基于图的映射核酸片段的系统和方法
1.相关申请的交叉引用
2.本技术要求于2019年1月25日提交的美国临时专利申请62/797,022的优先权,出于所有目的,将其公开内容通过引用整体并入本文。
技术领域
3.本说明书描述了使用基于图的映射将核酸片段组合成相干序列(coherent sequence)。
背景技术:
4.生物分子测序的进步,特别是在核酸和蛋白质样品方面,已经彻底改变了细胞和分子生物学领域。由于自动化测序系统的发展,现在可以对全基因组进行测序。然而,使用这些新测序技术进行测序的基因组的数量和复杂性也相应增加。尽管现在可以获得大量的高保真核酸序列,但将这些序列组装和组织成完整的基因组仍然存在许多问题。
5.从序列读段组装这些复杂的基因组,特别是二倍体和更高倍性的基因组是一个必然植根于计算技术中的难题。这是由于多种因素造成的,包括基因组的庞大规模、传统测序技术的读段大小限制以及多倍体基因组同源区域的序列变异。例如,虽然相对
‘
简单’的大肠杆菌单倍体基因组在单个环状基因组中仅包含460万个碱基对,但人类基因组包含二十三条染色体的两个副本,每个副本包含超过32亿个碱基对。
6.假设平均测序读段长度为250个碱基对,并且序列读段之间的平均重叠仅为15%,那么人类基因组的1倍覆盖将需要近3000万个序列读段。此外,人类基因组包括每个基因座的两个拷贝,其序列变化约0.1%,这意味着250个碱基对序列读段中超过22%将与用于协助组装过程的任何特定参考基因组不同。此外,代表大约650万个序列读段的不同的22%的序列读段需要相对于彼此进行定相,以便为二倍体基因组组装单倍群。这只是例如蕨类植物的高阶多倍体基因组中更加复杂(平均2n=121.0;2n范围=18
–
1440;clark j等人,new phytologist,210:1072
–
82(2016))。因此,很明显,不使用计算技术实际上无法进行基因组组装。参见kelly mj.computers:the best friends a human genome ever had,genome,31(2):1027
‑
33(1989)。
7.然而,仅通用计算机的计算能力并不是高效基因组组装所需的全部,人类基因组计划所需的大量努力证明了这一点,即使在计算技术的帮助下(与全球合作需要13年才能完成);chial,h.,dna sequencing technologies key to the human genome project,nature education,1(1):219(2008))。相反,需要复杂的比对算法来处理基因组组装所需的非常大量测序数据。相对于第一个组装的人类基因组,更贪心的比对算法的发展大大加快了基因组组装过程。
8.映射测序读段体现在从blast到blasr、bwa
‑
mem、damapper、ngmlr、graphmap、minimap等工具中。这些方法中的每一种都有其自身特定的要求和用途。然而,它们都有一个共同的设计,用于映射到线性目标序列,例如参考基因组。在另一方面,映射器如bgreat
(如在limasset等人2016,bmc bioinformatics 17:237中所述)和debga(例如,如由liu等人2016,bioinformatics 32(21):3224
‑
3232所述)旨在处理第二代测序数据和de bruijn图,而不是线性目标序列。blastgraph使用blast映射结果来聚类比对并执行比较基因组分析(如ye等人,2013,bioinformatics 29(24):3222
‑
3224中所述)。然而,根据limasset等人进行的实验,blastgraph即使在简单的非线性数据集上也表现不佳,例如大肠杆菌基因组(bmc bioinformatics 17:237,2016)。gramtools将短读段映射到人口参考图(例如,如maciuca等人,2016年,在互联网上dx.doi.org/10.1101/059170)。一般来说,所有这些方法都专门设计用于短读段序列。
9.虽然对于组装相对简单的线性基因组区域有用,但短读段序列有许多缺点。例如,短读段序列在具有高gc含量的基因组区域、重复区域和表现出结构变异(例如重复)的区域中提供的信息有限(例如,在pollard等人,2018,hum molecul genetics 27(r2):r234
–
r241中)。随着更复杂的基因组被测序和组装,能够准确有效地使用长读段序列的映射器将变得越来越必要。目前能够将长噪声读段映射到任意序列图的一种方法是变异图(例如,“vg”)工具包,它提供了构建、查看和操作变异图的工具,包括诸如图上全长poa对齐等特征、映射读段和来自读段比对的调用变体(例如,如garrison等人,2018,nat.biotech.36,875
‑
879中所述)。它最初是为短读段而设计和优化的,后来通过将它们分成短的重叠块(基本上模拟短读段)来修改以支持更长的序列。然而,为了实现基于图的功能,vg使用基于bwt的有向图索引方法(gcsa2),该方法仅对任何图中长度最大为256的路径进行索引(例如,如sir
é
n等人在proc.alenex 2017,siam,第13
‑
27页,西班牙巴塞罗那,2017年1月17
‑
18日.doi:10.1137/1.9781611974768.2中所述)。路径长度的上限旨在作为一种启发式方法,以防止在具有大量分支的区域中出现“组合爆炸”。vg还展开和打开循环有向图以制作可以应用偏序对齐的有向无循环图(“dag化”的过程)(例如,在garrison等人,2018,nat.biotech.36,875
‑
879中)。在图中展开循环也是一种昂贵的组合操作,它可大大增加图的大小(例如,如kavya等人,2019,j comp biol 26:1
‑
15和naga等人,2019,j comp biol 26(1)中所述)。此外,尽管gcsa2索引方法提供独立于图大小的线性时间精确匹配查询以找到超最大精确匹配种子,但基于bwt和fm索引的算法通常可能在查找过程中携带大的隐藏常数因子以及重要的预构建索引所需的时间,在考虑到大序列和图的最大映射率时需要考虑到这一点。因此,虽然vg的范式非常适合许多应用,但还有许多其他类型的基因组需要不同的方法。
10.基于图映射的用例的一个例子是在环状基因组的背景下。当面对环状基因组的序列时,大多数映射器或者证明在参考环状序列的端部附近的覆盖的显著下降(例如blasr,如chaisson和tesler bmc bioinformatics 13:238,2012所述),或者通过输出不直接依赖于主比对的辅助比对维持高测序覆盖(它们也可以放置在重复区)(例如minimap2,如li,bioinformatics 15;34(18):3094
‑
3100,2018所述)。克服这一缺陷的一种方法是使用基于图的映射方法。将参考限定为图可为广泛的重要应用提供看似透明的支持。事实上,minimap2和graphmap确实寻求解决环形基因组映射问题(例如,参见id.和sovi等人,2016,nat comm7:11307)。然而,特别是graphmap的一个重要限制是该方法预期参考集中的所有序列都是环形或线性的。它无法处理线性和环形参考基因组的混合。
11.除了环状基因组之外,还有许多其他测序情况可以从改进的映射方法中受益。例
如,许多映射器难以识别基因组拓扑,例如倒置。大多数映射方法也无法从短读段序列中识别单倍型。此外,对映射到多基因组图(例如,用于变异检测和/或诊断目的的由多个人类基因组组成的图,用于物种识别的细菌数据库,或用于进一步组装改进和物种分离的宏基因组组装)越来越感兴趣(例如在vollmers等人,2017,plos one 12(1):e0169662中讨论的)。
12.鉴于对复杂基因组的基因组测序和组装的需求不断增长,本领域需要改进的基于图的长读段映射方法。
技术实现要素:
13.本公开通过提供基于长序列读段确定核酸序列的改进方法来解决背景中提出的基于图的映射方法的缺点。该方法开始于获得具有杂合基因组的物种的单个二倍体或多倍体生物的有向图,其中该有向图代表杂合基因组的全部或部分并且包括一个或多个非线性拓扑组件。在一些实施方案中,一个或多个非线性拓扑组件中的每个相应的非线性拓扑组件表示相对于一组目标序列的变化。
14.在一些实施方案中,所述一个或多个非线性拓扑组件中的每个相应的非线性拓扑组件具有对应的起始节点和对应的终止节点,所述起始节点和终止节点至少由对应的第一分支和对应的第二分支连接,其中对应的第一分支和对应的第二分支中的一个对应于目标序列。在一些实施方案中,通过重叠多个序列读段中的相应序列读段,使用来自单个生物的生物样品的多个序列读段形成有向图,这些序列读段相对于目标序列具有大于百分之十的随机误差,其中重叠的第一序列读段和第二序列之间的突出的量是不受限制的,前提是第一和第二序列读段之间存在最小的共有区。
15.该方法通过从电子格式的生物样品中获取查询序列来进行。查询序列是(i)多个序列读段中的序列读段,或(ii)由多个序列读段形成的支架。此外,查询序列至少包含一个或多个非线性拓扑组件中的第一非线性拓扑组件的对应起始节点或对应终止节点。
