本发明公开了包含发光荧光体化合物的技术发光材料、包括安全特征的制品以及用于制备发光荧光体化合物的方法。更具体地讲,技术领域涉及掺入包含发光苯并噻唑的发光颗粒的发光材料和安全制品,以及用于制备这种发光颗粒的方法。
背景技术:
发光荧光体化合物是在通过外部能源诸如电磁辐射激发化合物时能够发射红外、可见和/或紫外光谱中可检测量的辐射的化合物。发光荧光体化合物广泛用于多种领域,包括但不限于照明装置、阴极射线管、暗发光制品和安全标记。发光荧光体化合物通常可表征为无机或有机发光荧光体化合物。无机发光荧光体化合物通常包括主体材料(如晶格)、发射离子(如稀土金属的发射离子),并且在一些情况下,包括“敏化”离子(如过渡金属或不同稀土金属的发射离子,其可吸收能量并将能量转移到发射稀土金属离子)。多种有机发光荧光体化合物是已知的,其中许多基于芳族化学。例如,各种有机发光荧光体化合物可为羟基喹啉盐;对苯二甲酸的衍生物;邻氨基苯甲酸的衍生物,诸如苯并咪唑、苯并噻唑、苯并噁嗪酮或喹唑啉酮;噻吨;水杨酸衍生物;或稀土金属的有机络合物。
发光荧光体化合物的所选化学性质可使该化合物具有特定的发射性质,包括具有给定光谱的发射以及各种时间性质。一些荧光体化合物的独特光谱特性使其非常适用于验证或识别具有特定价值或重要性的制品(例如,纸币、护照、生物样品等)。因此,已将具有已知光谱特征的发光荧光体化合物掺入到各种类型的制品中,以增强检测此类制品的假冒或伪造复制品,或识别和追踪该制品的能力。例如,发光荧光体化合物已以添加剂、涂层和印刷的或以其他方式施加的特征的形式掺入到各种类型的制品中,该特征可在验证或追踪制品的过程中进行分析。
可使用专门设计的验证设备通过已知的验证技术来验证包含发光荧光体化合物的制品。虽然此类验证技术在检测和阻止相对知识水平较低的假冒和伪造活动方面非常有效,但它们确实表现出缺点。例如,具有适当资源和设备的个体可能能够采用光谱分析技术以确定一些荧光体化合物的组分。然后可将荧光体化合物复制并与不真实的制品一起使用,从而损害原本可由特定荧光体化合物提供的验证有益效果。
对于某些应用,期望可见光谱内的发射,其中期望特定可见颜色的光谱发射。已识别出在各种发射波长下产生可检测发射的各种无机和有机发光荧光体。然而,希望在电磁波谱的某些未充分发展的部分内进一步发展发光荧光体化学物质。具体地讲,在电磁波谱的可见黄色至橙色部分(诸如约555nm至约630nm)内发射的有机发光荧光体化合物的已知化学性质在一定程度上受到限制。
已开发出各种脲/硫脲衍生物,包括含有脲基键的含苯并噻唑的化合物,以用于各种用途。例如,已提出将含苯并噻唑的化合物用于抗微生物应用。然而,先前生产此类材料的努力已经表现出相对低的收率,并且没有建议在抗微生物剂领域之外使用此类材料。
因此,尽管已开发出多种在可见电磁波谱的各个部分内发射电磁辐射的发光荧光体化合物,但希望开发出另外的化合物,这可使得假冒和伪造活动更加困难,这可证明有利于识别和跟踪特别关注的制品,和/或这可证明可用于其中需要电磁波谱的可见部分中的发射的其他应用中。另外,根据随后的本发明的具体实施方式和所附权利要求,结合附图和本发明背景,本发明的其他期望特征和特性将变得显而易见。
技术实现要素:
本文提供了包含发光荧光体化合物的发光材料、包括安全特征的制品以及用于制备发光荧光体化合物的方法。在一个实施方案中,发光材料包含介质和发光颗粒。发光颗粒包含具有脲基键的发光苯并噻唑。发光苯并噻唑响应约366nm波长的电磁辐射的激发而具有约555nm至约630nm波长内的峰强度。
在另一个实施方案中,提供了包括基底和基底中和/或基底上的验证特征的制品。验证特征包括发光颗粒。发光颗粒包含具有脲基键的发光苯并噻唑。发光苯并噻唑响应约366nm波长的电磁辐射的激发而具有约555nm至约630nm波长内的峰强度。
在一个实施方案中,提供了形成发光颗粒的方法。该方法包括将包含氨基官能苯并噻唑反应物和有机溶剂的混合物加热至大于约60℃的温度。在加热至大于约60℃的温度后,将含异氰酸酯化合物添加至所述混合物中以形成反应混合物。将反应混合物在大于约60℃的温度处保持一段反应期,以形成包含具有脲基键的发光苯并噻唑的发光颗粒。
附图说明
下文将结合以下附图描述本发明,其中类似的数字表示类似的元件,并且
图1是示出根据实施方案的发光颗粒的示例性样品的激发和发射光谱的曲线图;
图2是示出根据实施方案的发光颗粒的另一示例性样品的激发和发射光谱的曲线图;
图3是根据实施方案的包含介质和发光颗粒的发光材料的示意图;以及
图4是根据实施方案的包括验证特征的制品的示意性侧视图。
具体实施方式
