5g)。(产率:67%,ms:[m h]
=494)
[0280][0281]
在氮气气氛下将化合物subc
‑
3(15g,30.4mmol)和化合物a(8g,30.4mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(12.6g,91.1mmol)溶解在水(38ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。在反应10小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物31(15.6g)。(产率:76%,ms:[m h]
=676)
[0282]
制备例1
‑
32
[0283][0284]
在氮气气氛下将化合物sub4(15g,43.6mmol)和化合物c(9g,43.6mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(18.1g,130.9mmol)溶解在水(54ml)中,添加至混合物中,并将混合物充分搅拌,然后添加四(三苯基膦)钯(0)(0.5g,0.4mmol)。在反应8小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物subc
‑
4(16.4g)。(产率:80%,ms:[m h]
=470)
[0285][0286]
在氮气气氛下将化合物subc
‑
4(15g,31.9mmol)和化合物a(8.4g,31.9mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(13.2g,95.8mmol)溶解在水(40ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。在反应8小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物32(13.5g)。(产率:65%,ms:[m h]
=652)
[0287]
制备例1
‑
33
[0288][0289]
在氮气气氛下将化合物sub10(15g,38.1mmol)和化合物c(7.9g,38.1mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(15.8g,114.3mmol)溶解在水(47ml)中,添加至混合物中,并将混合物充分搅拌,然后添加四(三苯基膦)钯(0)(0.4g,0.4mmol)。在反应12小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物subc
‑
5(14.2g)。(产率:72%,ms:[m h]
=520)
[0290][0291]
在氮气气氛下将化合物subc
‑
5(15g,28.8mmol)和化合物a(7.6g,28.8mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(12g,86.5mmol)溶解在水(36ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.1g,0.3mmol)。在反应9小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物33(12.1g)。(产率:60%,ms:[m h]
=702)
[0292]
制备例1
‑
34
[0293][0294]
在氮气气氛下将化合物sub27(15g,40.8mmol)和化合物c(8.4g,40.8mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(16.9g,122.3mmol)溶解在水(51ml)中,添加至混合物中,并将混合物充分搅拌,然后添加四(三苯基膦)钯(0)(0.5g,0.4mmol)。在反应12小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物subc
‑
6(15.7g)。(产率:78%,ms:[m h]
=494)
[0295][0296]
在氮气气氛下将化合物subc
‑
6(15g,30.4mmol)和化合物a(8g,30.4mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(12.6g,91.1mmol)溶解在水(38ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。在反应10小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物34(15.2g)。(产率:74%,ms:[m h]
=676)
[0297]
制备例1
‑
35
[0298][0299]
在氮气气氛下将化合物sub34(15g,39.1mmol)和化合物c(8.1g,39.1mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(16.2g,117.2mmol)溶解在水(49ml)中,添加至混合物中,并将混合物充分搅拌,然后添加四(三苯基膦)钯(0)(0.5g,0.4mmol)。在反应9小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物subc
‑
7(15.9g)。(产率:80%,ms:[m h]
=510)
[0300][0301]
在氮气气氛下将化合物subc
‑
7(15g,29.4mmol)和化合物a(7.7g,29.4mmol)添加
至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(12.2g,88.2mmol)溶解在水(37ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。在反应9小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物35(14.2g)。(产率:70%,ms:[m h]