16.该方法继续使用有向图来形成查询序列到有向图的映射。查询序列的映射包括(i)第一比对组件到有向图的第一部分,(ii)第二比对组件到有向图的第二部分和(iii)描述有向图中在有向图的第一部分和第二部分之间的关系的路径。在这种情况下,第一部分和第二部分中的一个在第一非线性拓扑组件的第一或第二分支中,而第一部分和第二部分中的另一个不在第一非线性拓扑组件的分支中。
17.其他实施方案涉及与这里描述的方法相关联的系统、便携式消费者装置和计算机可读介质。
18.在适用时,本文公开的任何实施方案可以应用于任何其他方面。
19.根据以下详细描述,本公开的其他方面和优点对于本领域技术人员将变得显而易见,其中仅示出和描述了本公开的说明性实施方案。应当理解,本公开可以有其他的和不同的实施方案,并且其几个细节可以在各种明显的方面进行修改,所有这些都不偏离本公开。因此,附图和描述在本质上被认为是说明性的,而不是限制性的。
20.通过引用并入
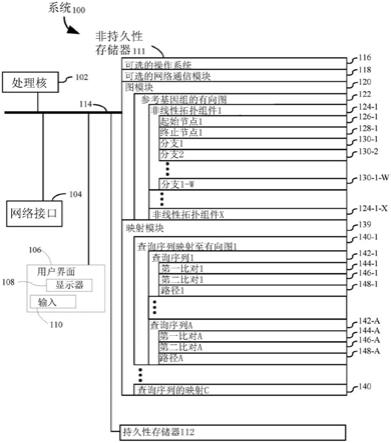
21.本文中所引用的所有出版物、专利和专利申请以其全文通过引用并入。如果本文中的术语与并入的参考文献中的术语发生冲突,则以本文中的术语为准。
附图说明
22.在附图的图中,通过实例而非限制的方式示出了本文公开的实施方式。贯穿附图的若干视图,相似的附图标记指代对应的部分。
23.图1是图示根据本公开的一些实施方案的计算装置的示例框图。
24.图2a和2b图示了根据本公开的一些实施方案的使用有向图来映射查询序列的方法的示例流程图,其中在虚线框中描绘了可选步骤。
25.图3图示了根据本公开的一些实施方案的跨组装的基因组比对定相单倍群的实例。
26.图4a、4b和4c共同示出了根据本公开的一些实施方案的示例图。
27.图5图示了根据本公开的一些实施方案的映射到环状基因组的实例。
28.图6a和6b共同示出了根据本公开的一些实施方案的具有基因组倒位的映射的实例。
29.图7a和7b共同示出了根据本公开的一些实施方案,使用已知插入序列(例如质粒、线粒体dna或靶向基因组区域)进行映射的实例。
30.图8a和8b共同示出了根据本公开的一些实施方案的经由限定剪接位点的边缘的转录组映射的实例。
具体实施方式
31.现将详细参照实施方案,在附图中展示其实例。在以下详细描述中,阐述了许多具体细节以便彻底理解本公开。然而,对本领域普通技术人员而言将显而易见的是,本公开可以在没有这些具体细节的情况下实施。在其它例子中,并未详细描述熟知的方法、程序、组件、电路以及网络,以免不必要地模糊实施例的各个方面。
32.本文描述的实施方式提供了使用长读段序列读段的各种技术方案。对于具有杂合基因组的单个二倍体或多倍体生物,获得代表基因组全部或部分的有向图。还获得了来自相应生物物种的生物样品的查询序列(例如,感兴趣的序列)。然后使用有向图形成查询序列到有向图的映射。因此,该方法能够检测结构变体以及单核苷酸变体。本文提供的方法需要使用长序列读段。
33.定义
34.如本文所用,术语“约”或“大约”可表示在由本领域普通技术人员确定的特定值的可接受误差范围内,这可部分取决于如何测量或确定该值(例如,测量系统的局限性)。例如,根据本领域的实践,“约”可以表示1个标准偏差以内或超过1个标准偏差。“约”可以表示给定值的
±
20%、
±
10%、
±
5%或
±
1%的范围。术语“约”或“大约”可表示值的一个数量级内、5倍内或2倍内。在申请和权利要求中描述特定值的地方,除非另有说明,否则应假定术语“约”的含义在特定值的可接受误差范围内。术语“约”可以具有本领域普通技术人员通常理解的含义。术语“约”可以指
±
10%。术语“约”可以指
±
5%。
35.如本文所公开,术语“受试者”是指任何有生命的或无生命的生物,包括但不限于人(例如,男人、女人、胎儿、怀孕女性、儿童等)、非人类动物、植物、细菌、真菌或原生生物。任何人类或非人类动物可选自,包括但不限于哺乳动物、爬行动物、鸟类、两栖动物、鱼类、有蹄动物、反刍动物、牛科动物(例如家牛(cattle))、马科动物(例如马)、山羊类和绵羊类
(例如绵羊、山羊)、猪类(例如猪)、骆驼科动物(例如骆驼、美洲驼、羊驼)、猴类、猿类(例如大猩猩、黑猩猩)、熊科动物(例如熊)、家禽类、狗类、猫类、鼠类、鱼类、海豚类、鲸类和鲨鱼类。受试者可以是任何阶段的雄性或雌性(例如,男性、女性或儿童)。
36.如本文所用,术语“生物样品”、“患者样品”或“样品”是指取自受试者的任何样品,其可反映与受试者相关的生物状态。生物样品的实例包括但不限于受试者的血液、全血、血浆、血清、尿液、脑脊液、粪便、唾液、汗液、眼泪、胸水、心包液或腹膜液。生物样品可以包括源自活的或死的受试者的任何组织或材料。生物样品可以是无细胞样品。生物样品可以包括核酸(例如,dna或rna)或其片段。样品可以是液体样品或固体样品(例如,细胞或组织样品)。生物样品可以是体液,例如血液、血浆、血清、尿液、阴道液、来自鞘膜积液的流体(例如睾丸的)、阴道冲洗液、胸水、腹水、脑脊液、唾液、汗液、泪、痰、支气管肺泡灌洗液、从乳头排出的流体、从身体的不同部分(例如,甲状腺、乳腺)抽吸的流体等等。生物样品可以是粪便样品。生物样品可被处理以物理地破坏组织或细胞结构(例如,离心和/或细胞裂解),从而释放细胞内成分为可进一步含有酶、缓冲液、盐、洗涤剂溶液等的溶液,其可用于制备用于分析的样品。
37.如本文所用,术语“核酸”和“核酸分子”可互换使用。该术语指任何组成形式的核酸,例如脱氧核糖核酸(dna,例如互补dna(cdna)、基因组dna(gdna)等)、核糖核酸(rna,例如信息rna(mrna)、短抑制性rna(sirna)、核糖体rna(rrna)、转移rna(trna)、微rna、由胎儿或胎盘高度表达的rna等)和/或dna或rna类似物(例如,其含有碱基类似物、糖类似物和/或非天然主链等)、rna/dna杂交体和聚酰胺核酸(pna),所有这些都可以是单链或双链形式。除非另有限制,核酸可以包含已知的天然核苷酸类似物,其中一些可以以与天然存在的核苷酸类似的方式起作用。核酸可以是用于进行本文的过程的任何形式(例如,线性、环状、超螺旋、单链、双链等)。核酸可以是或可以来自质粒、噬菌体、自主复制序列(ars)、着丝粒、人工染色体、染色体或能够在体外或宿主细胞、细胞、在某些实施方案中细胞核或细胞质中复制或被复制的其他核酸。在一些实施方案中,核酸可以来自单个染色体或其片段(例如,核酸样品可以来自从二倍体生物获得的样品的一个染色体)。在某些实施方案中,核酸包括核小体、核小体的片段或部分或核小体样结构。核酸有时包含蛋白质(例如,组蛋白、dna结合蛋白等)。通过本文所述方法分析的核酸有时基本上是分离的并且基本上不与蛋白质或其他分子相关联。核酸还包括从单链合成、复制或扩增的rna或dna的衍生物、变体和类似物(“有义”或“反义”、“正”链或“负”链、“正向”阅读框或“反向”阅读框)和双链多核苷酸。脱氧核糖核苷酸包括脱氧腺苷、脱氧胞苷、脱氧鸟苷和脱氧胸苷。对于rna,碱基胞嘧啶被尿嘧啶取代,糖2'位置包括羟基部分。可以使用从受试者获得的核酸作为模板来制备核酸。
38.如本文所用,术语“终止位置”或“结束位置”(或仅“末端”)是指最外碱基的基因组坐标或基因组身份或核苷酸身份,例如在无细胞dna分子例如血浆dna分子的末端。结束位置可以对应于dna分子的任一末端。以这种方式,如果指的是dna分子的起点和末端,两者都可以对应终止位置。在某些情况下,一个结束位置是无细胞dna分子的一个末端上最外碱基的基因组坐标或核苷酸身份,其通过分析方法检测或确定,例如,大规模平行测序或新一代测序、单分子测序、双链或单链dna测序文库制备方案、聚合酶链反应(pcr)或微阵列。在某些情况下,这种体外技术可以改变无细胞dna分子真正的体内物理末端。因此,每个可检测的末端可以代表生物学真实末端或末端是一个或多个核苷酸向内或从分子的原始末端延
伸的一个或多个核苷酸,例如klenow片段对非平端双链dna分子的突出端的5'平端和3'填充。结束位置的基因组身份或基因组坐标可以从序列读段与人类参考基因组例如hg19的比对结果得出。它可以来自代表人类基因组原始坐标的索引或代码的目录。它可以指无细胞dna分子上的位置或核苷酸身份,通过但不限于目标特异性探针、微型测序、dna扩增读段。术语“基因组位置”可以指多核苷酸(例如,基因、质粒、核酸片段、病毒dna片段)中的核苷酸位置。术语“基因组位置”不限于在基因组内的核苷酸位置(例如,在配子或微生物的单倍体染色体组中,或在多细胞生物中的每个细胞中)。
39.如本文所用,术语“片段”(例如,dna片段)指的是多核苷酸或多肽序列的包含至少三个连续核苷酸的部分。核酸片段可以保留亲本多核苷酸的生物学活性和/或一些特征。在一个例子中,鼻咽癌细胞可以将爱泼斯坦
‑
巴尔病毒(ebv)dna的片段沉积到受试者例如患者的血流中。这些片段可包含一个或多个bamhi
‑
w序列片段,可用于检测血浆中肿瘤来源的dna的水平。bamhi
‑
w序列片段对应于可以使用bam
‑
hi限制酶识别和/或消化的序列。bamhi
‑
w序列可以指序列5'
‑
ggatcc
‑
3'。