以下具体实施方式本质上仅为示例性的,并非旨在限制发光材料、包括安全特征的制品、以及用于制备如本文所述的发光荧光体化合物的方法。另外,不意图受前述背景技术或以下详细描述中呈现的任何理论的束缚。
下文详细讨论的实施方案涉及包含发光苯并噻唑的发光颗粒,以及发光材料、制品和形成包含发光苯并噻唑的发光颗粒的方法。下述发光颗粒的实施方案增加了可用于验证或鉴别的可用材料的多样性,发光颗粒响应于用波长为约366nm的电磁辐射的激发而提供电磁波谱的黄色至橙色部分(诸如约555nm至约630nm)中的发射光谱。来自本文所讨论的发光颗粒的发射的光谱特征可用作用于验证目的的可测量量,并且可发现其中期望在可见光谱的黄色至橙色部分中的发光发射的其他用途。此外,本文提供了形成发光颗粒的方法,所述方法能够实现相对高收率的发光苯并噻唑,从而使发光颗粒具有最小的颗粒脱色和优异的发射性能。
如本文所提及的,术语“约”旨在涵盖预期将与所述值有效相同或产生类似特性的数值,诸如在所述值的10%内或另选地在所述值的5%内的数值。例如,“约555nm”的值也可有效涵盖550nm的值,或者553nm的值,前提条件是差值的影响不表示基础物理特性(例如,人眼可见的颜色)的材料差异。也如本文所用,术语“平均d99粒度”被定义为质量体积99%(d99)粒度平均直径,如通过激光衍射型测量装置诸如由microtrac公司(montgomritville,pa)生产的装置所测量的。
现在将描述发光颗粒的实施方案,其中发光颗粒适于包括在发光材料或制品中,如下文进一步详细描述。发光颗粒包含发光苯并噻唑,并且可任选地包含在发光颗粒形成后留下的痕量的其他化学物质。在实施方案中,发光颗粒包含基于发光颗粒的总重量计至少99重量%,诸如至少99.5重量%的量的发光苯并噻唑。在实施方案中,发光苯并噻唑的上述浓度是在发光颗粒形成后实现的,而无需进一步纯化作为干燥沉淀物获得的发光颗粒。在实施方案中,发光颗粒具有小于或等于约5μm的平均d99粒度,并且此类平均粒度的发光颗粒适用于胶版印刷应用。此类平均粒度可在发光颗粒形成后实现,而无需进一步加工在形成后作为干燥沉淀物获得的发光颗粒。另选地,可进一步处理沉淀物以获得目标平均粒度值。
发光颗粒中的发光苯并噻唑具有脲基键,脲基键也可被认为是二价脲键或碳酰二胺键。更具体地讲,脲基键将发光苯并噻唑的含苯并噻唑部分与化合物的另外部分接合。在实施方案中,脲基键接合发光苯并噻唑中的含苯并噻唑基的基团和任选的卤代苯基基团。在实施方案中,含苯并噻唑基的基团是苯基苯并噻唑基基团,并且脲基键将苯基苯并噻唑基基团接合到发光苯并噻唑中的任选的卤代苯基基团。更具体地讲,在实施方案中,脲基键直接键合至苯基苯并噻唑基基团的苯基,并且还直接键合至任选卤代的苯基基团的苯基。在实施方案中,脲基键在苯基的碳2处键合。在实施方案中,苯基苯并噻唑基基团不含羟基侧基,其中苯基仅含有侧挂于其上的氢原子,并且据信这些特征有助于观察到的本主题发光颗粒的发射光谱。
在具体的实施方案中,发光苯并噻唑具有以下结构:
其中r1为h、卤素自由基、烷基基团、酯基团、醚基团、芳族基团或烷氧基基团;r2为h、卤素自由基、烷基基团、酯基基团、醚基基团、芳族基团或烷氧基基团,其中r1和r2可表示用以形成多环芳族基团的芳族基团与苯基基团的键;并且r3为h、卤素自由基或烷基基团。在一个实施方案中,r1、r2和r3均为h。在一个实施方案中,r2和r3均为h,并且r1为如上所述除h之外的基团。例如,r1为卤素自由基,作为一个具体实施方案,为氯。又如,r1为烷基基团,诸如甲基。又如,r1为酯基团,诸如-cooch3。又如,r1为醚基团,诸如-coch3。在另一个实施方案中,r1和r2是如上所述的除h之外的基团。例如,r1和r2表示用以形成多环芳族基团(诸如萘基基团)的芳族基团与苯基基团的键。又如,r1和r2均为卤素自由基,诸如氯。在另一个实施方案中,r3为卤素自由基或烷基基团。例如,r3为卤素自由基,诸如氯;r1为h;并且r2为h。又如,r1和r3为卤素自由基,诸如氯,并且r2为h。
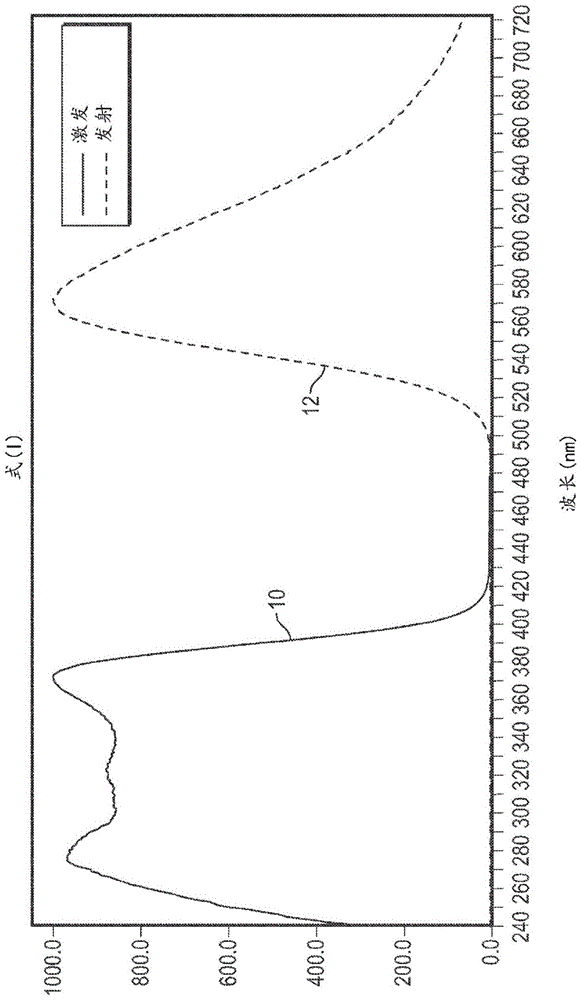
在实施方案中,并且参考图1和图2,本文所述的发光苯并噻唑响应于用约366nm波长的电磁辐射激发而具有约555nm至约630nm(诸如约555nm至约600nm,或诸如约558nm至约589nm)波长内的峰强度。应当理解,可使用在一定波长范围内产生电磁辐射的光源来实现激发,如图1和图2的曲线10所示,前提条件是产生约366波长的发射。参见图1和图2,示出了包含分别为式(i)和式(ii)的发光苯并噻唑的发光颗粒的激发和发射光谱。具体地讲,参见图1,曲线12示出了式(i)的发光苯并噻唑的发射光谱,并且峰值发射为约572nm /-2nm。图2的曲线14示出了式(ii)的发光苯并噻唑的发射光谱,其中卤素自由基存在且为氯,并且峰值发射为约573nm /-2nm。