=692)
[0302]
制备例1
‑
36
[0303][0304]
在氮气气氛下将化合物sub28(15g,34.6mmol)和化合物c(7.2g,34.6mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(14.4g,103.9mmol)溶解在水(43ml)中,添加至混合物中,并将混合物充分搅拌,然后添加四(三苯基膦)钯(0)(0.4g,0.3mmol)。在反应8小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物subc
‑
8(13.3g)。(产率:69%,ms:[m h]
=559)
[0305][0306]
在氮气气氛下将化合物subc
‑
8(15g,26.8mmol)和化合物a(7g,26.8mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(11.1g,80.5mmol)溶解在水(33ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.1g,0.3mmol)。在反应12小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物36(15.5g)。(产率:78%,ms:[m h]
=741)
[0307]
制备例1
‑
37
[0308][0309]
在氮气气氛下将化合物sub19(15g,34.6mmol)和化合物c(7.2g,34.6mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(14.4g,103.9mmol)溶解在水(43ml)中,添加至混合物中,并将混合物充分搅拌,然后添加四(三苯基膦)钯(0)(0.4g,0.3mmol)。在反应10小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物subc
‑
9(13.9g)。(产率:72%,ms:[m h]
=559)
[0310][0311]
在氮气气氛下将化合物subc
‑
9(15g,26.8mmol)和化合物a(7g,26.8mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(11.1g,80.5mmol)溶解在水(33ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.1g,0.3mmol)。在反应11小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物37(14.5g)。(产率:73%,ms:[m h]
=741)
[0312]
制备例1
‑
38
[0313][0314]
在氮气气氛下将化合物sub12(15g,41.9mmol)和化合物c(8.7g,41.9mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(17.4g,125.8mmol)溶解在水(52ml)中,添加至混合物中,并将混合物充分搅拌,然后添加四(三苯基膦)钯(0)(0.5g,
0.4mmol)。在反应9小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物subc
‑
10(14.2g)。(产率:70%,ms:[m h]
=484)
[0315][0316]
在氮气气氛下将化合物subc
‑
10(15g,31mmol)和化合物a(8.1g,31mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(12.9g,93mmol)溶解在水(39ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。在反应10小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物38(13.4g)。(产率:65%,ms:[m h]
=666)
[0317]
制备例1
‑
39
[0318][0319]
在氮气气氛下将化合物sub14(15g,36.8mmol)和化合物c(7.6g,36.8mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(15.2g,110.3mmol)溶解在水(46ml)中,添加至混合物中,并将混合物充分搅拌,然后添加四(三苯基膦)钯(0)(0.4g,0.4mmol)。在反应12小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物subc
‑
11(12.9g)。(产率:66%,ms:[m h]
=534)
[0320][0321]
在氮气气氛下将化合物subc
‑
11(15g,28.1mmol)和化合物a(7.4g,28.1mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(11.6g,84.3mmol)溶解在水(35ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.1g,0.3mmol)。在反应9小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物39(14.5g)。(产率:72%,ms:[m h]
=716)
[0322]
制备例1
‑
40
[0323][0324]
在氮气气氛下将化合物sub29(15g,36.8mmol)和化合物c(7.6g,36.8mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(15.2g,110.3mmol)溶解在水(46ml)中,添加至混合物中,并将混合物充分搅拌,然后添加四(三苯基膦)钯(0)(0.4g,0.4mmol)。在反应12小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物subc