40.如本文所用,“局部最大值”可指当与邻近位置相比时获得感兴趣参数的最大值的基因组位置(例如,核苷酸)或指在这样的基因组位置的感兴趣参数的值。例如,相邻位置的范围可以从50bp到2000bp。感兴趣的参数的例子包括但不限于在基因组位置终止的片段的数量、与该位置重叠的片段的数量或大于阈值大小的覆盖基因组位置的片段的比例。当感兴趣的参数具有周期性结构时,可能出现许多局部最大值。全局最大值是局部最大值的特定一个。类似地,“局部最小值”是指在与邻近位置相比时获得感兴趣参数的最小值的基因组位置,或者是指在这样的基因组位置的感兴趣参数的值。
41.如本文所用,在一个位置终止的核酸分子(例如,dna或rna)的术语“比率”可涉及核酸分子在该位置上终止的频率。该比率可以基于相对于所分析的核酸分子的数量归一化的在该位置上终止的核酸分子的数量。该比率可以基于相对于在不同的位置终止的核酸分子的数量归一化的在该位置上终止的核酸分子的数量。该比率可以基于相对于来自第二样品(例如参考样品)的在该位置终止的核酸分子的数量归一化的在该位置上终止的来自第一样品的核酸分子的数量。该比率可以基于相对于来自第二样品(例如参考样品)的在第二组位置终止的核酸分子的数量归一化的在第一组位置(例如基因组位置)上终止的来自第一样品的核酸分子的数量。因此,该比率可以对应于有多少核酸分子在某个位置终止的频率,并且在一些情况下与在该位置终止的核酸分子的数量中具有局部最大值的位置的周期性无关。
42.如本文所使用的,术语“相对丰度”可以指具有特定特性的核酸片段的第一量(例如终止于一个或多个指定的坐标/终止位置或者与基因组中的特定区域对齐的指定长度)与具有特定特性的核酸片段的第二量(例如终止于一个或多个指定的坐标/终止位置或者与基因组中的特定区域对齐的指定长度)的比率。在一个实例中,相对丰度可以指终止于第一组基因组位置的dna片段的数量与终止于第二组基因组位置的dna片段的数量的比率。在一些方面,“相对丰度”可以是一种分离值,其将在基因组位置的一个窗口内终止的dna分子的量(一个值)与在基因组位置的另一个窗口内终止的dna分子的量(其他值)相关联。两个窗口可以重叠,但大小可以不同。在其他实现方式中,两个窗口不能重叠。此外,窗口可以是一个核苷酸的宽度,因此相当于一个基因组位置。
43.如本文所用,术语“参考基因组”是指任何生物体或病毒的任何特定已知的、测序的或表征的基因组,无论是部分的还是完整的,其可用于参考来自受试者的已识别序列。用于人类受试者以及许多其他生物体的示例性参考基因组在由国家生物技术信息中心(“ncbi”)或加州大学圣克鲁兹分校(ucsc)主办的在线基因组浏览器中提供。“基因组”是指以核酸序列表达的生物体或病毒的完整遗传信息。如本文所用,参考序列或参考基因组通常是来自个体或多个个体的组装或部分组装的基因组序列。在一些实施方案中,参考基因组是来自一个或多个人类个体的组装或部分组装的基因组序列。参考基因组可以被视为一个物种基因集的代表性例子。在一些实施方案中,参考基因组包含分配给染色体的序列。示例性人类参考基因组包括但不限于ncbi build 34(ucsc等效物:hg16)、ncbi build 35(ucsc等效物:hg17)、ncbi build 36.1(ucsc等效物:hg18)、grch37(ucsc等效物:hg19)和grch38(ucsc等效物:hg38)。
44.如本文所用,术语“序列读段”或“读段”是指通过本文所述的或本领域已知的任何测序过程产生的核苷酸序列。可以从核酸片段的一端生成读段(“单端读段”),有时从核酸片段的两端生成读段(例如,双端读段、双端读段或配对读段)。序列读段的长度通常与特定的测序技术相关。在一些实施方案中,序列读段的均值、中值或平均长度为约5,000bp至50,000bp长(例如,约5,000bp、约7,500bp、约10,000bp、约12,500bp、约15,000bp、约20,000bp、约25,000bp、约30,000bp、约35,000bp、约40,000bp、约45,000bp、约50,000bp、约55,000、约60,000、约65,000、约70,000、约75,000或约80,000)。在一些实施方案中,序列读段具有约1000bp、2000bp、5000bp、10,000bp、50,000bp或更多的均值、中值或平均长度。序列读段(或测序读段)可以指对应于核酸分子的序列信息(例如,一串核苷酸)。例如,序列读段可以对应于核酸片段的一部分的一串核苷酸(例如,最多约50,000bp),可以对应于核酸片段的一端或两端的一串核苷酸,或可以对应于整个核酸片段的核苷酸。可以通过多种方式获得序列读段,例如,使用测序技术或使用探针,例如,在杂交阵列或捕获探针中,或扩增技术,例如聚合酶链反应(pcr)或使用单一引物或等温扩增的线性扩增。
45.如本文所用,本文所用的术语“测序”、“序列确定”等一般指可用于确定诸如核酸或蛋白质的生物大分子的顺序的任何和所有生化方法。例如,测序数据可以包括核酸分子例如dna片段中的全部或部分核苷酸碱基。
46.如本文所用,术语“单核苷酸变体”、“单核苷酸多态性”或“snp”是指在核苷酸序列例如个体的序列读段的位置(例如位点)处一个核苷酸替换为不同的核苷酸。从第一个核碱基x到第二个核碱基y的取代可以表示为“x>y”。例如,胞嘧啶到胸腺嘧啶snp可以表示为“c>t”。术语“het
‑
snp”是指杂合snp,其中基因组至少是二倍体并且两个或多个同源序列中的至少一个——但不是全部——表现出特定的snp。类似地,“hom
‑
snp”是同源snp,其中多倍体基因组的每个同源序列与参考基因组相比具有相同的变体。如本文所用,术语“结构变体”或“sv”是指基因组的大(例如,大于1kb)区域已经经历物理转化,例如倒位、插入、缺失或重复(例如,参见spielmann等人,2018,nat rev genetics 19:453
–
467的人类基因组sv综述)。
47.如本文所用,术语“折叠读段”、“折叠序列读段”或“折叠区域”是指源自多核苷酸分子的相似区域并已组合成一个所得序列的多个序列读段。例如,作为序列确定的一部分,来自多核苷酸的多个读段可以相互比较(例如,以提供原始多核苷酸的共有序列)。此外,所
得折叠读段可包括关于核苷酸错配、插入或缺失的信息。在核苷酸测序中使用折叠读段提供了更高程度的测序结果确定性(例如,而不是依赖于正被测序的多核苷酸的每个区域的单个序列读段)。
48.如本文所用,术语“节点”是指序列读段,而术语“分支”是指两个单独的序列读段(例如,节点)之间的重叠。有向图包括多个节点和多个分支。折叠读段与有向图对齐,使得任何折叠读段可以由边和相应顶点(例如,节点)的子集按顺序表示。如compeau等人所述,分支
49.(例如,有向边)可以指示节点之间重叠的方向性,从而有助于确定多个节点的整体拓扑组织(nat biotechnol 29(11):987
‑
991,2011)。这是形成有向图的基础。de bruijn图通常源自具有预限定重叠的序列读段,这种方法非常适合短读段测序。这可以通过将读段分成指定的长度(例如,k聚体)或通过限定边的长度(id.)来实现。通常,对于读段被切割成k聚体的情况,重叠将是k
‑
1。本文使用的术语“k聚体”是指dna序列的设定长度。有向图的单向边代表目标区域中k个核苷酸碱基(例如“k聚体”)的序列,并且这些边通过顶点(或节点)连接。
50.如本文所用,术语“非线性拓扑组件”或“非线性组件”是指复杂的核酸序列。特别地,此类组件可以是包含一种或多种变体的序列读段,其中指示了在一个或多个位置的映射序列的不同选项。或者,非线性组件可包括环状核酸。在某些情况下,这些变体可以是结构变异(sv)、单核苷酸变异(snp)、小核苷酸变异,或被模拟为更大的替代事件的具有升高数量的het
‑
snp的区域。
51.如本文所用,术语“单倍群(haplotig)”是指单倍型重叠群。重叠群包含一组连续的重叠核酸区段,代表核酸(例如基因组)的共有区域。并且单倍型包括几个通常一起遗传的多态性(例如,在基因组的一组区域内彼此靠近的那些,例如染色体)。形式上,单倍群是“具有相同单倍型的克隆的重叠群”,如makoff和flomen,2007,genome bio 8(6):r114中所述。
52.如本文所用,术语“定相”是指鉴定二倍体或多倍体基因组中的等位基因。特别是,定相用于确定哪些等位基因一起属于单个染色体或基因组的特定区域。choi等人,2018,plos genetics 14(4):e1007308中描述了对人类基因组进行定相的方法,该文献通过引用整体并入本文。
53.本文所使用的术语仅出于描述特定情况的目的,并且所述术语不旨在是限制性的。如在本文中所使用,除非上下文另外明确指示,否则单数形式“一”和“所述”意图也包括复数形式。此外,如果在详细说明和/或权利要求中使用了术语“包括”、“包含”、“具有”、“具备”、“带有”或它们的变体,则此类术语旨在以类似于术语“包括”的方式是包含性的。
54.下面参考示例应用描述几个方面以进行说明。应当理解,阐述了许多具体细节、关系和方法以提供对这里描述的特征的全面理解。然而,相关领域的普通技术人员将容易地认识到,可以在没有一个或多个具体细节或通过其他方法的情况下实践这里描述的特征。在此描述的特征不受所说明的动作或事件的顺序的限制,因为一些动作可以以不同的顺序发生和/或与其他动作或事件同时发生。此外,并非所有说明的动作或事件都需要用于实施根据这里描述的特征的方法。