现在将描述形成发光颗粒的方法的实施方案。可将氨基官能苯并噻唑反应物和有机溶剂装入合适的反应容器中以形成混合物并加热,然后在加热后向混合物中加入含异氰酸酯的化合物。在实施方案中,氨基官能苯并噻唑反应物是氨基苯基苯并噻唑,特别是2-氨基苯基苯并噻唑。合适的有机溶剂包括(但不限于)非质子溶剂,诸如甲基乙基酮。在实施方案中,将氨基官能苯并噻唑反应物和有机溶剂的混合物加热到显著高于环境温度的升高的温度。例如,在实施方案中,将混合物加热至大于约60℃的混合物温度。在实施方案中,在适当的压力下将混合物加热至足够高的混合物温度,以实现稳定的回流条件,其中来自混合物的蒸气在添加含异氰酸酯的化合物之前冷凝并返回至混合物。应当理解,本领域的技术人员可基于混合物的特定组分容易地识别适当的回流条件。在将混合物加热至混合物温度后,加入含异氰酸酯的化合物以形成反应混合物。在实施方案中,含异氰酸酯的化合物为任选地卤代的苯基异氰酸酯。将反应混合物在大于约60℃的温度下保持一段反应期,以形成包含发光苯并噻唑的发光颗粒。在实施方案中,在反应期后,发光颗粒作为沉淀物从反应混合物的液体部分中分离出来,任选洗涤并干燥沉淀物以去除残余的有机溶剂和未反应的反应物。在实施方案中,发光颗粒包含基于发光颗粒的总重量计至少99重量%,诸如至少99.5重量%的量的发光苯并噻唑。在实施方案中,发光苯并噻唑的上述浓度是在发光颗粒形成后实现的,而无需进一步纯化作为干燥沉淀物获得的发光颗粒。据信,上述方法使发光颗粒的收率最大化,其中基于来自反应的发光颗粒的理论收率,发光颗粒以至少约75重量%的收率、诸如至少约78重量%的收率或诸如至少约90重量%的收率形成。
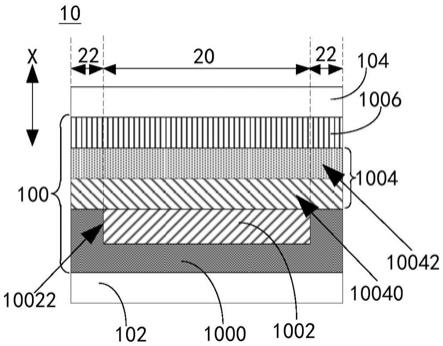
参见图3,包含如本文所述的发光苯并噻唑的发光颗粒100可以用在发光材料300中,该发光材料除了发光颗粒100之外还包含介质302。该介质302可选自油墨、油墨添加剂、胶水、液体、凝胶、聚合物、浆液、塑料、塑性基础树脂、玻璃、陶瓷、金属、纺织物、木材、纤维、纸浆和纸材。由于在介质302中包含发光颗粒100,发光颗粒100可用于通常利用此类介质302作为认证特征的基体材料或作为基材材料本身的安全应用中。例如但不限于,介质302可对应于用于形成制品的基底的材料,或者介质可对应于可施加到(例如,印刷、涂覆、喷涂、或以其他方式粘附到或粘合到)制品基底的表面的材料作为验证特征,或者介质可对应于用于形成嵌入基底内的特征(例如,嵌入验证特征、安全线等)的材料。在前一种情况下,例如,可通过将发光颗粒100与介质302组合,然后与介质302一起形成基底,和/或通过用发光颗粒100的胶态分散体浸渍介质302,来将发光颗粒100掺入到基底材料中。浸渍可例如通过印刷、滴涂、涂覆或喷涂工艺来进行。
现在将参考图4来描述包含发光颗粒的制品的实施方案。图4示出了根据示例性实施方案的包括发光颗粒100的制品400的剖视图。制品400包括基底402和验证特征404、406,该验证特征位于基底402的表面408上或集成在基底402内,其中该验证特征404、406包含发光颗粒100。例如,这可通过将包含介质和发光颗粒100的发光材料掺入到制品400之中或之上来实现。作为另外一种选择,发光材料实际上可用作基底402的基体材料。相反,在发光材料适用于基底402的表面408的实施方案中,发光材料可在预定位置中印刷到基底402的一个或多个表面408上。相反,当发光材料对应于嵌入的验证特征406时,嵌入的验证特征406在基底材料为延展性形式时(例如,当材料为浆液、熔融或非固化形式时)与基底材料集成在一起。以上述方式中的任一种,可将本文所述的发光材料或发光荧光体化合物掺入到制品400中。
如上所述,发光颗粒可掺入到制品400之中或之上。具体地讲,在该实施方案中,制品400可包括表面施加的和/或嵌入的验证特征404、406,该验证特征包含发光颗粒100,和/或制品400可包含发光颗粒100,该颗粒均匀或不均匀地分散在制品400本身的一个或多个部件内(例如,在基底402和/或制品400的一个或多个层或其他部件内)。验证特征404、406和发光颗粒100的的各种相对尺寸在图4中可不按比例绘制。尽管制品400被示为包括表面施加的和/或嵌入的验证特征404、406以及发光颗粒100两者,但另一制品可包括嵌入的验证特征406、表面施加的验证特征404和分散的发光颗粒100的中的一者或组合。最后,尽管图4中仅示出了一个表面施加的验证特征404和一个嵌入的验证特征406,但制品可包括多于一种的任一类型的验证特征404、406。