‑
12(12.9g)。(产率:66%,ms:[m h]
=534)
[0325]
[0326]
在氮气气氛下将化合物subc
‑
12(15g,28.1mmol)和化合物a(7.4g,28.1mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(11.6g,84.3mmol)溶解在水(35ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.1g,0.3mmol)。在反应9小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物40(12.7g)。(产率:63%,ms:[m h]
=716)
[0327]
制备例1
‑
41
[0328][0329]
在氮气气氛下将化合物sub30(15g,35.5mmol)和化合物c(7.3g,35.5mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(14.7g,106.4mmol)溶解在水(44ml)中,添加至混合物中,并将混合物充分搅拌,然后添加四(三苯基膦)钯(0)(0.4g,0.4mmol)。在反应12小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物subc
‑
13(14.6g)。(产率:75%,ms:[m h]
=550)
[0330][0331]
在氮气气氛下将化合物subc
‑
13(15g,27.3mmol)和化合物a(7.1g,27.3mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(11.3g,81.8mmol)溶解在水(34ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.1g,0.3mmol)。在反应8小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物41(13.6g)。(产率:68%,ms:[m h]
=732)
[0332]
制备例1
‑
42
[0333][0334]
在氮气气氛下将化合物sub17(15g,40.1mmol)和化合物c(8.3g,40.1mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(16.6g,120.4mmol)溶解在水(50ml)中,添加至混合物中,并将混合物充分搅拌,然后添加四(三苯基膦)钯(0)(0.5g,0.4mmol)。在反应8小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物subc
‑
14(13g)。(产率:65%,ms:[m h]
=500)
[0335][0336]
在氮气气氛下将化合物subc
‑
14(15g,30mmol)和化合物a(7.9g,30mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(12.4g,90mmol)溶解在水(37ml)中,添加至混合物中,并将混合物充分搅拌,然后添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。在反应8小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物42(14.7g)。(产率:72%,ms:[m h]
=682)
[0337]
制备例2
‑1[0338][0339]
在氮气气氛下将化合物a(15g,58.3mmol)和化合物b(10g,64.2mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(24.2g,175mmol)溶解在水(73ml)中,添加至混合物中,并将混合物充分搅拌,然后添加四(三苯基膦)钯(0)(0.7g,0.6mmol)。在反应11小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将
[0346][0347]
在氮气气氛下将化合物suba
‑
1(10g,34.6mmol)、化合物sub3(14.6g,34.6mmol)和叔丁醇钠(6.7g,69.3mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。当反应在3小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
3(14g)。(产率:60%,ms:[m h]
=674)
[0348]
制备例2
‑4[0349][0350]
在氮气气氛下将化合物suba
‑
1(10g,34.6mmol)、化合物sub4(13.8g,34.6mmol)和叔丁醇钠(6.7g,69.3mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。当反应在3小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
4(12.4g)。(产率:55%,ms:[m h]
=650)
[0351]
制备例2
‑5[0352][0353]
在氮气气氛下将化合物suba
‑
1(10g,34.6mmol)、化合物sub5(12.9g,34.6mmol)和叔丁醇钠(6.7g,69.3mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。当反应在3小时之后完成时,将反应混合物冷
[0361][0362]
在氮气气氛下将化合物suba
‑
1(10g,34.6mmol)、化合物sub8(11.6g,34.6mmol)和叔丁醇钠(6.7g,69.3mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。当反应在2小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
