1.本发明涉及一种偏振分束器用树脂制棱镜。
背景技术:
2.偏振分束器(pbs:polarizing beam splitter)是能够将入射光分割成p偏振光分量和s偏振光分量的偏振光分离元件,用于光学拾取器、反射型液晶投影仪等。近年来,被称为vr(virtual reality;虚拟现实感)、ar(augmented reality;增强现实感)的各种电子技术得到迅速发展,作为其图像显示装置开始普及头戴式显示器(hmd:head mounted display)产品,提出了在其图像投影部或图像显示部配置有偏振分束器的光学系统(专利文献1~2)。
3.另一方面,头戴式显示器是穿戴在头部而使用的图像显示装置,因此要求小型、轻量且在穿戴时的不快感较少。因此,以减轻棱镜形状的偏振分束器的目的,提出了将以往的玻璃制棱镜改变成树脂的方案。由于树脂在成型加工时产生的取向和应力畸变而容易产生双折射,因此提出了使用如下树脂制棱镜的方案,所述树脂制棱镜通过使用难以产生应力畸变的棱镜形成方法或难以产生双折射的树脂材料来降低了双折射(专利文献3~4)。
4.现有技术文献
5.专利文献
6.专利文献1:日本特开2002-116409号公报。
7.专利文献2:日本特开2015-161737号公报。
8.专利文献3:日本特表2015-532729号公报。
9.专利文献4:日本特开2018-156081号公报。
技术实现要素:
10.但是,本发明人们进行了研究,其结果发现,在使用树脂制棱镜的偏振分束器中,仅通过减少棱镜的双折射无法得到足够清晰的影像。
11.因此,本发明的目的在于,提供一种获得清晰的影像的偏振分束器用树脂制棱镜。
12.本发明人们为了解决上述课题而进行了深入研究,其结果发现,在使用由低双折射性树脂形成的棱镜而形成的偏振分束器中,意外地包含于棱镜中的荧光发光性物质的含量会影响到通过偏振分束器而获得的影像的清晰度,从而完成了本发明。
13.即,本发明为如下所示。
14.[1]一种偏振分束器用树脂制棱镜,其特征在于,
[0015]
相位差的绝对值的平均值为10nm以下,
[0016]
根据对溶解于三氯甲烷而获得的2.0质量%的溶液以激发波长436nm测定时的波长530nm处的荧光强度,使用荧光素的乙醇溶液的浓度-强度换算式来求出的荧光发光性物质的含量为0.1
×
10-9
~4.0
×
10-9
mol/l。
[0017]
[2]根据[1]所述的偏振分束器用树脂制棱镜,其中,玻璃化转变温度tg为大于120
℃并且在160℃以下。
[0018]
[3]根据[1]或[2]所述的偏振分束器用树脂制棱镜,其中,光弹性系数的绝对值为3.0
×
10-12
pa-1
以下。
[0019]
[4]根据[1]~[3]中任一项所述的偏振分束器用树脂制棱镜,其中,包含甲基丙烯酸系树脂。
[0020]
[5]根据[4]所述的偏振分束器用树脂制棱镜,其中,所述甲基丙烯酸系树脂包含具有结构单元x的甲基丙烯酸系树脂,所述结构单元x是主链上具有环结构的结构单元。
[0021]
[6]根据[5]所述的偏振分束器用树脂制棱镜,其中,所述结构单元x包含从由源自n-取代马来酰亚胺单体的结构单元、戊二酰亚胺系结构单元以及内酯环结构单元所构成的组中选出的至少一种的结构单元。
[0022]
[7]根据[5]所述的偏振分束器用树脂制棱镜,其中,所述结构单元x包含源自n-取代马来酰亚胺单体的结构单元。
[0023]
[8]根据[5]所述的偏振分束器用树脂制棱镜,其中,所述结构单元x包含戊二酰亚胺系结构单元。
[0024]
根据本发明,能够提供一种获得清晰的影像的偏振分束器用树脂制棱镜。
附图说明
[0025]
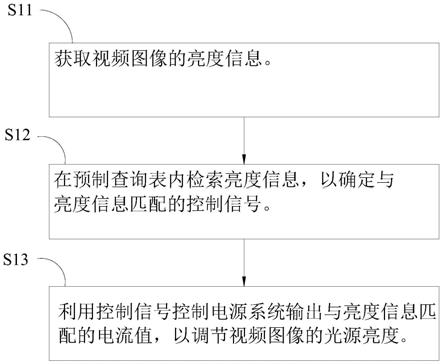
图1是用于评价在实施例中使用了本发明的偏振分束器用树脂制棱镜的偏振分束器的特性而制造的实验系统的示意图。
[0026]
其中,附图标记的说明如下。
[0027]
1:蓝色led;2:准直透镜;3:偏振分束器;4:反射型液晶面板(lcos);5:投影透镜;6:屏幕;31、32:树脂制棱镜;33:反射型偏光膜。
具体实施方式
[0028]
以下,对本具体实施方式(以下,称为“本实施方式”。)进行详细说明,但本发明并不限定于以下的记载,在其主旨的范围内能够进行各种变形而实施。
[0029]
[偏振分束器用树脂制棱镜]
[0030]
本实施方式的偏振分束器用树脂制棱镜的特征在于,棱镜的相位差的绝对值的平均值为10nm以下,根据对溶解于三氯甲烷而获得的2.0质量%的溶液以激发波长436nm测定时的波长530nm处的荧光强度,使用荧光素的乙醇溶液的浓度-强度换算式来求出的荧光发光性物质的含量为0.1