在各种实施方案中,制品400可为选自以下的任何类型的制品,其包括但不限于,身份证、驾驶执照、护照、身份证明文件、钞票、支票、文档、纸、股票证书、包装部件、信用卡、银行卡、标签、密封件、令牌、卡波诺芯片、邮戳、动物和生物样品。
在各种实施方案中,可为刚性或柔性的基底402可由一个或多个层或部件形成。基底402的多种构型太多而不能提及,因为各个实施方案的发光颗粒100可与大量不同类型的制品结合使用。因此,尽管图4中示出了简单的一体式基底402,但应当理解,基底402可具有多种不同构型中的任一种。例如,基底402可为包括相同或不同材料的多个层或部分的“复合”基底。例如但不限于,基底402可包括一个或多个纸层或部分和一个或多个塑料层或部分,其层合或以其他方式联接在一起以形成复合基底(例如,纸层/塑料层/纸层或塑料层/纸层/塑料层复合基底)。此外,虽然本文讨论了无生命的固体制品,但应当理解,“制品”还可包括人、动物、生物标本、液体样品,以及实施方案的发光材料可包括在其中或其上的实际上任何其他物体或材料。
表面施加的验证特征404可为例如但不限于印刷的验证特征或包括一种或多种刚性或柔性材料的验证特征,如本文所述的发光颗粒100包括在该一种或多种刚性或柔性材料中或包括在该一种或多种刚性或柔性材料上。例如但不限于,表面施加的验证特征404可包括包含发光颗粒100的油墨、颜料、涂层或油漆。另选地,表面施加的验证特征404可包括一种或多种刚性或柔性材料,发光颗粒100包括在该一种或多种刚性或柔性材料中或包括在该一种或多种刚性或柔性材料上,其中表面施加的验证特征404随后粘附或以其他方式附接到基底402的表面408。根据各种实施方案,表面施加的验证特征404可具有约1微米或更大的厚度412,并且表面施加的验证特征404可具有小于或等于基底402的宽度和长度的宽度和长度。
嵌入的验证特征406可包括一种或多种刚性或柔性材料,如本文所述的发光颗粒100包括在该一种或多种刚性或柔性材料中或包括在该一种或多种刚性或柔性材料上。例如但不限于,嵌入的验证特征406可被配置成离散的、刚性或柔性基底,安全线或另一种类型的结构的形式。根据各种实施方案,嵌入的验证特征406可具有在约1微米最多至基底402的厚度416的范围内的厚度422,并且嵌入的验证特征406可具有小于或等于基底402的宽度和长度的宽度和长度。
如上所述,在其他实施方案中,发光颗粒100可均匀地或不均匀地分散在基底402内,如图4所示,或在制品400的一个或多个其他部件内(例如,在制品400的一个或多个层或其他部件内)。发光颗粒100可分散在基底402或其他部件内,其例如但不限于通过将发光颗粒100混合到用于形成基底402或其他部件的材料中,和/或通过用发光颗粒100的胶态分散体浸渍基底402或其他部件来进行,如此前所述的。
本文所讨论的发光苯并噻唑的实施方案的吸收和发射特性与其结合安全性和验证特征的使用一致。例如,使用相对常规的验证设备,发光材料的实施方案可易于激发,并且通过常规技术检测发射。
以下实施例旨在补充而不是限制如上所述的发光颗粒和及其制备方法的描述。
实施例
n-[2-(2-苯并噻唑基)苯基]-n′-苯基-脲(式i)
作为根据本文所述方法制备的发光颗粒的第一示例,根据以下反应方案制备n-[2-(2-苯并噻唑基)苯基]-n′-苯基-脲:
为制备发光颗粒,在室温下向1l三颈烧瓶(配备有温度计、搅拌器和滴液漏斗)中加入200g的2-丁酮(即甲基乙基酮)和40g的2-氨基苯基苯并噻唑(0.176mol)以形成混合物。将混合物加热至平稳回流(混合物变成浅褐色不透明的溶液)。在回流和搅拌下,在75分钟内加入23.1g的苯基异氰酸酯(0.194mol)以形成反应混合物。在添加约1/3的苯基异氰酸酯之后,从溶液中沉淀出亮米色材料。为了更好地搅拌,将反应混合物用150g的2-丁酮稀释。在平稳回流下将反应混合物再搅拌2.5小时。此后,将反应混合物冷却至25℃,并且通过nutsch抽吸分离沉淀物并用300ml丙酮在室温下洗涤。将沉淀物在干燥烘箱中于60℃干燥。干燥后,称量沉淀物为47.9g,基于理论收率,其收率为约78%。通过hplc测定,沉淀物的熔点测定为约212℃至约214℃,dta峰(吸热峰)测定为约228℃,并且纯度(即,发光苯并噻唑含量)测定为99.8%。沉淀物在366nm波长激发下表现出亮黄橙色荧光,其中峰值发射在约572nm处。cie颜色坐标确定为使用得自horibajobinyvongmbh的光谱荧光计fluoromax-4所测量的x=0.508,y=0.486。图1是示出根据该实施例的发光颗粒的激发和发射光谱的曲线图。