8(13.8g)。(产率:68%,ms:[m h]
=588)
[0363]
制备例2
‑9[0364][0365]
在氮气气氛下将化合物suba
‑
1(10g,34.6mmol)、化合物sub9(11.6g,34.6mmol)和叔丁醇钠(6.7g,69.3mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。当反应在2小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
9(10.6g)。(产率:52%,ms:[m h]
=588)
[0366]
制备例2
‑
10
[0367][0368]
在氮气气氛下将化合物suba
‑
1(10g,34.6mmol)、化合物sub10(12.5g,34.6mmol)和叔丁醇钠(6.7g,69.3mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其
中添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。当反应在3小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
10(11.5g)。(产率:54%,ms:[m h]
=614)
[0369]
制备例2
‑
11
[0370][0371]
在氮气气氛下将化合物suba
‑
1(10g,34.6mmol)、化合物sub11(15.2g,34.6mmol)和叔丁醇钠(6.7g,69.3mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。当反应在2小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
11(13.6g)。(产率:57%,ms:[m h]
=690)
[0372]
制备例2
‑
12
[0373][0374]
在氮气气氛下将化合物suba
‑
1(10g,34.6mmol)、化合物sub12(13.9g,34.6mmol)和叔丁醇钠(6.7g,69.3mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。当反应在3小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
12(15.8g)。(产率:70%,ms:[m h]
=654)
[0375]
制备例2
‑
13
[0376][0377]
在氮气气氛下将化合物suba
‑
1(10g,34.6mmol)、化合物sub13(115.5g,34.6mmol)和叔丁醇钠(6.7g,69.3mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。当反应在2小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
13(13.8g)。(产率:68%,ms:[m h]
=588)
[0378]
制备例2
‑
14
[0379][0380]
在氮气气氛下将化合物suba
‑
1(10g,34.6mmol)、化合物sub14(13.8g,34.6mmol)和叔丁醇钠(6.7g,69.3mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。当反应在2小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
14(14.8g)。(产率:66%,ms:[m h]
=650)
[0381]
制备例2
‑
15
[0382][0383]
在氮气气氛下将化合物suba
‑
1(10g,34.6mmol)、化合物sub15(13.8g,34.6mmol)和叔丁醇钠(6.7g,69.3mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。当反应在2小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物
通过硅胶柱色谱法纯化以得到化合物2
‑
15(14.4g)。(产率:64%,ms:[m h]
=650)
[0384]
制备例2
‑
16
[0385][0386]
在氮气气氛下将化合物suba
‑
1(10g,34.6mmol)、化合物sub16(16.4g,34.6mmol)和叔丁醇钠(6.7g,69.3mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。当反应在3小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
16(13.1g)。(产率:52%,ms:[m h]
=726)
[0387]
制备例2
‑
17
[0388][0389]
在氮气气氛下将化合物suba
‑
1(10g,34.6mmol)、化合物sub17(16.4g,34.6mmol)和叔丁醇钠(6.7g,69.3mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。当反应在2小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
17(16.6g)。(产率:66%,ms:[m h]
=726)
[0390]
制备例2
‑
18
[0391][0392]
在氮气气氛下将化合物suba
‑
1(10g,34.6mmol)、化合物sub18(11.1g,34.6mmol)和叔丁醇钠(6.7g,69.3mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。当反应在2小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