55.示例性系统实施方案
56.现在结合图1描述示例性系统的细节。图1是说明根据一些实施方式的系统100的框图。在一些实施方案中,装置100包含一个或多个处理单元(cpu)102(也称为处理器或处理核)、一个或多个网络接口104、用户接口106、非永久性存储器111、永久性存储器112和一个或多个用于互连这些组件的通信总线114。一个或多个通信总线114可选地包含互连并控制系统组件之间的通信的电路系统(有时称为芯片组)。非永久性存储器111通常包含高速随机存取存储器,如dram、sram、ddr ram、rom、eeprom、闪速存储器,而永久性存储器112通常包括cd
‑
rom、数字多功能盘(dvd)或其它光学存储装置、磁带盒、磁带、磁盘存储装置或其它磁性存储装置、磁盘存储装置、光盘存储装置、闪速存储器装置或其它非易失性固态存储装置。永久性存储器112可选地包含一个或多个布置的远离cpu 102的存储装置。永久性存储器112和非永久性存储器112内的一个或多个非易失性存储装置包括非暂时性计算机可读存储介质。在一些实施方案中,非永久性存储器111或者(替代性地)所述非暂时性计算机可读存储介质(有时与永久性存储器112结合)存储以下程序、模块和数据结构或者其子集:
57.·
任选的操作系统116,所述操作系统包含用于处理各种基本系统服务和用于执行硬件相关任务的程序;
58.·
任选的网络通信模块(或指令)118,所述网络通信模块(或指令)用于将系统100与其它装置连接,或与通信网络连接;
59.·
用于将长序列读段映射到有向图的图模块120,其包括具有杂合基因组的生物体的有向图122,该杂合基因组包括至少一个非线性拓扑组件124,每个非线性拓扑组件包括起始节点126和终止节点128,且每对节点至少由对应的第一分支130
‑
1和对应的第二分支130
‑
2连接;和
60.·
映射模块139,其包括查询序列140到有向图122的映射图,每个映射图包括一个或多个查询序列142,每个查询序列至少包括第一比对144、第二比对146以及第一和第二比对之间的拓扑关系的路径148。
61.在一些实施方案中,有向图122源自序列读段或重叠群的从头组装。在一些实施方案中,有向图122源自参考基因组(例如,一个或多个现有注释)。在一些实施方案中,用于形成有向图的序列读段源自生物体的生物样品,并且相对于目标序列集,序列读段总体具有随机误差。
62.在一些实施方案中,查询序列142包含多个序列读段中的序列读段,或由多个序列读段形成的支架(例如,重叠群、单元群(unitig)或单倍群)。在一些实施方案中,查询序列142至少描述了相应非线性拓扑组件124的起始节点或终止节点。
63.在各种实施方案中,一个或多个上述元件存储在一个或多个先前提及的存储装置中,并且对应于用于执行上述功能的指令组。上述模块、数据或程序(例如指令集)不需实施为单独分开的软件程序、过程、数据集或模块,并且因此这些模块和数据的各个子集可以在各种实施方案中被组合或以其它方式重新布置。在一些实施方案中,非永久性存储器111可选地存储上述模块和数据结构的子集。此外,在一些实施方案中,所述存储器存储以上未描述的另外的模块和数据结构。在一些实施方案中,上述元件中的一个或多个存储在可视化系统100的计算机系统之外的计算机系统中,所述计算机系统可由可视化系统100寻址,使得可视化系统100可在需要时检索所有或部分这样的数据。
64.尽管图1描绘了“系统100”,但是该图更多地旨在作为可能存在于计算机系统中的
各种特征的功能描述,而不是作为本文所述的实施方案的结构示意图。在实践中,并且如本领域普通技术人员所认识的,可以将单独示出的项目组合,并且可以将一些项目分离。此外,尽管图1描绘了非永久性存储器111中的某些数据和模块,但是这些数据和模块中的一些或全部可以存在于永久性存储器112中。
65.虽然已经参照图1公开了根据本公开的系统,但是现在参照图2a和2b详细描述根据本公开的方法。
66.在将长序列读段或重叠群映射到目标序列时使用图
67.方框202.提供了一种通过基于图的映射对核酸进行测序的方法。
68.在一些实施方案中,目标序列将是查询被映射到的序列。目标序列可以包括任何类型的序列,例如参考基因组,或组装的重叠群、单元群或支架。
69.方框204.使用计算机系统100,获得具有杂合基因组的物种的单个二倍体或多倍体生物的有向图,其中该有向图代表杂合基因组的全部或部分并且包括一个或多个非线性拓扑组件(例如气泡或环)。在一些实施方案中,一个或多个非线性拓扑组件中的每个相应的非线性拓扑组件代表相对于一组目标序列的变异(例如,结构变异、单核苷酸变异(snp)、小核苷酸变异或在het
‑
snp中重的且模拟为更大的替换事件的区域)。
70.在一些实施方案中,有向图代表杂合基因组的至少百分之十,并且有向图包括十个或更多个非线性拓扑组件。
71.在一些实施方案中,有向图代表杂合基因组的至少百分之十五,并且有向图包括二十五个或更多个非线性拓扑组件。
72.在一些实施方案中,有向图代表杂合基因组的至少百分之五、杂合基因组的至少百分之十、杂合基因组的至少百分之二十、杂合基因组的至少百分之三十、杂合基因组的至少百分之四十、杂合基因组的至少百分之五十、杂合基因组的至少百分之七十五、杂合基因组的至少百分之九十、或杂合基因组的至少百分之百。
73.在一些实施方案中,有向图包括多个节点,例如,每个节点可以表示序列读段。因此,有向图中的节点总数将与基因组或被映射的基因组部分的大小成正比。例如,在其中大比例的复杂二倍体和更高阶多倍体基因组被映射的一些实施方案中,有向图将具有至少10,000个节点。在一些实施方案中,有向图将具有至少100,000个节点。在一些实施方案中,有向图将具有至少500,000个节点。在其他实施方案中,有向图将具有至少1百万、两百万、三百万、四百万、五百万或更多节点。
74.在一些实施方案中,有向图包括两个或更多个非线性拓扑组件、五个或更多个非线性拓扑组件、十个或更多个非线性拓扑组件、二十五个或更多个非线性拓扑组件、五十个或更多个非线性拓扑组件、或者一百个或更多个非线性拓扑组件。
75.在一些实施方案中,例如在映射大比例的复杂二倍体或更高阶多倍体基因组的情况下,有向图将具有至少1,000个非线性拓扑组件。在一些实施方案中,有向图将具有至少10,000个非线性拓扑组件。在一些实施方案中,有向图将具有至少100,000个非线性拓扑组件。在其他实施方案中,有向图将具有至少500,000、1百万、两百万、三百万、四百万、五百万或更多非线性拓扑组件。在一些实施方案中,对于具有映射到每个可能等位基因的至少一个序列读段的受试者中的每个杂合等位基因,图中存在非线性元素。也就是说,观察到每个snp和每个indel中至少一个序列读段已映射到等位基因1,至少一个读段已映射到等位基
因2。
76.在一些实施方案中,有向图包括杂合基因组的上述百分比、节点的数量和非线性拓扑组件的数量的任意组合。
77.在一些实施方案中,所述一个或多个非线性拓扑组件中的每个相应的非线性拓扑组件具有对应的起始节点和对应的终止节点,所述起始节点和终止节点至少由对应的第一分支和对应的第二分支连接。在这样的实施方案中,对应的第一分支和对应的第二分支对应于(例如映射到)目标序列。在一些实施方案中,该组目标序列代表以下中的一个或多个:(i)物种的参考序列或(ii)组装的重叠群、单元群、单倍群或支架。组装可以从头进行,也可以通过将多个测序读段映射到另一个目标序列来进行。
78.在一些实施方案中,一个或多个非线性拓扑组件中非线性拓扑组件的对应第一和第二分支分别代表杂合基因组中变异的第一等位基因和第二等位基因。在一些实施方案中,可能有更多分支和/或更多等位基因。例如,在一些实施方案中,生物体的基因组是多倍体而不仅仅是二倍体,并且可能存在与染色体对一样多的等位基因。在一些实施方案中,一个或多个非线性拓扑组件包括三个对应的分支和三个相应的等位基因、四个对应的分支和四个相应的等位基因、五个相应的分支和五个相应的等位基因、或六个相应的分支和六个相应的等位基因。
79.通过重叠多个序列读段中的相应序列读段,使用来自单个生物的生物样品的多个序列读段形成有向图,这些序列读段相对于目标序列具有大于百分之十的随机误差,其中重叠的第一序列读段和第二序列之间的突出的量是不受限制的,前提是第一和第二序列读段之间存在最小的共有区(例如,从而将本文描述的方法与de bruijn图区分开)。在一些实施方案中,多个序列读段具有大于10,000个碱基对的平均序列长度。
80.在一些实施方案中,多个序列读段具有大于2,500个碱基对、大于5,000个碱基对、大于10,000个碱基对、大于15,000个碱基对、大于20,000个碱基对、大于50,000个碱基对、或大于100,000个碱基对的平均长度。
81.方框218.使用计算机系统100,从电子格式的生物样品中获得查询序列。查询序列是(i)多个序列读段中的序列读段,或(ii)由多个序列读段形成的支架(例如重叠群、单元群或单倍群)。这里,查询序列至少包含一个或多个非线性拓扑组件中的第一非线性拓扑组件的对应起始节点或对应终止节点。