×
10-9
~4.0
×
10-9
mol/l。
[0031]-棱镜的相位差-[0032]
关于本实施方式中的偏振分束器用树脂制棱镜,棱镜的相位差的绝对值的平均值为10nm以下,优选为8nm以下,更优选为6nm以下。相位差的绝对值的平均值为10nm以下,由此能够获得消光系数高且提供清晰的影像的偏振分束器。
[0033]
此处,棱镜的相位差是通过与制成偏振分束器的情况下透射光在棱镜内的光程长度相等的光程长度来测定的值,具体而言,可以利用后述的实施例所述的方法来测定。
[0034]-荧光发光性物质的含量-[0035]
关于本实施方式中的偏振分束器用树脂制棱镜,从对在三氯甲烷中溶解棱镜而获
得的2.0质量%的溶液以激发波长436nm测定时的波长530nm处的荧光强度,使用荧光素的乙醇溶液的浓度-强度换算式来求出的荧光发光性物质的含量为0.1
×
10-9
~4.0
×
10-9
mol/l,优选为0.2
×
10-9
~3.5
×
10-9
mol/l,更优选为0.3
×
10-9
~3.0
×
10-9
mol/l,进一步优选为0.3
×
10-9
~2.5
×
10-9
mol/l。荧光发光性物质的含量为上述上限以下,由此能够获得色调再现度高的影像,影像能够清晰地辨别而不模糊。另外,荧光发光性物质的含量为上述下限以上,由此能够抑制对肉眼有害的低波长的光以必要以上进入肉眼。
[0036]
当荧光强度高(荧光发光性物质的含量多)时模糊地看见影像的原因未明确,但推定为通过透过棱镜中的光来产生荧光的情况下,光也朝向原来的透过光的进行方向以外的方向,由此模糊地看见影像。
[0037]
需要说明的是,关于荧光发光性物质的含量,在树脂制棱镜的表面上被实施有硬涂层或防反射涂层的情况下,去除这些涂层之后,进行测定。具体而言,能够利用后述的实施例所述的方法来测定。
[0038]-玻璃化转变温度-[0039]
本实施方式中的偏振分束器用树脂制棱镜的玻璃化转变温度(tg),优选大于120℃且在160℃以下。
[0040]
从针对从头戴式显示器等的显示设备类发热的耐热性的观点、尺寸变化所伴随的光弹性双折射的发生的方面出发,优选偏振分束器用树脂制棱镜的玻璃化转变温度大于120℃。玻璃化转变温度(tg),从使用环境温度下的尺寸稳定性的方面出发,更优选为125℃以上,进一步优选为130℃以上。
[0041]
另一方面,在玻璃化转变温度(tg)为160℃以下的情况下,能够避免在极端高温的熔融加工,抑制树脂等的热分解,能够获得良好的产品。玻璃化转变温度(tg),从更加获得上述的效果的观点出发,优选为150℃以下,更优选为140℃以下。
[0042]
需要说明的是,玻璃化转变温度(tg)可以依据jis-k7121进行测定来确定。具体而言,可以利用后述的实施例所述的方法来求出。
[0043]-光弹性系数c
r-[0044]
本实施方式的偏振分束器用树脂制棱镜的光弹性系数c
r
的绝对值|c
r
|,优选为3.0
×
10-12
pa-1
以下,更优选为2.0
×
10-12
pa-1
以下,进一步优选为1.5
×
10-12
pa-1
以下,进一步优选为1.0
×
10-12
pa-1
以下。关于光弹性系数,在各种文献中有记载(例如,参照《化学总说》,no.39,1998(学会出版中心发行)),并由下述式(i-a)和(i-b)定义。可知光弹性系数c
r
的值越接近零,因外力的双折射变化越小。
[0045]
|c
r
|=|δn|/σ
r
(i-a)
[0046]
|δn|=|nx-ny|(i-b)
[0047]
(式中,c
r
表示光弹性系数,σ
r
表示拉伸应力,|δn|表示双折射的绝对值,nx表示拉伸方向的折射率,ny表示在面内与拉伸方向相垂直的方向的折射率。)
[0048]
只要本实施方式的树脂制棱镜的光弹性系数c
r
的绝对值|c
r
|为3.0
×
10-12
pa-1
以下,能够获得在固定棱镜时产生的应力、随着尺寸温度变化而产生的光弹性双折射充分小、可获得清晰的影像的树脂制棱镜。
[0049]
需要说明的是,关于光弹性系数c
r
的测定,细切割树脂制棱镜之后,使用真空压缩成型机使其成为压膜来进行。在树脂制棱镜的表面上被实施有硬涂层或防反射涂层的情况
下,去除这些涂层之后,进行测定。具体而言,由后述的实施例所述的方法来求出。
[0050]-透光率-[0051]
关于本实施方式的偏振分束器用树脂制棱镜,对于使用分光色彩计以d65光源10