18(11.1g)。(产率:56%,ms:[m h]
=572)
[0393]
制备例2
‑
19
[0394][0395]
在氮气气氛下将化合物suba
‑
1(10g,34.6mmol)、化合物sub19(15g,34.6mmol)和叔丁醇钠(6.7g,69.3mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。当反应在2小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
19(16.4g)。(产率:69%,ms:[m h]
=687)
[0396]
制备例2
‑
20
[0397][0398]
在氮气气氛下将化合物suba
‑
1(10g,34.6mmol)、化合物sub20(13.7g,34.6mmol)和叔丁醇钠(6.7g,69.3mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。当反应在3小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
20(15g)。(产率:67%,ms:[m h]
=648)
[0399]
制备例2
‑
21
[0400][0401]
在氮气气氛下将化合物suba
‑
1(10g,34.6mmol)、化合物sub21(11.1g,34.6mmol)和叔丁醇钠(6.7g,69.3mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。当反应在3小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
21(10.5g)。(产率:53%,ms:[m h]
=572)
[0402]
制备例2
‑
22
[0403][0404]
在氮气气氛下将化合物a(15g,58.3mmol)和化合物c(10g,64.2mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(24.2g,175mmol)溶解在水(73ml)中,添加至混合物中,并将混合物充分搅拌,然后添加四(三苯基膦)钯(0)(0.7g,0.6mmol)。在
反应8小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物suba
‑
2(12.4g)。(产率:74%,ms:[m h]
=289)
[0405][0406]
在氮气气氛下将化合物suba
‑
2(10g,34.6mmol)、化合物sub22(12g,34.6mmol)和叔丁醇钠(6.7g,69.3mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。当反应在2小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
22(14.1g)。(产率:68%,ms:[m h]
=598)
[0407]
制备例2
‑
23
[0408][0409]
在氮气气氛下将化合物suba
‑
1(10g,34.6mmol)、化合物sub23(12g,34.6mmol)和叔丁醇钠(6.7g,69.3mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。当反应在2小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
23(11g)。(产率:53%,ms:[m h]
=598)
[0410]
制备例2
‑
24
[0411][0412]
在氮气气氛下将化合物suba
‑
2(10g,34.6mmol)、化合物sub24(17.7g,34.6mmol)和叔丁醇钠(6.7g,69.3mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。当反应在3小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
24(15.3g)。(产率:58%,ms:[m h]
=763)
[0413]
制备例2
‑
25
[0414][0415]
在氮气气氛下将化合物sub25(10g,59.1mmol)、化合物suba
‑
1(34.1g,118.2mmol)和叔丁醇钠(17g,177.3mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.6g,1.2mmol)。当反应在3小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
25(27.1g)。(产率:68%,ms:[m h]
=674)
[0416]
制备例2
‑
26
[0417][0418]
在氮气气氛下将化合物sub26(10g,51.7mmol)、化合物suba
‑
1(29.9g,103.5mmol)和叔丁醇钠(14.9g,155.2mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.5g,1mmol)。当反应在3小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
26(18g)。(产率:50%,ms:[m h]
=698)
[0419]
制备例2
‑
27
[0420][0421]
在氮气气氛下将化合物sub27(10g,30mmol)、化合物suba
‑
1(17.3g,60mmol)和叔丁醇钠(8.6g,90mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.3g,0.6mmol)。当反应在2小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
27(14.6g)。(产率:58%,ms:[m h]
=838)
[0422]
制备例2
‑
28
[0423][0424]
在氮气气氛下将化合物suba