82.在一些实施方案中,查询序列是多个序列读段中的序列读段。在一些实施方案中,查询序列是由多个序列读段中的两个或更多个序列读段的共有物形成的支架。
83.在一些实施方案中,查询序列通过单分子实时(smrt)测序获得,其通常提供长序列读段。在一些实施方案中,smrt测序如rhoads和au(genomics proteomics bioinformatics13:278
‑
289,2015)所述进行。这种长读段测序提供了足以单独用于从头全基因组测序的信息(例如,如chin等人,2013,nat methods 10(6):563
‑
569中所述)。
84.方框224.该方法继续使用有向图形成查询序列到有向图的映射。查询序列的映射包括(i)第一比对组件到有向图的第一部分,(ii)第二比对组件到有向图的第二部分和(iii)描述有向图中在有向图的第一部分和第二部分之间的关系的路径。在这种情况下,第一部分和第二部分中的一个在第一非线性拓扑组件的第一或第二分支中,而第一部分和第二部分中的另一个不在第一非线性拓扑组件的分支中。在一些实施方案中,每个非线性拓
扑组件有两个以上的分支,并且相同的查询也可以跨越多个非线性拓扑组件(例如,映射到参考图的大重叠群)。
85.在一些实施方案中,非线性拓扑组件包括至少2个分支、至少3个分支、至少4个分支、至少5个分支、至少6个分支、至少7个分支、至少8个分支、至少9个分支、或至少10个分支。
86.在一些实施方案中,映射为查询序列提供目标序列中的起始位置和结束位置。在一些实施方案中,一个或多个程序单独地或共同地进一步包括用于使用序列比对算法以及起始位置和结束位置将查询序列与目标序列进行比对的指令。
87.在一些实施方案中,序列比对算法是动态编程全局比对算法、动态编程半全局比对算法或动态编程局部比对算法。
88.不同的比对算法具有不同的效率或经过调整以更好地检测特定基因组特征。事实上,有多种不同类型的算法,如下讨论。gassst(全局比对短序列搜索工具)旨在改进插入缺失的检测(例如,如rizk和lavenier,bioinformatics 26(20):2534
–
2540,2010所述)。burrows
‑
wheeler aligner的smith
‑
waterman比对(bwa
‑
sw)方法旨在使用长读段序列提高效率(例如,如li和durbin,bioinformatics 26(5):589
–
595,2010所述)。parasail旨在提供更快的序列内局部成对比对(例如,如daily,bmc bioinformatics 17:81,2016所述)。skewer特别专注于使用下一代测序配对末端读段(例如,如jiang等人,2014,bmc bioinformatics 15:182中所述)。dnaclust旨在提高高度相似dna区域的序列的聚类效率(例如,如ghodsi等人,2011,bmc bioinformatics 12:271所述)。局部比对算法的历史悠久,其包括诸如blast和smith
‑
waterman的普遍方法(altschul等人,1990,j mol biol 215:403
‑
410;smith和waterman,1981,j mol biol 147(1):195
‑
197)。然而,也有最近开发的方法试图改进初始算法。例如,bowtie 2改进了间隙比对的性能(例如,如langmead和salzberg,nat methods 9(4):357
‑
359,2012所述)。li和homer提供了不同序列比对算法的比较,特别是得出的结论是,与基于短读段的测序相比,长读段测序能够获得更详细的基因组信息(briefings in bioinformatics 11(5):473
‑
483,2010)。
89.在将长序列读段或重叠群映射到目标序列时形成有向图。在本公开的一些实施方案中,提供了一种用于构建具有杂合基因组的物种的单个二倍体或多倍体生物的有向图的核酸测序方法。该方法通过将从生物体的生物样品获得的多个序列读段映射到物种的目标序列上来进行。多个序列读段相对于目标序列共同具有大于百分之十的随机误差。有向图表示杂合基因组的全部或一部分并且包括一个或多个非线性拓扑组件(例如,气泡或环)。一个或多个非线性拓扑组件中的每个相应的非线性拓扑组件代表相对于参考序列的变异(其中变异是例如结构变异、单核苷酸变异(snp)、小核苷酸变异或在het
‑
snp中重的且模拟为更大的替换事件的区域的集中的一个)。一个或多个非线性拓扑组件中的每个相应的非线性拓扑组件具有对应的起始节点和对应的终止节点,所述起始节点和终止节点至少由对应的第一分支和对应的第二分支连接。对应的第一分支和对应的第二分支中的一个对应于目标序列,并且对应的第一分支和对应的第二分支中的另一个对应于在多个序列读段中的一个或多个序列读段中通过映射识别的变异。
90.在一些实施方案中,映射为多个序列读段中的序列读段提供目标序列中的起始位置和结束位置。在此类实施方案中,使用序列比对算法和序列读段的起始位置和结束位置
将序列读段与目标序列比对。
91.在一些实施方案中,有向图代表物种基因组的至少百分之十、物种基因组的至少百分之二十、物种基因组的至少百分之三十、物种基因组的至少百分之四十、物种基因组的至少百分之五十、物种基因组的至少百分之六十、或物种基因组的至少百分之七十。
92.在一些实施方案中,有向图包括多个节点,例如,每个节点可以表示序列读段。因此,有向图中的节点总数将与基因组或被映射的基因组部分的大小成正比。例如,在其中大比例的复杂二倍体和更高阶多倍体基因组被映射的一些实施方案中,有向图将具有至少10,000个节点。在一些实施方案中,有向图将具有至少100,000个节点。在一些实施方案中,有向图将具有至少500,000个节点。在其他实施方案中,有向图将具有至少1百万、两百万、三百万、四百万、五百万或更多节点。
93.在一些实施方案中,例如在映射大比例的复杂二倍体或更高阶多倍体基因组的情况下,有向图将具有至少1,000个非线性拓扑组件。在一些实施方案中,有向图将具有至少10,000个非线性拓扑组件。在一些实施方案中,有向图将具有至少100,000个非线性拓扑组件。在其他实施方案中,有向图将具有至少500,000、1百万、两百万、三百万、四百万、五百万或更多非线性拓扑组件。在一些实施方案中,对于具有映射到每个可能等位基因的至少一个序列读段的受试者中的每个杂合等位基因,图中存在非线性元素。也就是说,观察到每个snp和每个indel中至少一个序列读段已映射到等位基因1,至少一个读段已映射到等位基因2。
94.用例1:从长测序读段的分阶段从头组装形成图。
95.典型的映射方法通常无法处理单单倍群。使用常规映射器和仅主要组装作为参考,单倍群会在结构变化的区域
‑
图气泡
‑
中分解为主要的和一个或多个补充的比对。例如,某些映射器(例如,选择了
“‑‑
bestn 1”选项的blasr)将仅报告主要比对(参见chaisson和tesler bmc bioinformatics 13:238,2012)。然而,这掩盖了一些潜在的遗传变异。
96.提供了一种核酸测序方法,其包括为具有杂合基因组的物种的单个二倍体或多倍体生物构建单倍群组装图。在一些实施方案中,单倍群组装图代表杂合基因组并包含一种或多种非线性拓扑组件(例如,气泡或环)。一个或多个非线性拓扑组件中的每个相应的非线性拓扑组件具有对应的起始节点和对应的终止节点,所述起始节点和终止节点至少由对应的第一分支和对应的第二分支连接。对应的第一和第二分支分别代表对应于杂合基因组中的第一和第二同源染色体的第一和第二定相单倍群。
97.在一些实施方案中,可能有更多分支和/或更多等位基因。例如,在一些实施方案中,生物体的基因组是多倍体而不仅仅是二倍体,并且可能存在与染色体对一样多的等位基因。在一些实施方案中,一个或多个非线性拓扑组件包括三个对应的分支和三个相应的等位基因、四个对应的分支和四个相应的等位基因、五个相应的分支和五个相应的等位基因、或六个相应的分支和六个相应的等位基因。
98.测序方法继续为通过从以电子格式的单一二倍体或多倍体生物体获得的生物样品中获得源自多个序列读段的单倍群。单倍群至少包含一个或多个非线性拓扑组件中的第一非线性拓扑组件的对应起始节点或对应终止节点。该方法继续使用单倍群组装图来形成单倍群的映射。单倍群的映射包括(i)第一比对成分到单倍群组装图的第一部分,(ii)第二比对成分到单倍群组装图的第二部分,以及(iii)描述单倍群组装图中在单倍群组装图的
第一部分和第二部分之间的关系的路径。第一部分和第二部分中的一个在第一非线性拓扑组件的第一或第二分支中,而第一部分和第二部分中的另一个不在第一非线性拓扑组件的分支中。
99.在一些实施方案中,该映射为单倍群提供了在目标序列中的起始位置和结束位置。一个或多个程序单独地或共同地进一步包括用于使用序列比对算法以及起始位置和结束位置将单倍群与物种的目标序列进行比对的指令。
100.在一些实施方案中,单倍群组装图代表杂合基因组的至少百分之十,并且单倍群组装图包含十个或更多个非线性拓扑组件。在一些实施方案中,单倍群组装图代表杂合基因组的至少百分之十五,并且单倍群组装图包含二十五个或更多个非线性拓扑组件。
