1.本发明涉及油水分离材料领域,尤其涉及一种高效油水分离纳米纤维膜及其制备方法和应用。
背景技术:
2.油水分离是解决工业含油废水、溢油污染和环境保护的重要方法。它不仅具有科学研究价值,而且具有实际应用价值。因此,开发高效处理含油污水的功能材料势在必行。
3.对于含油废水的处理,传统的分离技术主要有重力分离、气浮、混凝、脱乳化等,但存在分离效率低、能耗高、操作过程复杂、二次污染严重、乳化液分离效果差等缺点。膜分离技术具有分离效率高和操作简单的特点,受到广泛关注。
4.目前,超亲水膜主要是通过表面改性来制备的,如表面涂覆和表面接枝。然而,改性后的膜表面不稳定,易脱落,亲水性逐渐减弱。对于油水分离,需要更稳定的水化层来阻止油滴接触膜基质表面,使膜具有较高的分离效率和稳定性。
技术实现要素:
5.本发明提供了一种高效油水分离纳米纤维膜及其制备方法和应用,本发明提供的高效油水分离纳米纤维膜的膜通量高、油水分离效率高,且循环多次后分离效率仍然较高,稳定性较好。
6.本发明提供了一种高效油水分离纳米纤维膜的制备方法,包括以下步骤:
7.(1)在保护气氛下,将丙烯腈、甲基丙烯酸甲酯和二甲基亚砜混合后加热至聚合反应温度,然后加入引发剂mebr和偶氮二异丁腈,进行聚合反应,得到纺丝原液;
8.所述引发剂mebr的结构如式i所示:
[0009][0010]
(2)采用静电纺丝法将步骤(1)得到的纺丝原液制备成膜,得到纤维膜;
[0011]
(3)在保护气氛下,将亲水单体、2,2'-联吡啶、cubr和溶剂混合,得到混合溶液;将所述步骤(2)得到的纤维膜浸没在混合溶液中进行atrp聚合反应,得到高效油水分离纳米纤维膜。
[0012]
优选的,所述步骤(1)中丙烯腈、甲基丙烯酸甲酯、引发剂mebr和偶氮二异丁腈的质量比为10~20:0.1~0.5:1~3:0.1~0.3,所述纺丝原液中引发剂mebr的质量浓度为5~35%。
[0013]
优选的,所述步骤(1)中聚合反应温度为60~76℃,所述聚合反应的时间为4~10h。
[0014]
优选的,所述步骤(2)中静电纺丝法的电压为5~30kv,注射速率为0.2~2ml/h,接
收距离为15~30cm。
[0015]
优选的,所述步骤(2)得到的纤维膜中纤维的直径为100~1000nm。
[0016]
优选的,所述步骤(3)中亲水单体包括2[-(甲基丙烯酰基氧基)乙基]二甲基(3-磺酸丙基)氢氧化铵单体、n-异丙基丙烯酰胺单体、3-磺酸丙基甲基丙烯酸钾单体或甲基丙烯酰氧乙基三甲基氯化铵单体。
[0017]
优选的,所述步骤(3)中亲水单体、2,2'-联吡啶、cubr和溶剂的用量比为2~6g:80~160mg:10~50mg:8ml。
[0018]
优选的,所述atrp聚合反应的温度为15~35℃,时间为1~6h。
[0019]
本发明还提供了上述技术方案所述制备方法制备得到的高效油水分离纳米纤维膜。
[0020]
本发明还提供了上述技术方案所述高效油水分离纳米纤维膜作为油水分离膜的应用。
[0021]
有益效果:
[0022]
本发明通过将引发剂mebr以共价键方式键入到高分子聚合物中,然后通过静电纺丝的方法制备纤维膜,再将亲水单体与纤维膜表面及内部进行atrp聚合反应,实现亲水单体对纤维膜的亲水改性,使纤维膜的比表面积增大,水化作用增强,大大提高了纤维膜的膜通量、油水分离效率和稳定性。
附图说明
[0023]
图1为实施例1中间步骤得到的纳米纤维膜的扫描电镜图;
[0024]
图2为实施例1由atrp聚合改性得到的高效油水分离纳米纤维膜吸水前后的扫描电镜图;
[0025]
图3为实施例1和对比例1制备的油水分离纳米纤维膜在空气中与水滴的接触角图;
[0026]
图4为对比例1制备的油水分离纳米纤维膜的循环实验图。
具体实施方式
[0027]
本发明提供了一种高效油水分离纳米纤维膜的制备方法,包括以下步骤:
[0028]
(1)在保护气氛下,将丙烯腈、甲基丙烯酸甲酯和二甲基亚砜混合后加热至聚合反应温度,然后加入引发剂mebr和偶氮二异丁腈,进行聚合反应,得到纺丝原液;
[0029]
所述引发剂mebr的结构如式i所示:
[0030][0031]
(2)采用静电纺丝法将步骤(1)得到的纺丝原液制备成膜,得到纤维膜;
[0032]
(3)在保护气氛下,将亲水单体、2,2'-联吡啶、cubr和溶剂混合,得到混合溶液;将所述步骤(2)得到的纤维膜浸没在混合溶液中进行atrp聚合反应,得到高效油水分离纳米纤维膜。
[0033]
在本发明中,所有原料如无特殊说明,均为市售商品。
[0034]
本发明在保护气氛下,将丙烯腈、甲基丙烯酸甲酯和二甲基亚砜混合后加热至聚合反应温度,然后加入引发剂mebr和偶氮二异丁腈,进行聚合反应,得到纺丝原液。
[0035]
在本发明中,所述聚合反应的方程式如式ii所示:
[0036][0037]
在本发明中,所述丙烯腈、甲基丙烯酸甲酯、引发剂mebr和偶氮二异丁腈的质量比优选为10~20:0.1~0.5:1~3:0.1~0.3,更优选为22.5:0.5:2:0.18;所述纺丝原液中引发剂mebr的质量浓度优选为5~35%,进一步优选为5%~30%,更优选为5%~25%。
[0038]
在本发明中,所述引发剂mebr的制备方法优选包括以下步骤:
[0039]
在氮气保护下,将2-溴代异丁酰溴、2-羟乙基甲基丙烯酸酯和三乙胺在二氯甲烷中进行反应,得到引发剂mebr;所述2-溴代异丁酰溴、2-羟乙基甲基丙烯酸酯的摩尔比优选为1:1,所述反应的温度优选为0℃~20℃,所述反应的时间优选为3~7h。反应完成后,优选将反应产物水洗得到引发剂mebr。
[0040]
在本发明中,所述聚合反应温度优选为60~76℃,更优选为60~70℃;所述聚合反应的时间优选为4~10h,更优选为6~8h。在本发明中,所述保护气氛优选为氮气气氛。本发明通过上述聚合反应,将引发剂mebr以共价键的形式引入聚合物内部,然后通过后续亲水单体与引发剂反应,将亲水单体牢固地固定在纤维膜表面和内部,有利于提高制备得到的高效油水分离纳米纤维膜的亲水性能和稳定性能。
[0041]
得到纺丝原液后,本发明采用静电纺丝法将纺丝原液制备成膜,得到纤维膜。
[0042]
在本发明中,所述静电纺丝法的电压优选为5~30kv,更优选为10~25kv,注射速率优选为0.2~2ml/h,更优选为0.5~1.5ml/h,接收距离优选为15~30cm,更优选为20~30cm。在本发明中,所述纤维膜中纤维的直径优选为100~1000nm。
[0043]
得到纤维膜后,本发明在保护气氛下,将亲水单体、2,2'-联吡啶、cubr和溶剂混合,得到混合溶液。
[0044]
在本发明中,所述保护气氛优选为氮气气氛,所述亲水单体优选包括2[-(甲基丙烯酰基氧基)乙基]二甲基(3-磺酸丙基)氢氧化铵单体、n-异丙基丙烯酰胺单体、3-磺酸丙基甲基丙烯酸钾单体或甲基丙烯酰氧乙基三甲基氯化铵单体。本发明优选采用上述亲水单体,有利于提高最终制备得到的高效油水分离纳米纤维膜的亲水性能和稳定性能。
[0045]
在本发明中,所述亲水单体、2,2'-联吡啶、cubr和溶剂的用量比优选为2~6g:80~160mg:10~50mg:8ml,更优选为3~6g:100~160mg:20~50mg:8ml。在本发明中,所述溶剂优选为水和甲醇的混合溶剂或者水和异丙醇的混合溶剂,所述水和甲醇的混合溶剂中水和甲醇的体积比优选为3:2;所述水和异丙醇的混合溶剂中水和异丙醇的体积比优选为1:3。
[0046]
在本发明中,所述亲水单体、2,2'-联吡啶、cubr和溶剂的混合方式优选为:将亲水单体与溶剂混合,然后通氮气除氧,再加入2,2'-联吡啶和cubr,继续通氮气,得到混合溶液;所述通氮气除氧的时间优选为20~25min,所述继续通氮气的时间优选为10~15min。本发明优选采用上述混合方式,有利于充分去除混合溶液中的氧气,避免氧气对atrp聚合反应产生影响。