‑
2(10g,34.6mmol)、化合物sub28(7.2g,34.6mmol)和叔丁醇钠(6.7g,69.3mmol)添加至甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添
加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。当反应在3小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物suba
‑2‑
1(11.2g)。(产率:70%,ms:[m h]
=462)
[0425][0426]
在氮气气氛下将化合物suba
‑2‑
1(10g,21.7mmol)、化合物suba
‑
1(6.3g,21.7mmol)和叔丁醇钠(4.2g,43.3mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.1g,0.2mmol)。当反应在3小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
28(10.7g)。(产率:69%,ms:[m h]
=714)
[0427]
制备例2
‑
29
[0428][0429]
在氮气气氛下将化合物suba
‑
2(10g,34.6mmol)、化合物sub29(8.5g,34.6mmol)和叔丁醇钠(6.7g,69.3mmol)添加至甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.2g,0.3mmol)。当反应在2小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物suba
‑2‑
2(9.8g)。(产率:57%,ms:[m h]
=498)
[0430][0431]
在氮气气氛下将化合物suba
‑2‑
2(10g,20.1mmol)、化合物suba
‑
1(5.8g,20.1mmol)和叔丁醇钠(3.9g,40.2mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.1g,0.2mmol)。当反应在2小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
29(10.1g)。(产率:67%,ms:[m h]
=750)
[0432]
制备例2
‑
30
[0433][0434]
在氮气气氛下将化合物d(15g,45mmol)和化合物b(7.7g,49.5mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(18.7g,135mmol)溶解在水(56ml)中,添加至混合物中,并将混合物充分搅拌,然后添加四(三苯基膦)钯(0)(0.5g,0.5mmol)。在反应10小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物subd
‑
1(13.1g)。(产率:80%,ms:[m h]
=365)
[0435][0436]
在氮气气氛下将化合物subd
‑
1(10g,27.4mmol)、化合物sub22(9.5g,27.4mmol)和
叔丁醇钠(5.3g,54.8mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.1g,0.3mmol)。当反应在2小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
30(9.6g)。(产率:52%,ms:[m h]
=674)
[0437]
制备例2
‑
31
[0438][0439]
在氮气气氛下将化合物subd
‑
1(10g,27.4mmol)、化合物sub30(11.5g,27.4mmol)和叔丁醇钠(5.3g,54.8mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.1g,0.3mmol)。当反应在2小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
31(13.9g)。(产率:68%,ms:[m h]
=748)
[0440]
制备例2
‑
32
[0441][0442]
在氮气气氛下将化合物d(15g,45mmol)和化合物c(7.7g,49.5mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(18.7g,135mmol)溶解在水(56ml)中,添加至混合物中,并将混合物充分搅拌,然后添加四(三苯基膦)钯(0)(0.5g,0.5mmol)。在反应9小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物subd
‑
2(9.3g)。(产率:72%,ms:[m h]
=289)
[0443][0444]
在氮气气氛下将化合物subd
‑
2(10g,27.4mmol)、化合物sub31(12.4g,27.4mmol)和叔丁醇钠(5.3g,54.8mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.1g,0.3mmol)。当反应在2小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
32(15g)。(产率:70%,ms:[m h]
=780)
[0445]
制备例2
‑
33
[0446][0447]
在氮气气氛下将化合物a(15g,58.3mmol)和化合物e(14.9g,64.2mmol)添加至thf(300ml)中,并将混合物搅拌并回流。然后,将碳酸钾(24.2g,175mmol)溶解在水(73ml)中,添加至混合物中,并将混合物充分搅拌,然后添加四(三苯基膦)钯(0)(0.7g,0.6mmol)。在反应10小时之后,将反应混合物冷却至室温,并分离有机层和水层,然后将有机层蒸馏。将其再次溶解在氯仿中,用水洗涤两次,然后分离有机层,向其中添加无水硫酸镁,将混合物搅拌并过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物suba