101.在一些实施方案中,单倍群组装图代表杂合基因组的至少百分之五、杂合基因组的至少百分之十、杂合基因组的至少百分之二十、杂合基因组的至少百分之三十、杂合基因组的至少百分之四十、杂合基因组的至少百分之五十、杂合基因组的至少百分之七十五、杂合基因组的至少百分之九十、或杂合基因组的至少百分之百。
102.在一些实施方案中,单倍群组装图包括两个或更多个非线性拓扑组件、五个或更多个非线性拓扑组件、十个或更多个非线性拓扑组件、二十五个或更多个非线性拓扑组件、五十个或更多个非线性拓扑组件、或者一百个或更多个非线性拓扑组件。
103.在一些实施方案中,单倍群组装图包括杂合基因组的上述百分比和非线性拓扑组件的数量的任意组合。
104.在一些实施方案中,通过重叠多个序列读段中的相应序列读段,使用多个序列读段形成单倍群组装图,其中重叠的第一序列读段和第二序列之间的突出的量是不受限制的,前提是第一和第二序列读段之间存在最小的共有区(例如,以区分具有预限定重叠长度的de bruijn图)。
105.在一些实施方案中,最小共有区包含至少1个碱基、至少2个碱基、至少3个碱基、至少4个碱基、至少5个碱基、至少6个碱基、至少7个碱基、至少8个碱基、至少9个碱基、至少10个碱基、至少15个碱基、至少20个碱基、至少30个碱基、至少40个碱基、至少50个碱基、至少60个碱基、至少70个碱基、至少80个碱基、至少90个碱基、至少100个碱基、至少500个碱基或至少1000个碱基。在一些实施方案中,每个共有区具有相同的重叠长度。在一些实施方案中,每个共有区具有不同的重叠长度。在一些实施方案中,第一组共有区具有第一重叠长度并且第二组共有区具有第二重叠长度。
106.图3说明了在包含单倍型之间结构变异的组装基因组中比对定相单倍群的输出的实例。单倍群定相适用于falcon
‑
unzip组装方法,其旨在作为二倍体组装器(例如,如chin等人,2016,nat methods 13(12):1050
‑
1054中所述)。在一些实施方案中,公开的方法在本文中应用以通过实现准确的单倍群放置和较少片段化单倍群图(以及更长的定相单倍群)的构建来增强falcon
‑
unzip组装。在一些实施方案中,所得组装体以图形格式输出以保留尽可能多的关于发散单倍型的信息(例如,与仅提供线性重叠群的fasta输出格式相反)。
107.在图3中,主要重叠群302显示为具有两个相关的重叠群304和306,产生各自的气泡310和312。在一些实施方案中,在单个重叠群内最佳映射的序列读段产生连续比对。在一些实施方案中,映射跨越一个或多个分支位置的读段被报告为具有伴随比对路径的分裂比对。在一些实施方案中,将分数分配给每个比对。在一些实施方案中,每个分数指示相应比
对的质量。在一些实施方案中,仅报告分数高于阈值的比对。在一些实施方案中,如果有一个以上的比对具有高于阈值的相关分数,则报告每个这样的比对。
108.在一些实施方案中,质量分数以sam格式报告(例如,如在
‘
sequence alignment/map format specification’中描述,互联网上在samtools.github.io/hts
‑
specs/samv1.pdf。
109.在一些实施方案中,分数的阈值为至少1、至少5、至少10、至少15、至少20、至少25、至少30、至少35、至少40、至少45、至少50、或至少55。
110.在一些实施方案中,使用给定序列读段中所有比对的每个碱基质量分数的总和以及每个错配碱基的质量分数的总和计算质量分数(例如,如li等人,2008,genome res18(11):1851
‑
1858中所述,在此通过引用整体并入)。在一些实施方案中,质量分数是phred分数,其是碱基判读不正确的对数缩放概率(例如,如ruffalo等人,2012,bioinformatics28(5):i349
‑
i355中所述)。
111.这种方法提供了在重叠群末端附近增加的覆盖率(从而能够更好地精修)、改进的读段定相以及提高解析组装中复杂重复结构的能力。
112.用例2:通过映射到单倍群图对读段进行定相。
113.a:使用单倍群图对长读段进行定相。
114.通过falcon
‑
unzip单倍群组装和长距离测序数据(例如,10x、dovetail、bionano)的组合解析单倍型支架是脊椎动物基因组计划(vgp)联盟等团体使用的常用程序。当组装图的背景可用时,映射长距离读段应该更加准确。解压缩组装是高度连续的,但在对齐长距离数据时通常会丢弃连接信息。通常,短的双端读段仅映射到重叠群/单倍群。在稍微重复的区域(在较大的基因组上丰富)的情况,映射可能会变得不明确。图背景可以将映射结果限制在候选目标序列之间存在路径(长度可接受)的一个区域。相关方法使用hi
‑
c数据与gfa格式的组装图相结合,以准确地搭建组装体(参见ghurye等人biorxiv 261149;互联网上doi.org/10.1101/261149)。
115.提供了另一种核酸测序方法,其包括为具有杂合基因组的物种的单个二倍体或多倍体生物构建单倍群组装图。单倍群组装图代表杂合基因组并包含一种或多种非线性拓扑组件(例如,气泡或环)。一个或多个非线性拓扑组件中的每个相应的非线性拓扑组件具有对应的起始节点和对应的终止节点,所述起始节点和终止节点至少由对应的第一分支和对应的第二分支连接。对应的第一和第二分支分别代表对应于杂合基因组中的第一和第二同源染色体的第一和第二定相单倍群。
116.在一些实施方案中,例如在多倍体基因组中,可以有2个或更多个分支。在一些实施方案中,可以有3个或更多个分支、4个或更多个分支、5个或更多个分支、6个或更多个分支、7个或更多个分支、8个或更多个分支、9个或更多个分支或10个或更多个分支。
117.该方法继续为通过从以电子格式的单一二倍体或多倍体生物体获得的生物样品中获得多个序列读段。多个序列读段中的第一序列读段至少包含一个或多个非线性拓扑组件中的第一非线性拓扑组件的对应起始节点或对应终止节点。在接下来的实施方案中,该方法还可以包括起始节点和终止节点以及整个气泡(或环或甚至多个附近气泡)。该方法继续使用单倍群组装图以形成多个序列读段中每个相应序列读段的映射。第一序列读段的映射被报告为分裂比对,其包括(i)第一比对成分到单倍群组装图的第一部分,(ii)第二比对
成分到单倍群组装图的第二部分,以及(iii)描述单倍群组装图中单倍群组装图的第一部分和第二部分之间的关系的路径,其中第一部分和第二部分之一在第一非线性拓扑组件的第一或第二分支中,并且第一部分和第二部分中的另一个不在第一非线性拓扑组件的分支中。
118.在一些实施方案中,映射提供了目标序列中相应序列读段的起始位置和结束位置,并且其中一个或多个程序单独地或共同地进一步包括用于使用序列比对算法以及起始位置和结束位置将相应序列读段与目标序列进行比对的指令。
119.在一些实施方案中,通过重叠多个序列读段中的相应序列读段,使用多个序列读段形成单倍群组装图。重叠的第一序列读段和第二序列之间的突出的量是不受限制的,前提是第一和第二序列读段之间存在最小的共有区。这与de bruijn图不同,该图对于每个序列读段具有特定的k聚体长度重叠。重叠长度的细节在上面讨论过,这里不再重复。
120.b:使用单倍群图对双端短序列读段进行定相。
121.本公开的一个方面提供了一种核酸测序方法,其包括为具有杂合基因组的单个二倍体或多倍体生物构建单倍群组装图。单倍群组装图代表杂合基因组并包含一种或多种非线性拓扑组件(气泡或环)。一个或多个非线性拓扑组件中的每个相应的非线性拓扑组件具有对应的起始节点和对应的终止节点,所述起始节点和终止节点至少由对应的第一分支和对应的第二分支连接。对应的第一和第二分支分别代表对应于杂合基因组中的第一和第二同源染色体的第一和第二定相单倍群。
122.该方法继续为通过从以电子格式的单一二倍体或多倍体生物体获得的生物样品中获得多个双端序列读段。多个双端序列读段中的第一组双端序列读段至少包含一个或多个非线性拓扑组件(例如,气泡或环)中的第一非线性拓扑组件的相应起始节点或相应终止节点)。该方法继续使用单倍群组装图以形成多个双端序列读段中每个相应双端序列读段的映射。第一组双端序列读段的映射被报告为分裂比对,其包括(i)第一比对成分到代表第一组双端序列读段中的第一序列读段的单倍群组装图的第一部分,(ii)第二比对成分到代表第一组双端序列读段中的第二序列读段的单倍群组装图的第二部分,以及(iii)描述在单倍群组装图中单倍群组装图的第一部分和第二部分之间关系的路径。第一部分和第二部分中的一个在第一非线性拓扑组件的第一或第二分支中,而第一部分和第二部分中的另一个不在第一非线性拓扑组件的分支中。
123.在一些实施方案中,映射为目标序列中的每个相应组的双端序列读段提供起始位置和结束位置。一个或多个程序单独地或共同地进一步包括用于使用序列比对算法以及起始位置和结束位置将每个相应组的双端序列读段与目标序列进行比对的指令。
124.在一些实施方案中,通过重叠多个双端序列读段中的相应双端序列读段组,使用多个双端序列读段形成单倍群组装图。重叠的第一组双端序列读段组和第二组双端序列读段组之间的突出的量不受限制,前提是第一组双端序列读段组和第二组双端序列读段组之间存在最小共有区。