[0047]
得到混合溶液后,本发明将所述纤维膜浸没在混合溶液中进行atrp聚合反应,得到高效油水分离纳米纤维膜。
[0048]
在本发明中,所述atrp聚合反应的温度优选为15~35℃,更优选为15~30℃,时间优选为1~6h,更优选为2~5h。本发明在atrp聚合反应过程中,亲水单体在溴化亚铜与配体2,2'-联吡啶的催化作用下进行聚合。
[0049]
本发明优选在atrp聚合反应完成后,将反应后的纤维膜取出后进行水洗,得到高效油水分离纳米纤维膜。
[0050]
本发明通过将引发剂mebr以共价键方式键入到高分子聚合物中,然后通过静电纺丝的方法制备纤维膜,再将亲水单体与纤维膜表面及内部进行atrp聚合反应,实现亲水单体对纤维膜的亲水改性,使纤维膜的比表面积增大,水化作用增强,大大提高了纤维膜的膜通量、油水分离效率和稳定性。
[0051]
本发明还提供了上述技术方案所述制备方法制备得到的高效油水分离纳米纤维膜。本发明提供的高效油水分离纳米纤维膜具有大的比表面积,比表面积可达420m2/g,大的比表面积有利于改性纳米纤维膜的亲水性,一滴5μl的水滴在膜上仅仅需要6s就完全被吸收。
[0052]
本发明还提供了上述技术方案所述高效油水分离纳米纤维膜作为油水分离膜的应用。本发明优选将所述高效油水分离纳米纤维膜用于分离十六烷和水的油水混合物。
[0053]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。
[0054]
实施例1
[0055]
首先制备含不饱和双键的引发剂2-((2-溴-2-甲基丙酰基)氧基)乙基甲基丙烯酸酯(mebr):在氮气保护下,等摩尔量的2-溴代异丁酰溴与2-羟乙基甲基丙烯酸酯和三乙胺在二氯甲烷中在冰水浴条件充分反应后,多次水洗纯化除去溶剂后得到引发剂mebr;
[0056]
然后制备键入引发剂的聚丙烯腈纺丝原液:在带有回流冷凝管的三口瓶中加入22.5g丙烯腈、0.5g甲基丙烯酸甲酯和溶剂dmso,在通氮气的状态下加热到60℃后将引发剂mebr与偶氮二异丁腈的混合液(2g mebr,0.18g偶氮二异丁腈)滴加到体系中,继续反应10h得到纺丝原液;
[0057]
然后在电压20kv、注射速率0.7ml/h、接收距离20cm下通过静电纺丝法在铝箔上接收到了平均直径为520nm的纳米纤维膜;
[0058]
最后通过atrp反应进行聚合物刷的改性:将4g 2[-(甲基丙烯酰基氧基)乙基]二甲基(3-磺酸丙基)氢氧化铵单体溶解于8ml水/甲醇的混合溶剂(水和甲醇的体积比3:2)中,通氮气除氧20min,加入120mg 2,2'-联吡啶和40mg cubr,继续通氮气10min后加入纳米纤维膜进行atrp聚合反应,反应温度为20℃,反应4.5小时取出样品后用去离子水冲洗后得到高效油水分离纳米纤维膜。
[0059]
对实施例1中间过程得到的纳米纤维膜进行扫描电镜测试,结果如图1所示。由图1
可知本发明通过静电纺丝的方法成功制备得到了纳米纤维膜,图1中a和b分别为不同放大倍数下的纳米纤维膜sem图。
[0060]
对实施例1由atrp聚合改性得到的高效油水分离纳米纤维膜吸水前后进行扫描电镜测试,结果如图2所示,图2中a为吸水前高效油水分离纳米纤维膜的sem图,图2中a1为吸水后高效油水分离纳米纤维膜的sem图,图2中a2为吸水后1μm下高效油水分离纳米纤维膜的sem图,图2中a3为吸水后100nm下高效油水分离纳米纤维膜的sem图。由图2可知亚表面改性后的纳米纤维膜有明显的凸起结构,比表面积增大。
[0061]
测试了所制备的高效油水分离纳米纤维膜在分离十六烷/水(体积比1:1)的油水混合物时的通量变化和分离效率,结果表明,制备的高效油水分离纳米纤维膜在1bar下的膜通量可达3620l m-2