‑
3(14.9g)。(产率:70%,ms:[m h]
=365)
[0448][0449]
在氮气气氛下将化合物suba
‑
3(10g,27.4mmol)、化合物sub32(2.6g,27.4mmol)和叔丁醇钠(5.3g,54.8mmol)添加至甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添
加双(三叔丁基膦)钯(0)(0.1g,0.3mmol)。当反应在2小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物suba
‑3‑
1(5.8g)。(产率:50%,ms:[m h]
=422)
[0450][0451]
在氮气气氛下将化合物suba
‑3‑
1(10g,23.7mmol)、化合物suba
‑
2(6.9g,23.7mmol)和叔丁醇钠(4.6g,47.4mmol)添加至甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.1g,0.2mmol)。当反应在2小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
33(8.9g)。(产率:56%,ms:[m h]
=674)
[0452]
制备例2
‑
34
[0453][0454]
在氮气气氛下将化合物suba
‑
3(10g,27.4mmol)、化合物sub33(4.6g,27.4mmol)和叔丁醇钠(5.3g,54.8mmol)添加至甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.1g,0.3mmol)。当反应在3小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物suba
‑3‑
2(9.1g)。(产率:67%,ms:[m h]
=498)
[0455][0456]
在氮气气氛下将化合物suba
‑3‑
2(10g,20.1mmol)、化合物suba
‑
2(5.8g,20.1mmol)和叔丁醇钠(3.9g,40.2mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.1g,0.2mmol)。当反应在2小时之后完成时,将反
应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
34(9.6g)。(产率:64%,ms:[m h]
=750)
[0457]
制备例2
‑
35
[0458][0459]
在氮气气氛下将化合物suba
‑3‑
2(10g,20.1mmol)、化合物suba
‑
1(5.8g,20.1mmol)和叔丁醇钠(3.9g,40.2mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.1g,0.2mmol)。当反应在3小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
35(8.6g)。(产率:57%,ms:[m h]
=750)
[0460]
制备例2
‑
36
[0461][0462]
在氮气气氛下将化合物suba
‑
3(10g,27.4mmol)、化合物sub34(4.6g,27.4mmol)和叔丁醇钠(5.3g,54.8mmol)添加至甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.1g,0.3mmol)。当反应在3小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物suba
‑3‑
3(8.6g)。(产率:63%,ms:[m h]
=498)
[0463][0464]
在氮气气氛下将化合物suba
‑3‑
3(10g,20.1mmol)、化合物suba
‑
2(5.8g,20.1mmol)和叔丁醇钠(3.9g,40.2mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.1g,0.2mmol)。当反应在2小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
36(10.4g)。(产率:69%,ms:[m h]
=750)
[0465]
制备例2
‑
37
[0466][0467]
在氮气气氛下将化合物sub35(10g,51.7mmol)、化合物suba
‑
2(29.9g,103.5mmol)和叔丁醇钠(14.9g,155.2mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(0.5g,1mmol)。当反应在2小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
37(23.8g)。(产率:66%,ms:[m h]
=698)
[0468]
制备例2
‑
38
[0469][0470]
在氮气气氛下将化合物sub33(10g,107.4mmol)、化合物subd
‑
1(78.4g,
214.8mmol)和叔丁醇钠(31g,322.1mmol)添加至二甲苯(200ml)中,并将混合物搅拌并回流。然后,向其中添加双(三叔丁基膦)钯(0)(1.1g,2.1mmol)。当反应在2小时之后完成时,将反应混合物冷却至室温,并在减压下除去溶剂。然后,将化合物再次完全溶解在氯仿中,用水洗涤两次,然后分离有机层,用无水硫酸镁处理,然后过滤,并将滤液在减压下蒸馏。将浓缩的化合物通过硅胶柱色谱法纯化以得到化合物2
‑
38(53.9g)。(产率:67%,ms:[m h]
=750)
[0471]
[实施例]
[0472]
实施例1
[0473]