125.在一些实施方案中,最小共有区包含至少1个碱基、至少2个碱基、至少3个碱基、至少4个碱基、至少5个碱基、至少6个碱基、至少7个碱基、至少8个碱基、至少9个碱基、至少10个碱基、至少15个碱基、至少20个碱基、至少30个碱基、至少40个碱基、至少50个碱基、至少60个碱基、至少70个碱基、至少80个碱基、至少90个碱基、至少100个碱基、至少500个碱基或
至少1000个碱基。在一些实施方案中,每个共有区具有相同的重叠长度。在一些实施方案中,每个共有区具有不同的重叠长度。在一些实施方案中,第一组共有区具有第一重叠长度并且第二组共有区具有第二重叠长度。
126.如图4a、4b和4c所示,单倍群组装图可以包括一个或多个代表定相单倍群的气泡(例如420、422和432)。每个相位单倍群(例如406、412和418)之间的区域通常是不包含足够信息以成功定相的折叠序列。
127.解压缩组装后(例如,使用falcon
‑
unzip),可以使用单倍群图作为中间输出。在一些实施方案中,单倍群图是有向无环图(dag),由具有至少两个分支(每个单倍型一个)和折叠区域(例如,基因组中没有足够信息进行定相的区域)的气泡组成。每个图中的气泡分支(例如420、422)代表二倍体基因组的定相部分。读段可以映射到falcon
‑
unzip单倍群图,并通过选择图中具有最高比对分数的分支作为最可能的目的地(例如,相位)来定相。
128.图4b和4c说明了相同组装的两个可能的图,以及两个长距离双端读段402和404的潜在映射。在一些实施方案中,两个双端读段402和404的方向是已知的。在这样的实施方案中,可以确定图4b中所示的图表,其中每个气泡420和422的位置是关于彼此已知的。在一些实施方案中,由于402和404的已知位置提供的连通性信息,第一映射位置可能是优选的。在一些实施方案中,402和404之间的连通性是未知的,因此仅确定每个单独的气泡420和422。在一些实施方案中,读段404是重复的并且还被映射到第二图位置(例如,在图4c中的区域430上)。
129.在一些实施方案中,该方法用于改善解析单倍型支架的结果,提供更高质量的组装,并改善falcon解压缩结果。
130.用例3:映射到环形基因组。
131.环形基因组可以在图背景中模拟,只需添加一条边,将染色体的末端连接到同一染色体的开头。在这种情况下,图形感知映射器会知道比对可以在序列结束后安全地扩展,并从它自己的开始继续。通过图形定义指定环形背景允许输入fasta序列集(例如,真核基因组)中线性和环形染色体的混合参考。
132.因此,本公开的一个方面提供了一种核酸测序方法,其中获得具有环状基因组的物种的图。该图表示环状基因组并包括指定环状基因组的序列的片段线。序列的开头限定了片段线的开头,序列的结尾限定了片段线的结尾,和表示图中的环状边界的边线,边线指定片段线的结尾连接到片段线的开头。该方法继续从电子格式的物种的生物体获得的生物样品获得多个序列读段。多个测序读段中的第一序列读段跨越环状边界。该方法继续使用该图来形成多个测序读段中的每个测序读段的映射。第一测序读段的映射被报告为分裂比对,包括(i)第一比对成分到序列的末端部分,(ii)第二比对成分到序列的初始部分,和(iii)描述图中序列的末端部分和图中序列的初始部分之间的关系的路径。
133.本公开的另一个方面提供了一种核酸测序方法,其中获得具有环状基因组的物种的图。该图表示环状基因组并包括指定环状基因组的序列的片段线。序列的开头限定了片段线的开头,序列的结尾限定了片段线的结尾。边线表示图形中的环状边界,边线指定片段线的结尾链接到片段线的开头。该方法继续从电子格式的物种的生物体获得的生物样品获得多个双端测序读段。多个双端测序读段中的第一组双端测序读段包含环状边界。该方法继续使用该图来形成多个双端序列读段中的每个双端测序读段的映射。第一组双端序列读
段的映射报告为分裂比对,包括(i)第一组双端序列读段中的第一序列读段与序列末端部分的比对,(ii)第一组双端序列读段中的第二序列读段与序列的初始部分的比对,和(iii)描述图中末端部分和初始部分之间关系的路径。
134.图5说明了环状参考基因组。环状参考是最简单的图形式,只有一条边504将序列508的结尾连接到其开头506。基于图的映射器可以检测应该跨间隙比对的读段并构建正确的比对。这确保了映射读段的高覆盖率保留在此类重叠群的前部和后部,从而能够更好地精修环状重叠群。在一些实施方案中,falcon组装器包含一个特征以天然地输出环状重叠群的环状边缘。这意味着,如果基于图的映射器与此类重叠群图一起使用,arrow或其他精修工具将在它们处置时具有几乎统一的可用读段覆盖范围(这是当前使用blasr作为默认比对器时的一个问题)。
135.在一些实施方案中,跨越环状边界的读段将报告两个映射位置。在一些实施方案中,第一映射位置出现在染色体502的开头506附近,一直到达结尾508。在一些实施方案中,第二映射位置出现在染色体的结尾508附近,理想情况下到达开头506。在一些附加实施方案中,提供映射路径来描述第一和第二映射之间的关系。这种方法确保了整个环状基因组中映射读段的良好覆盖,并允许更好地精修序列。
136.用例4:使用基因组倒位进行映射。
137.倒位作为不同基因组之间的自然结构变异(sv)出现。许多当前的映射器对于包含倒位的序列表现不佳。例如,chen等人描述了各种映射工具的结构变化预测,分析的每种方法对于倒位的成功率都相对较低(nat methods 6(9):677
‑
681,2009)。倒位的检测无缝地从本文描述的基于图的映射的定义中出现。在本公开的一些实施方案中,映射到参考的反向互补链的序列的一部分可以通过隐式边缘连接到查询片段的其余部分。
138.因此,本公开的一个方面提供了一种核酸测序方法,其中以电子格式获得表示包含基因组倒位的物种的生物体的基因组序列的一部分的查询序列。查询序列包括第一查询部分和第二查询部分。在该方法中,查询序列被映射到物种的目标序列。映射包括将第一查询部分映射到目标序列的第一链的第一目标部分和将第二查询部分映射到目标序列的第二链的第二目标部分,其中第一链是第二链的倒位互补,从而形成分裂比对,其包括(i)第一比对成分,其包括第一查询部分与第一链的第一目标部分的比对,(ii)第二比对成分,其包括第二查询部分与第二链的第二目标部分的比对,以及(iii)描述第一目标部分和第二目标部分之间关系的比对路径。
139.在一些实施方案中,查询序列包括第三查询部分。在一些实施方案中,查询序列包括第四查询部分、第五查询部分、第六查询部分、第七查询部分、第八查询部分、第九查询部分或第十查询部分。在一些实施方案中,在同一个查询中可以有多个倒位。在一些实施方案中,一个查询可以包括1个或更多个倒位、2个或更多个倒位、3个或更多个倒位、4个或更多个倒位、5个或更多个倒位、10个或更多个倒位、15个或更多个倒位、或20个或更多个倒位。
140.映射包括将第三查询部分映射到第一链的第三参考部分,并且分裂比对还包括第三比对成分,其包含第三查询部分与第一链的第三参考部分的比对,并且比对路径进一步描述第一参考部分和第二参考部分与第三参考部分的关系。
141.在一些实施方案中,查询序列是从生物体的生物样品获得的单一序列读段。在一些实施方案中,查询序列是由从生物体的生物样品获得的多个序列读段形成的重叠群。
142.图6a和6b图示了倒位映射的实例。查询序列600具有参考基因组的一个或多个锚。如图6a所示,在一些实施方案中,查询序列600包括三个锚序列602、604和606。
143.在一些实施方案中,查询序列包含至少2个锚序列、至少3个锚序列、至少4个锚序列、至少5个锚序列、至少6个锚序列、至少7个锚序列、至少8个锚序列、至少9个锚序列、或至少10个锚序列。
144.如图6b所示,锚序列602和606中的两个映射到参考序列的正向链610,第三锚定序列604映射到参考序列的相应反向链608。与本文针对其他用例所描述的不同,该实施方案的图不能通过参考序列或其他输入来预先确定。相反,在该实施方案中,该图是隐含地确定的。在一些实施方案中,随后针对结构变体分析通过该方法确定的图。
145.用例5:将smrtbell
tm
参考模拟为图。
146.在一些实施方案中,使用smrtbell模板。如travers等人所述,在一些实施方案中,smrtbell模板由感兴趣的序列、衔接子序列和任选的条形码序列组成(nucleic acids res38(15):e159,2010)。在一些实施方案中,基因组的已知区域被靶向用于测序。在这样的实施方案中,密切相关的参考序列用于对插入物建模并构建环状图。在一些实施方案中,使用smrtbell模板提高测序效率,因为衔接子和/或条形码序列有助于比对展开的长读段序列(例如,多次穿过环状模板的长读段序列)。在一些实施方案中,长读段序列穿过环状模板至少1次、至少2次、至少5次、至少10次、至少20次或至少50次。