h-1
,通过用卡尔
·
菲舍尔滴定仪(metrohm 813kf,switzerland)检测收集到的油的纯度,由分离后油中的水含量可知,本发明提供的高效油水分离纳米纤维膜的分离效率大于99.90%;在对油水乳液(十二烷基硫酸钠稳定的十六烷/水乳液,其中十二烷基硫酸钠、十六烷和水的用量比为(10mg:1ml:100ml)进行分离时,仅重力下的膜通量大于60lm-2
h-1
,且滤液中cod含量为75mg/l。
[0062]
实施例2
[0063]
首先按照实施例1的方法制备引发剂mebr;
[0064]
然后制备键入引发剂的聚丙烯腈纺丝原液:在带有回流冷凝管的三口瓶中加入22.5g丙烯腈、0.5g甲基丙烯酸甲酯和溶剂dmso,在通氮气的状态下加热到60℃后将引发剂mebr和偶氮二异丁腈的混合液(2g mebr,0.18g偶氮二异丁腈)滴加到体系中,继续反应10h得到纺丝原液;
[0065]
然后在电压15kv、注射速率0.6ml/h、接收距离25cm下通过静电纺丝法在铝箔上接收到了平均直径为480nm的纳米纤维膜,真空干燥备用;
[0066]
最后通过atrp反应进行亲水聚合物刷表面改性:将3.5g异丙基丙烯酰胺单体溶解于8ml水/甲醇(水和甲醇的体积比3:2)的混合溶剂中,通氮气除氧20min,加入100mg 2,2'-联吡啶和35mg cubr,继续通氮气10min后加入纳米纤维膜进行atrp聚合反应,反应温度为25℃,反应3小时取出样品后用去离子水冲洗后得到高效油水分离纳米纤维膜。
[0067]
测试了所制备的高效油水分离纳米纤维膜在分离十六烷/水(体积比1:1)的油水混合物时的通量变化和分离效率,结果表明,制备的高效油水分离纳米纤维膜的膜通量可达3500l m-2
h-1
,通过用卡尔
·
菲舍尔滴定仪(metrohm813kf,switzerland)检测收集到的油的纯度,由分离后油中的水含量可知,本发明提供的高效油水分离纳米纤维膜的分离效率大于98.50%;在对十二烷基硫酸钠稳定的十六烷/水(体积比1:100)乳液进行油水乳液进行分离时,仅重力下的膜通量大于58lm-2
h-1
,且滤液中cod含量为72mg/l。
[0068]
实施例3
[0069]
首先按照实施例1的方法制备引发剂mebr;
[0070]
然后制备键入引发剂的聚丙烯腈纺丝原液:在带有回流冷凝管的三口瓶中加入22.5g丙烯腈、0.5g甲基丙烯酸甲酯和溶剂dmso,在通氮气的状态下加热到60℃后将引发剂mebr和偶氮二异丁腈的混合液(2g mebr,0.18g偶氮二异丁腈)滴加到体系中,继续反应10h得到纺丝原液;
[0071]
然后在电压10kv、注射速率1.0ml/h、接收距离30cm下通过静电纺丝法在铝箔上接
收到了平均直径为680nm的纳米纤维膜,真空干燥备用;
[0072]
最后通过atrp反应进行pnipam聚合物刷表面改性:在n2气氛下将6g 3-磺酸丙基甲基丙烯酸钾单体溶解在水和异丙醇的混合溶液中(水和异丙醇的体积比为3:1)除氧20min后,加入160mg 2,2'-联吡啶和50mg cubr,继续通气10min后,继续通氮气10min后对纳米纤维膜进行atrp聚合反应,反应温度为25℃,反应2h后取出样品后用去离子水冲洗后得到高效油水分离纳米纤维膜。
[0073]
测试了所制备的高效油水分离纳米纤维膜在分离十六烷/水(体积比1:1)的油水混合物时的通量变化和分离效率,结果表明,制备的高效油水分离纳米纤维膜的膜通量在温度20℃
±
2时可达1850lm-2
h-1
,而温度为45℃
±
3达2867l m-2
h-1
,通过用卡尔
·
菲舍尔滴定仪(metrohm 813kf,switzerland)检测收集到的油的纯度,由分离后油中的水含量可知,本发明提供的高效油水分离纳米纤维膜在温度20℃