将其上涂覆有的厚度的ito(铟锡氧化物)薄膜的玻璃基底放入包含溶解在其中的清洁剂的蒸馏水中并通过超声波洗涤。在这种情况下,使用的清洁剂为市售的来自fisher co.的产品,并且蒸馏水为通过使用市售的来自millipore co.的过滤器过滤两次的蒸馏水。将ito洗涤30分钟,然后通过使用蒸馏水重复两次超声洗涤10分钟。在用蒸馏水洗涤完成之后,用异丙醇、丙酮和甲醇溶剂对基底进行超声洗涤,并干燥,之后将其输送至等离子体清洗器。然后,用氧等离子体清洗基底5分钟,然后转移至真空蒸镀器。
[0474]
在由此准备的ito透明电极上形成厚度为的以下化合物hi
‑
1作为空穴注入层,但是以下化合物a
‑
1以1.5重量%的浓度进行p掺杂。在空穴注入层上真空沉积以下化合物ht
‑
1以形成膜厚度为的空穴传输层。然后,在空穴传输层上真空沉积以下化合物eb
‑
1以形成膜厚度为的电子阻挡层。然后,在电子阻挡层上以49:49:2的重量比分别真空沉积先前制备的化合物1和化合物2
‑
1作为主体、以及以下化合物dp
‑
7作为掺杂剂以形成膜厚度为的发光层。在发光层上真空沉积以下化合物hb
‑
1以形成膜厚度为的空穴阻挡层。在空穴阻挡层上以2:1的比真空沉积以下化合物et
‑
1和以下化合物liq以形成膜厚度为liq以形成膜厚度为的电子注入和传输层。在电子注入和传输层上顺序地沉积氟化锂(lif)和铝至厚度分别为和从而形成阴极。
[0475][0476]
在上述过程中,将有机材料的沉积速率保持在秒至秒,将阴极的氟化锂和铝的沉积速率分别保持在秒和秒,并且将沉积期间的真空度保持在2
×
10
‑7托至5
×
10
‑6托,从而制造有机发光器件。
[0477]
实施例2至100
[0478]
以与实施例1中相同的方式制造有机发光器件,不同之处在于使用以下表1至表3中所示的化合物作为发光层的主体。
[0479]
比较例1至85
[0480]
以与实施例1中相同的方式制造有机发光器件,不同之处在于使用以下表4至表7中所示的化合物作为发光层的主体。在以下表6和表7中,意指将单一化合物用作发光层的主体,并且表7中的化合物分别如下。
[0481][0482]
通过向实施例和比较例中制造的有机发光器件施加电流(15ma/cm2)来测量驱动电压、发光效率和寿命,并且结果示于以下表1至表7中。寿命t95意指亮度减小至初始亮度(6000尼特)的95%所需的时间(小时)。
[0483]
[表1]
[0484]
[0485]
[0486][0487]
[表2]
[0488]
[0489]
[0490][0491]
[表3]
[0492]
[0493][0494]
[表4]
[0495]
[0496]
[0497][0498]
[表5]
[0499][0500]
[表6]
[0501]
分类主体效率(cd/a)寿命t95(小时)发光颜色比较例49化合物120.3122红色比较例50化合物321.1135红色比较例51化合物523.2148红色比较例52化合物922.6127红色比较例53化合物1021.8143红色比较例54化合物1423.2157红色比较例55化合物1722.6145红色
比较例56化合物1921.4128红色比较例57化合物2124.5172红色比较例58化合物2319.4126红色比较例59化合物2520.2129红色比较例60化合物2921.3141红色比较例61化合物3021.5133红色比较例62化合物3120.2145红色比较例63化合物3221.6157红色比较例64化合物3322.3140红色比较例65化合物3421.6152纤色比较例66化合物3522.2143红色比较例67化合物3622.8142红色比较例68化合物3721.6158红色比较例69化合物3822.3141红色比较例70化合物3921.5151红色比较例71化合物4020.7160红色比较例72化合物4122.6159纤色比较例73化合物4223.8163红色
[0502]
[表7]
[0503]
分类主体效率(cd/a)寿命t95(小时)发光颜色比较例74c
‑
117.4107红色比较例75c
‑
216.183红色比较例76c
‑
316.494红色比较例77c
‑
416.087红色比较例78c
‑
518.7110红色比较例79c
‑
616.547红色比较例80c
‑
715.322红色比较例81c
‑
815.137红色比较例82c
‑
917.375红色比较例83c
‑
1017.592红色比较例84c
‑
1115.863红色比较例85c
‑
1216.178红色
[0504]
如以上的表所示,相比于其中仅使用由化学式1表示的化合物和化学式2表示的化合物中的一者(表6)或者两者均不使用(表7)的比较例的有机发光器件,其中将由化学式1表示的第一化合物和由化学式2表示的第二化合物同时用作发光层的主体材料的实施例的有机发光器件表现出优异的发光效率和显著改善的寿命特性。具体地,根据实施例的器件表现出比其中将由化学式1表示的化合物用作单一主体的比较例的器件更高的效率和更长的寿命。此外,甚至相比于其中将比较例的化合物c
‑
1至c
‑
12用作第一主体并且将由化学式2表示的化合物用作第二主体的比较例的器件,根据实施例的器件也表现出改善的效率和
寿命特性。由此,确定当将由化学式1表示的第一化合物和由化学式2表示的第二化合物的组合用作共主体时,在红色发光层中有效地进行向红色掺杂剂的能量转移。这可以判断是因为第一化合物具有高的电子和空穴稳定性,此外,因为随着同时使用第二化合物,空穴的量增加,因此红色发光层中的电子和空穴保持更稳定的平衡。
[0505]
因此,当将第一化合物和第二化合物同时用作有机发光器件的主体材料时,确定可以改善有机发光器件的驱动电压、发光效率和/或寿命特性。通常,考虑到有机发光器件的发光效率和寿命特性具有彼此折衷的关系,这可以被认为是采用本公开内容的化合物的组合的有机发光器件表现出相比于比较例的器件的显著改善的器件特性。
[0506]
附图标记说明
[0507]
1:基底 2:阳极
[0508]
3:发光层 4:阴极
[0509]
5:空穴传输层 6:电子传输层
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。