147.在根据用例5的一个方面,提供了包括获得环状图的核酸测序方法。环状图包括指定在第一方向上的插入序列的第一片段,指定在第一方向上的衔接子序列的第二片段,指定在第二方向上的插入序列的第三片段,指定在第二方向上的衔接子序列的第四片段,将第一片段的末端连接到第二片段的开头的第一边,将第二片段的末端连接到第三片段的开头的第二边,将第三片段的末端连接到第四片段的开头的第三边,以及将第四片段的末端连接至第一片段的开头的第四边。第二方向是第一方向的反补。
148.该方法通过从电子格式的生物样品中获取查询序列读段继续进行。序列读段跨越由环状图表示的序列。然后该方法继续使用该图形成序列读段的映射。序列读段的映射报告为序列读段中插入序列的每个实例的比对。
149.在一些实施方案中,映射还包括衔接子序列的每个实例的比对。
150.在一些实施方案中,序列读段多次跨越环状图。在一些实施方案中,多次是三次或更多次。在一些实施方案中,多次是五次或更多次。在一些实施方案中,多次是二十次或更多次。
151.smrtbell
tm
构建体,例如如图7a所示,由所需的插入序列(例如702和710)、衔接子(例如,706和714)和任选的条形码序列(例如,704、708、712和716)组成。在一些实施方案中,基因组的已知区域被靶向用于测序,并且密切相关的参考序列用于对插入物建模并从环形构建体构建环状图。在一些实施方案中,条形码序列是彼此的反向互补物(例如712/716和704/708对)。在一些实施方案中,条形码序列是对称的(例如,716将是708或712的反向互补)。在一些实施方案中,条形码序列不是对称的(例如,716不是708或712的反向互补)。在一些实施方案中,所需插入物将以正向(例如,702)和反向(例如,710)序列存在。在一些实施方案中,环状构建体的一个或多个区域是双链的(例如719)。在一些实施方案中,衔接子序列(例如714和706)是彼此的反向互补。在一些实施方案中,衔接子序列(例如,714
和706)不是彼此的反向互补。在一些实施方案中,每个衔接子序列在环状构建体的每个末端形成发夹区(例如,718和717)。
152.在本公开的一些实施方案中,在图背景中对沿已知扩增子参考展开和对齐zmw读段进行建模。在最简单的情况下(例如,没有可选的条形码序列704、708、712和716),基于环形构建体的图将包括第一片段(例如714)、第二片段(例如702)、第三片(例如706)、第四片段(例如710)以及这些片段中的每两个之间的边缘。在一些实施方案中,片段中的两个将是彼此的反向互补(例如702和710)。
153.在一些实施方案中,来自smrtbell
tm
构建体的测序读段包括zmw读段720,如图7b所示。从图7b中可以看出,在一些实施方案中——取决于zmw读段有多长——插入序列的多个拷贝(例如722
‑
a、722
‑
b和722
‑
c)将存在。在一些实施方案中,插入序列的每个拷贝被称为子读段,多个子读段相互比较,从而提高整体测序结果的准确性。
154.用例6:通过限定剪接位点的边进行转录组映射。
155.转录组映射是基于图的映射器的自然应用。因此,在一些实施方案中,公开的方法针对circrna映射。circrna是环状分子。在一些实施方案中,对此类环状分子进行测序产生看似无序但实际上是剪接外显子的环状排列的外显子。在一些实施方案中,向转录组添加环形边缘能够映射此类rna
‑
seq数据。li等人描述了circrna映射的一个例子,但该方法依赖于k聚体分析,并且不适用于本公开中使用的长序列读段(bmc genomics 19(suppl 6):572,2018)。
156.因此,根据用例6的方法提供了一种核酸测序方法,该方法包括获得转录组注释,该注释限定物种的参考序列中多个外显子中每个相应外显子的映射,并构建对应于转录组注释的图。该图包括物种的参考序列、第一片段、第二片段以及第一外显子的3'端和第二外显子的5'端之间的链接。
157.在一些实施方案中,第一片段代表多个外显子中的第一外显子。在一些实施方案中,外显子是在图中表示为节点的序列。在一些实施方案中,边代表剪接位点(例如,两个片段虚拟连接在一起的那些位置)。在一些实施方案中,第一片段还包括参考序列中第一外显子的5'端和3'端的相应坐标。在一些实施方案中,第二片段代表多个外显子中的第二外显子并且包括参考序列中第二外显子的5'端和3'端的相应坐标。
158.图8a和8b说明了转录组映射的一个例子。在一些实施方案中,转录组通过沿着参考基因组(例如810)标记剪接区域的图来描述。第一片段(例如,单个节点或一组节点)代表多个外显子(例如802、804、806和808)中的第一外显子802。在一些实施方案中,作为序列的外显子表示为图中的节点或节点集。边代表两个片段应该虚拟连接在一起的剪接位点。第一片段还包括参考序列中第一外显子的5'端和3'端的相应坐标。第二片段代表多个外显子中的第二外显子并且包括参考序列中第二外显子的5'端和3'端的相应坐标。
159.该方法通过以电子格式从物种的生物体的生物样品获得mrna的序列读段继续进行。序列读段包含第一外显子和第二外显子(例如802和804)。在一些实施方案中,序列读段包含至少2个外显子、至少3个外显子、至少4个外显子、至少5个外显子、至少6个外显子、至少7个外显子、至少8个外显子、至少9个外显子、或至少10个外显子。然后该方法继续使用该图形成序列读段到参考序列810的映射。
160.在一些实施方案中,即使当序列读段中的外显子的顺序与其在参考基因组中出现
的线性顺序不同时,所述链接也允许进行映射。在一些实施方案中,不在序列读段中的第三外显子介于参考序列中的第一外显子和第二外显子之间。
161.在一些实施方案中,查询序列是从环状rna分子(circrna)获得的,查询序列中外显子的顺序与其在参考序列中的相应顺序不是线性的。
162.转录组注释可以以图形的形式表示,连接相邻的剪接位点。通过应用基于图的映射器,在映射isoseq/rna
‑
seq读段之前不需要明确构建转录组序列。
163.在一些实施方案中,转录组映射的预期结果是在同一比对中具有预定义数量的cigar(简明特异缺口比对报告(concise idiosyncratic gapped alignment report))操作的剪接比对,或沿基因组的单个外显子区域的比对路径。在一些实施方案中,该组可能的cigar操作至少包括比对匹配(例如,序列匹配或错配)、对参考的插入、从参考的删除、从参考跳过的区域、序列匹配、或序列错配。
164.结束语
165.可以为本文描述为单个实例的组件、操作或结构提供多个实例。最后,各个组件、操作和数据存储之间的边界在某种程度上是任意的,并且在特定说明性配置的上下文中说明了特定操作。设想了其它功能分配,并且可以落入所述(多个)实施方案的范围内。总体上,在示例配置中作为单独分开的组件呈现的结构和功能可以实施为组合结构或组件。类似地,作为单个组件呈现的结构和功能可以实施为单独分开的组件。这些其它变型、修改、添加和改进落入所述(一个或多个)实施方案的范围内。
166.还将理解,虽然术语第一、第二等可在本文中用于描述各种元件,但这些元件不应受这些术语限制。这些术语仅用于区分一个元件与另一元件。例如,在不脱离本公开的范围的情况下,第一主题可以被称为第二主题,并且类似地,第二主题可以被称为第一主题。虽然第一主题和第二主题都为主题,但这些主题不是同一个主题。
167.在本公开中使用的术语仅用于描述特定实施例,并不旨在限制本发明。如在本发明的说明书和所附权利要求中所使用的,单数形式“一”、“一个”和“所述”也旨在包括复数形式,除非上下文另外清楚地指示。还应当理解,这里使用的术语“和/或”是指并涵盖相关联的所列项目中的一个或多个的任何和所有可能的组合。应进一步理解的是,当在本说明书中使用时,术语“包括(comprises和/或comprising)”指定所陈述的特征、整数、步骤、操作、要素和/或组件的存在,但不排除存在或添加一个或多个其它特征、整数、步骤、操作、要素、组件和/或其群组。
168.如本文中所使用,取决于上下文,术语“如果”可以被解释成意指“当
……
时”或“在
……
时”或者“响应于确定”或“响应于检测”。类似地,取决于上下文,短语“如果确定”或“如果检测到[所陈述的条件或事件]”可以被解释为意指“在确定
……
时”或“响应于确定”或“在检测到(所陈述的条件或事件)时”或“响应于检测到(所陈述的条件或事件)”。
[0169]
前述描述包含体现说明性实施方案的示例系统、方法、技术、指令序列和计算机器程序产品。出于解释的目的,阐述了许多具体细节,以便提供对本发明主题的各个实施方案的理解。然而对于本领域的技术人员将显而易见的是,本发明的主题可以在没有这些具体细节的情况下实践。总体而言,未详细示出众所周知的说明实例、协议、结构和技术。
[0170]
为了解释的目的,前面的描述已经参照特定的实施方案进行了描述。然而,上述说明性讨论并不旨在穷举或将所述实施方案限制于所公开的精确形式。鉴于以上教导,许多
修改和变化是可以的。选择和描述这些实施方案是为了最好地解释这些原理及其实际应用,由此使得本领域其它技术人员能够用适合预期的特定用途的多种修改方案来最好地使用这些实施方案和多种实施方案。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。