±
2℃下的分离效率为98.72%,而在45℃
±
3℃时为9.13%;在对十二烷基硫酸钠稳定的十六烷/水(体积比1:100)乳液进行油水乳液进行分离时,仅重力下的膜通量大于65l m-2
h-1
,且滤液中cod含量为74mg/l。
[0074]
实施例4
[0075]
首先按照实施例1的方法制备引发剂mebr;
[0076]
然后制备键入引发剂的聚丙烯腈纺丝原液:在带有回流冷凝管的三口瓶中加入22.5g丙烯腈、0.5g甲基丙烯酸甲酯和溶剂dmso,在通氮气的状态下加热到60℃后将引发剂mebr和偶氮二异丁腈的混合液(2g mebr,0.18g偶氮二异丁腈)滴加到体系中,继续反应10h得到纺丝原液;
[0077]
然后在电压20kv、注射速率0.7ml/h、接收距离20cm下通过静电纺丝法在铝箔上接收到了平均直径为310nm的纳米纤维膜,真空干燥备用;
[0078]
最后通过atrp反应进行聚合物刷的改性:将4g甲基丙烯酰氧乙基三甲基氯化铵单体溶解于8ml水/甲醇(水和甲醇的体积比3:2)的混合溶剂中,通氮气除氧20min,加入120mg 2,2'-联吡啶和40mg cubr,继续通氮气10min后加入纳米纤维膜进行atrp聚合反应,反应温度为20℃,反应一段时间后取出样品后用去离子水冲洗后得到高效油水分离纳米纤维膜。
[0079]
测试了所制备的高效油水分离纳米纤维膜在分离十六烷/水(体积比1:1)的油水混合物时的通量变化和分离效率,结果表明,制备的高效油水分离纳米纤维膜的膜通量可达3750l m-2
h-1
,通过用卡尔
·
菲舍尔滴定仪(metrohm813kf,switzerland)检测收集到的油的纯度,由分离后油中的水含量可知,本发明提供的高效油水分离纳米纤维膜的分离效率大于99.90%;在对十二烷基硫酸钠稳定的十六烷/水(体积比1:100)乳液进行油水乳液进行分离时,仅重力下的膜通量大于62lm-2
h-1
,且滤液中cod含量为75mg/l。
[0080]
对比例1
[0081]
采用氯硅烷引发剂,结构如下式所示:
[0082][0083]
然后制备键入引发剂的聚丙烯腈纺丝原液:在带有回流冷凝管的三口瓶中加入
22.5g丙烯腈、0.5g甲基丙烯酸甲酯和溶剂dmso,在通氮气的状态下加热到60℃后将0.18g偶氮二异丁腈滴加到体系中,继续反应10h得到纺丝原液;
[0084]
在电压20kv、注射速率1ml/h、接收距离20cm的条件下得到纤维膜;
[0085]
在膜表面通过气相沉积法使氯硅烷引发剂分子锚固在纤维膜上,氯硅烷引发剂在纤维膜中的质量分数为7%,得到表面改性的油水分离纳米纤维膜。
[0086]
对实施例1和对比例1油水分离纳米纤维膜在空气中与水滴的接触情况进行测试,结果如图3所示,图3中a为对比例1的测试结果,图3中b为实施例1的测试结果。由图3可知,实施例1的纳米纤维膜的水渗透速率更高。
[0087]
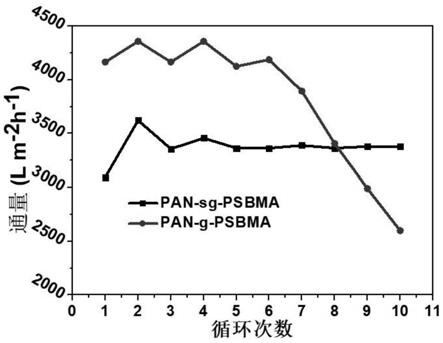
测试对比例1得到的油水分离纳米纤维膜分离水和石油醚油水混合物(水和石油醚的体积比为1:10)时,循环多次后的分离效果,结果如图4所示。图4中“pan-g-psbma”为对比例1的循环测试结果,“pan-sg-psbma”为实施例1的循环测试结果。由图4可知,对比例1提供的油水分离纳米纤维膜循环10次后,膜通量下降。然而实施例1提供的纳米纤维膜循环10次后,膜通量保持稳定。
[0088]
综上,本发明提供了一种高效油水分离纳米膜,本发明提供的高效油水分离纳米膜的膜通量高、油水分离效率高,且循环多次后分离效率仍然较高,稳定性较好。
[0089]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。