n-[2-(2-苯并噻唑基)苯基]-n′-(4-氯苯基)-脲(式ii)
作为根据本文所述方法制备的发光颗粒的另一个示例,根据以下反应方案制备n-[2-(2-苯并噻唑基)苯基]-n′-(4-氯苯基)-脲:
为制备发光颗粒,在室温下向1l三颈烧瓶(配备有温度计、搅拌器和滴液漏斗)中加入200g的2-丁酮(即甲基乙基酮)和40g的2-氨基苯基苯并噻唑(0.176mol)以形成混合物。将混合物加热至平稳回流(混合物变成浅褐色不透明的溶液)。在回流和搅拌下,在75分钟内加入29.8g的4-氯-苯基异氰酸酯(0.194mol)以形成反应混合物。在添加约1/3的4-氯-苯基异氰酸酯之后,从溶液中沉淀出亮米色材料。为了更好地搅拌,将反应混合物用150g的2-丁酮稀释。在平稳回流下将反应混合物再搅拌2.5小时。此后,将反应混合物冷却至25℃,并且通过nutsch抽吸分离沉淀物并用300ml丙酮在室温下洗涤。将沉淀物在干燥烘箱中于60℃干燥。干燥后,称量沉淀物为60.4g,基于理论收率,其收率为约90%。通过高效液相色谱法(hplc)测定,沉淀物的熔点测定为约220℃至约224℃,dta峰(吸热峰)测定为约235℃,并且纯度(即,发光苯并噻唑含量)测定为99.5%。沉淀物在366nm波长激发下表现出亮黄橙色荧光,其中峰值发射在约573nm处。cie颜色坐标确定为使用得自horibajobinyvongmbh的光谱荧光计fluoromax-4所测量的x=0.505,y=0.489。图2是示出根据该实施例的发光颗粒的激发和发射光谱的曲线图。
n-[2-(2-苯并噻唑基)苯基]-n′-(4-甲基苯基)-脲(式iii)
作为根据本文所述方法制备的发光颗粒的另一个示例,根据以下反应方案制备n-[2-(2-苯并噻唑基)苯基]-n′-(4-甲基苯基)-脲:
为制备发光颗粒,在室温下向500ml三颈烧瓶(配备有温度计、搅拌器和滴液漏斗)中加入150g的丁酮和36.9g的2-氨基苯基苯并噻唑(0.163mol)以形成混合物。将混合物加热至平稳回流(混合物变成浅褐色不透明的溶液)。在回流和搅拌下,在100分钟内加入24g的4-甲基-苯基异氰酸酯(0.194mol)以形成反应混合物。在添加约1/3的4-甲基-苯基异氰酸酯之后,从溶液中沉淀出亮米色材料。在平稳回流下将反应混合物再搅拌7小时。此后,将反应混合物冷却至45℃,并且通过nutsch抽吸分离沉淀物并用150ml丙酮在室温下洗涤。将产物在150ml丙酮中回流搅拌30分钟以去除杂质。将沉淀物在干燥烘箱中于60℃干燥。干燥后,称量沉淀物为49.7g,基于理论收率,其收率为约85%。通过高效液相色谱法(hplc)测定,沉淀物的熔点测定为约200℃至约203℃,dta峰(吸热峰)测定为约216℃,并且纯度(即,发光苯并噻唑含量)测定为99.9%。沉淀物在366nm波长激发下表现出亮黄色荧光,其中峰值发射在约569nm处。cie颜色坐标确定为使用得自horibajobinyvongmbh的光谱荧光计fluoromax-4所测量的x=0.502,y=0.493。
n-[2-(2-苯并噻唑基)苯基]-n′-(1-萘基)-脲(式iv)
作为根据本文所述方法制备的发光颗粒的另一个示例,根据以下反应方案制备n-[2-(2-苯并噻唑基)苯基]-n′-(1-萘基)-脲:
为制备发光颗粒,在室温下向500ml三颈烧瓶(配备有温度计、搅拌器和滴液漏斗)中加入150g的丁酮和29.1g的2-氨基苯基苯并噻唑(0.129mol)以形成混合物。将混合物加热至平稳回流(混合物变成浅褐色不透明的溶液)。在回流和搅拌下,在80分钟内加入24g的1-异氰酸萘酯(0.142mol)以形成反应混合物。在添加约2/3的1-异氰酸萘酯之后,从溶液中沉淀出亮米色材料。在平稳回流下将反应混合物再搅拌8.5小时。此后,将反应混合物冷却至25℃,并且通过nutsch抽吸分离沉淀物并用200ml丙酮在室温下洗涤。将产物在200ml丙酮中回流搅拌30分钟以去除杂质。将沉淀物在干燥烘箱中于60℃干燥。干燥后,称量沉淀物为46.4g,基于理论收率,其收率为约91%。通过高效液相色谱法(hplc)测定,沉淀物的熔点测定为约206℃至约250℃,dta峰(吸热峰)测定为约216℃,并且纯度(即,发光苯并噻唑含量)测定为95.3%。沉淀物在366nm波长激发下表现出亮黄色荧光,其中峰值发射在约566nm处。cie颜色坐标确定为使用得自horibajobinyvongmbh的光谱荧光计fluoromax-4所测量的x=0.487,y=0.504。
4-(3-(2-(苯并[d]噻唑-2-基)苯基)脲基)苯甲酸甲酯(式v)
作为根据本文所述方法制备的发光颗粒的另一个示例,根据以下反应方案制备4-(3-(2-(苯并[d]噻唑-2-基)苯基)脲基)苯甲酸甲酯:
为制备发光颗粒,在室温下向500ml三颈烧瓶(配备有温度计、搅拌器和滴液漏斗)中加入150g的丁酮和27.8g的2-氨基苯基苯并噻唑(0.123mol)以形成混合物。将混合物加热至平稳回流(混合物变成浅褐色不透明的溶液)。在回流和搅拌下,在75分钟内加入溶于25g丁酮中的24g的4-异氰酸根合苯甲酸甲酯(0.135mol),以形成反应混合物。在添加约1/3的4-异氰酸根合苯甲酸甲酯之后,从溶液中沉淀出亮米色材料。在平稳回流下将反应混合物再搅拌4小时。此后,将反应混合物冷却至40℃,并且通过nutsch抽吸分离沉淀物并用150ml丙酮在室温下洗涤。将产物在150ml丙酮中回流搅拌30分钟以去除杂质。将沉淀物在干燥烘箱中于60℃干燥。干燥后,称量沉淀物为37.8g,基于理论收率,其收率为约76%。通过高效液相色谱法(hplc)测定,沉淀物的熔点测定为约197℃至约199℃,dta峰(吸热峰)测定为约214℃,并且纯度(即,发光苯并噻唑含量)测定为99.6%。沉淀物在366nm波长激发下表现出亮黄色荧光,其中峰值发射在约566nm处。cie颜色坐标确定为使用得自horibajobinyvongmbh的光谱荧光计fluoromax-4所测量的x=0.484,y=0.507。
n-[2-(2-苯并噻唑基)苯基]-n′-(4-甲氧基苯基)-脲(式vi)
作为根据本文所述方法制备的发光颗粒的另一个示例,根据以下反应方案制备n-[2-(2-苯并噻唑基)苯基]-n′-(4-甲氧基苯基)-脲:
为制备发光颗粒,在室温下向500ml三颈烧瓶(配备有温度计、搅拌器和滴液漏斗)中加入150g的丁酮和33.2g的2-氨基苯基苯并噻唑(0.146mol)以形成混合物。将混合物加热至平稳回流(混合物变成浅褐色不透明的溶液)。在回流和搅拌下,在90分钟内加入24g的4-甲氧基-苯基异氰酸酯(0.161mol)以形成反应混合物。在添加约1/3的4-甲氧基-苯基异氰酸酯之后,从溶液中沉淀出亮米色材料。为了更好地搅拌,将反应混合物用50g的丁酮稀释。在平稳回流下将反应混合物再搅拌5.5小时。此后,将反应混合物冷却至50℃,并且通过nutsch抽吸分离沉淀物并用150ml丙酮在室温下洗涤。将产物在200ml丙酮中回流搅拌30分钟以去除杂质。将沉淀物在干燥烘箱中于60℃干燥。干燥后,称量沉淀物为48.9g,基于理论收率,其收率为约89%。通过高效液相色谱法(hplc)测定,沉淀物的熔点测定为约211℃至约216℃,dta峰(吸热峰)测定为约229℃,并且纯度(即,发光苯并噻唑含量)测定为97.4%。沉淀物在366nm波长激发下表现出亮黄色荧光,其中峰值发射在约565nm处。cie颜色坐标确定为使用得自horibajobinyvongmbh的光谱荧光计fluoromax-4所测量的x=0.487,y=0.506。
n-[2-(2-苯并噻唑基)苯基]-n′-(4-氯苯基)-脲(式vii)
作为根据本文所述方法制备的发光颗粒的另一个示例,根据以下反应方案制备n-[2-(2-苯并噻唑基)苯基]-n′-(4-氯苯基)-脲:
为制备发光颗粒,在室温下向500ml三颈烧瓶(配备有温度计、搅拌器和滴液漏斗)中加入200g的丁酮和40g的2-氨基苯基苯并噻唑(0.176mol)以形成混合物。将混合物加热至平稳回流(混合物变成浅褐色不透明的溶液)。在回流和搅拌下,在85分钟内加入29g的2-甲氧基-苯基异氰酸酯(0.194mol)以形成反应混合物。在平稳回流下将反应混合物再搅拌8.5小时。此后,将反应混合物冷却至40℃,并且通过nutsch抽吸分离沉淀物并用200ml丙酮在室温下洗涤。将产物在200ml丙酮中回流搅拌30分钟以去除杂质。将沉淀物在干燥烘箱中于60℃干燥。干燥后,称量沉淀物为35.7g,基于理论收率,其收率为约54%。通过高效液相色谱法(hplc)测定,沉淀物的熔点测定为约174℃至约176℃,dta峰(吸热峰)测定为约184℃,并且纯度(即,发光苯并噻唑含量)测定为99.5%。沉淀物在366nm波长激发下表现出亮黄橙色荧光,其中峰值发射在约589nm处。cie颜色坐标确定为使用得自horibajobinyvongmbh的光谱荧光计fluoromax一4所测量的x=0.544,y=0.450。
n-[2-(2-苯并噻唑基)苯基]-n′-(3-氯苯基)-脲(式viii)
作为根据本文所述方法制备的发光颗粒的另一个示例,根据以下反应方案制备n-[2-(2-苯并噻唑基)苯基]-n′-(3-氯苯基)-脲:
为制备发光颗粒,在室温下向500ml三颈烧瓶(配备有温度计、搅拌器和滴液漏斗)中加入135g的丁酮和32g的2-氨基苯基苯并噻唑(0.142mol)以形成混合物。将混合物加热至平稳回流(混合物变成浅褐色不透明的溶液)。在回流和搅拌下,在110分钟内加入22.3g的3-氯-苯基异氰酸酯(0.145mol)以形成反应混合物。通过加入80ml丁酮稀释反应混合物,并在平稳回流下再搅拌4.5小时。此后,将反应混合物冷却至45℃,并且通过nutsch抽吸分离沉淀物并用300ml丙酮在室温下洗涤。将产物在200ml丙酮中回流搅拌30分钟以去除杂质。将沉淀物在干燥烘箱中于65℃干燥。干燥后,称量沉淀物为39.8g,基于理论收率,其收率为约74%。通过高效液相色谱法(hplc)测定,沉淀物的熔点测定为约190℃,dta峰(吸热峰)测定为约204℃,并且纯度(即,发光苯并噻唑含量)测定为99.9%。沉淀物在366nm波长激发下表现出亮黄橙色荧光,其中峰值发射在约583nm处。cie颜色坐标确定为使用得自horibajobinyvongmbh的光谱荧光计fluoromax-4所测量的x=0.535,y=0.460。
n-[2-(2-苯并噻唑基)苯基]-n′-(4-氯苯基)-脲(式ix)
作为根据本文所述方法制备的发光颗粒的另一个示例,根据以下反应方案制备n-[2-(2-苯并噻唑基)苯基]-n′-(2-氯苯基)-脲:
为制备发光颗粒,在室温下向500ml三颈烧瓶(配备有温度计、搅拌器和滴液漏斗)中加入150g的丁酮和32.1g的2-氨基苯基苯并噻唑(0.142mol)以形成混合物。将混合物加热至平稳回流(混合物变成浅褐色不透明的溶液)。在回流和搅拌下,在70分钟内加入22.3g的2-氯-苯基异氰酸酯(0.145mol)以形成反应混合物。在添加约1/2的2-氯-苯基异氰酸酯之后,沉淀出亮米色材料。在平稳回流下将反应混合物再搅拌6.5小时。此后,将反应混合物冷却至50℃,并且通过nutsch抽吸分离沉淀物并用220ml丙酮在室温下洗涤。将沉淀物在干燥烘箱中于60℃干燥。干燥后,称量沉淀物为46g,基于理论收率,其收率为约85%。通过高效液相色谱法(hplc)测定,沉淀物的熔点测定为约190℃至约193℃,dta峰(吸热峰)测定为约202℃,并且纯度(即,发光苯并噻唑含量)测定为98.4%。沉淀物在366nm波长激发下表现出亮黄橙色荧光,其中峰值发射在约578nm处。cie颜色坐标确定为使用得自horibajobinyvongmbh的光谱荧光计fluoromax-4所测量的x=0.518,y=0.472。
n-[2-(2-苯并噻唑基)苯基]-n′-(3,5-二氯苯基)-脲(式x)
作为根据本文所述方法制备的发光颗粒的另一个示例,根据以下反应方案制备n-[2-(2-苯并噻唑基)苯基]-n′-(3,5-二氯苯基)-脲:
为制备发光颗粒,在室温下向250ml三颈烧瓶(配备有温度计、搅拌器和滴液漏斗)中加入50g的丁酮和11.4g的2-氨基苯基苯并噻唑(0.051mol)以形成混合物。将混合物加热至平稳回流(混合物变成浅褐色不透明的溶液)。在75-80℃的混合物温度下回流并搅拌下,在90分钟内加入10g的3,5-二氯-苯基异氰酸酯(0.053mol)以形成反应混合物。将反应混合物用另外的30ml丁酮稀释,并在平稳回流下再搅拌3.5小时。为了分解过量的异氰酸酯,加入15g乙醇并在回流下搅拌15分钟。此后,将反应混合物冷却至25℃,并且通过nutsch抽吸分离沉淀物并用100ml丙酮在室温下洗涤。将沉淀物在干燥烘箱中于65℃干燥。干燥后,称量沉淀物为19.1g,基于理论收率,其收率为约91%。通过高效液相色谱法(hplc)测定,沉淀物的熔点测定为约219℃至约221℃,dta峰(吸热峰)测定为约223℃,并且纯度(即,发光苯并噻唑含量)测定为97.1%。沉淀物在366nm波长激发下表现出亮绿黄色荧光,其中峰值发射在约558nm处。cie颜色坐标确定为使用得自horibajobinyvongmbh的光谱荧光计fluoromax-4所测量的x=0.453,y=0.537。
n-[2-(2-苯并噻唑基)苯基]-n′-(3,4-二氯苯基)-脲(式xi)
作为根据本文所述方法制备的发光颗粒的另一个示例,根据以下反应方案制备n-[2-(2-苯并噻唑基)苯基]-n′-(3,4-二氯苯基)-脲:
为制备发光颗粒,在室温下向250ml三颈烧瓶(配备有温度计、搅拌器和滴液漏斗)中加入50g的丁酮和11.4g的2-氨基苯基苯并噻唑(0.051mol)以形成混合物。将混合物加热至平稳回流(混合物变成浅褐色不透明的溶液)。在75-80℃的混合物温度下回流并搅拌下,在110分钟内加入溶解于25g丁酮中的10g的3,4-二氯-苯基异氰酸酯以形成反应混合物。将反应混合物用另外的30ml丁酮稀释,并在平稳回流下再搅拌4小时。为了分解过量的异氰酸酯,加入15g乙醇并在回流下搅拌15分钟。此后,将反应混合物冷却至25℃,并且通过nutsch抽吸分离沉淀物并用150ml丙酮在室温下洗涤。将沉淀物在干燥烘箱中于65℃干燥。干燥后,称量沉淀物为17g,基于理论收率,其收率为约81%。通过高效液相色谱法(hplc)测定,沉淀物的熔点测定为约202℃至约205℃,dta峰(吸热峰)测定为约213℃,并且纯度(即,发光苯并噻唑含量)测定为99.4%。沉淀物在366nm波长激发下表现出亮黄色荧光,其中峰值发射在约564nm处。cie颜色坐标确定为使用得自horibajobinyvongmbh的光谱荧光计fluoromax-4所测量的x=0.485,y=0.509。
1-(2-(苯并[d]噻唑-2-基)-4-氯苯基)-3-(3-氯苯基)脲(式xii)
作为根据本文所述方法制备的发光颗粒的另一个示例,根据以下反应方案制备1-(2-(苯并[d]噻唑-2-基)-4-氯苯基)-3-(3-氯苯基)脲:
为制备发光颗粒,在室温下向250ml三颈烧瓶(配备有温度计、搅拌器和滴液漏斗)中加入50g的丁酮和13g的2-氨基-5-氯苯基苯并噻唑(0.05mol)以形成混合物。将混合物加热至平稳回流(混合物变成浅褐色不透明的溶液)。在75-80℃的混合物温度下回流并搅拌下,在65分钟内加入溶解于20g丁酮中的8.4g的3-氯-苯基异氰酸酯以形成反应混合物。将反应混合物用另外的20ml丁酮稀释,并在平稳回流下再搅拌3.5小时。为了分解过量的异氰酸酯,加入15g乙醇并在回流下搅拌15分钟。此后,将反应混合物冷却至30℃,并且通过nutsch抽吸分离沉淀物并用100ml丙酮在室温下洗涤。将沉淀物在干燥烘箱中于65℃干燥。干燥后,称量沉淀物为19.4g,基于理论收率,其收率为约93%。通过高效液相色谱法(hplc)测定,沉淀物的熔点测定为约250℃,dta峰(吸热峰)测定为约253℃,并且纯度(即,发光苯并噻唑含量)测定为99.2%。沉淀物在366nm波长激发下表现出亮黄色荧光,其中峰值发射在约573nm处。cie颜色坐标确定为使用得自horibajobinyvongmbh的光谱荧光计fluoromax-4所测量的x=0.517,y=0.478。
1-(2-(苯并[d]噻唑-2-基)-4-氯苯基)-3-苯基脲(式xiii)
作为根据本文所述方法制备的发光颗粒的另一个示例,根据以下反应方案制备1-(2-(苯并[d]噻唑-2-基)-4-氯苯基)-3-苯基脲:
为制备发光颗粒,在室温下向250ml三颈烧瓶(配备有温度计、搅拌器和滴液漏斗)中加入50g的丁酮和13g的2-氨基-5-氯苯基苯并噻唑(0.05mol)以形成混合物。将混合物加热至平稳回流(混合物变成浅褐色不透明的溶液)。在75-80℃的混合物温度下回流并搅拌下,在70分钟内加入溶解于20g丁酮中的6.5g的苯基异氰酸酯以形成反应混合物。为完成反应,加入2ml的苯基异氰酸酯,并且在回流下再搅拌4.5小时。为了分解过量的异氰酸酯,加入10g乙醇并在回流下搅拌15分钟。此后,将反应混合物冷却至35℃,并且通过nutsch抽吸分离沉淀物并用100ml丙酮在室温下洗涤。将沉淀物在干燥烘箱中于65℃干燥。干燥后,称量沉淀物为16.2g,基于理论收率,其收率为约85%。通过高效液相色谱法(hplc)测定,沉淀物的熔点测定为约257℃,dta峰(吸热峰)测定为约257℃,并且纯度(即,发光苯并噻唑含量)测定为98.8%。沉淀物在366nm波长激发下表现出亮黄色荧光,其中峰值发射在约589nm处。cie颜色坐标确定为使用得自horibajobinyvongmbh的光谱荧光计fluoromax-4所测量的x=0.558,y=0.435。
包括用发光颗粒染色的纤维的发光材料
在染色工序中,将900g水中的50g聚酰胺微纤维(22dtex/3mm)用0.2g的式i的发光颗粒在60-85℃下处理4小时。
然后分离纤维并干燥,并且所得纤维为无色的。纤维显示具有582nm处最大荧光的红移。cie颜色坐标(x=0.503,y=0.445)
虽然在前述具体实施方式中已呈现至少一个示例性实施方案,但应当理解存在大量的变型形式。还应当理解,一个示例性实施方案或多个示例性实施方案仅是示例,并且不旨在以任何方式限制范围、适用性或配置。相反,前述具体实施方式将为本领域的技术人员提供一种用于实现示例性实施方案的便利路线图。应当理解,在不脱离如所附权利要求书中阐述的范围的情况下,可对示例性实施方案中描述的元件的功能和布置进行各种改变。
本文用于企业家、创业者技术爱好者查询,结果仅供参考。