°
视野测定的最厚部中的透光率,波长450nm的透过率(t450)相对于波长680nm的透过率(t680)的比例(t450/t680)优选为0.95~1.03,更优选为0.97~1.01,进一步优选为0.98~1.00。如果比例(t450/t680)为上述范围内,则能够获得良好的色调的影像。
[0052]
需要说明的是,关于透光率,具体而言,可以通过后述的实施例所述的方法来测定。
[0053]-分子量和分子量分布-[0054]
本实施方式中的偏振分束器用树脂制棱镜的由凝胶渗透色谱(gpc)测定的聚甲基丙烯酸甲酯换算的重均分子量(mw),优选为80,000~170,000的范围,更优选为100,000~160,000的范围,更加优选为100,000~150,000的范围,进一步优选为120,000~150,000的范围。如果重均分子量(mw)为上述范围,则机械强度和流动性的平衡也优异。
[0055]
需要说明的是,树脂制棱镜的重均分子量(mw)、数均分子量(mn)、z均分子量(mz)可以以下述的装置和条件测定。
[0056]
·
测定装置:东曹株式会社制凝胶渗透色谱(hlc-8320gpc)。
[0057]
·
测定条件如下所示。
[0058]
色谱柱:依次串联连接一根tskguardcolumn superh-h、两根tskgel superhm-m、一根tskgel superh2500来使用。
[0059]
色谱柱温度:40℃。
[0060]
展开剂:四氢呋喃,流速:0.6ml/min,作为内部标准,以0.1g/l添加2,6-二叔丁基-4-甲基苯酚(bht)。
[0061]
检测器:ri(差示折射)检测器。
[0062]
检测灵敏度:3.0mv/分钟。
[0063]
样品:0.02g的树脂制棱镜的四氢呋喃20ml溶液。
[0064]
进样量:10μl。
[0065]
检量线用标准样品:使用单分散的重量峰分子量已知且分子量不同的以下十种聚甲基丙烯酸甲酯(聚合物实验室(polymerlaboratories)制;pmma calibration kit m-m-10)。
[0066]
重量峰分子量(mp)
[0067]
标准试样11,916,000标准试样2625,500标准试样3298,900标准试样4138,600标准试样560,150标准试样627,600标准试样710,290标准试样85,000标准试样92,810
标准试样10850
[0068]
在上述的条件下,测定相对于树脂制棱镜的洗脱时间的ri检测强度。
[0069]
以上,基于通过检量线用标准样品的测定来获得的各检量线,求出树脂制棱镜的重均分子量(mw)、数均分子量(mn)和z均分子量(mz),并使用其值而确定分子量分布(mw/mn)和(mz/mw)。需要说明的是,在树脂制棱镜的表面上被实施有硬涂层或防反射涂层的情况下,去除这些涂层之后,进行测定。
[0070]-甲醇不溶组分-[0071]
本实施方式中的偏振分束器用树脂制棱镜的甲醇不溶组分的量,相对于甲醇不溶组分的量与甲醇可溶组分的量的总量100质量%的比例,优选为95质量%以上,更优选为95.5质量%以上,进一步优选为96质量%以上,更进一步优选为96.5质量%以上,特别优选为97质量%以上,最优选为97.5质量%以上。通过将甲醇不溶组分的量的比例设为95质量%以上,能够抑制注射成型时的银纹发生等成型时的麻烦。
[0072]
需要说明的是,甲醇不溶组分和甲醇可溶组分是指,将偏振分束器用树脂制棱镜溶解于三氯甲烷而制成三氯甲烷溶液以后,通过将溶液滴加到大过量的甲醇中来进行再沉淀,区分滤液和滤渣,此后通过使各自干燥来获得的组分。在树脂制棱镜的表面上被实施有硬涂层或防反射涂层的情况下,去除这些涂层之后,进行测定。
[0073]
具体而言,可以如下求出。将树脂制棱镜5g溶解于三氯甲烷100ml以后,将溶液加入滴液漏斗,历时约1小时滴加到用搅拌子进行搅拌的1l的甲醇中,进行再沉淀。滴加全部量之后,静置1小时,然后将膜过滤器(先科东洋株式会社(
アドバンテック
东洋株式会社)制造,t050a090c)用作过滤器,进行吸滤。滤渣在60℃真空干燥16小时而作为甲醇不溶组分。另外,关于滤液,将旋转蒸发器的浴温设为40℃,将真空度从初期设定的390torr逐渐下降而最终设为30torr,去除溶剂以后,回收在茄型烧瓶中残留的可溶组分,作为甲醇可溶组分。称量甲醇不溶组分的质量和甲醇可溶组分的质量的各自质量,算出甲醇可溶组分的量相对于甲醇可溶组分的量与甲醇不溶组分的量的总量(100质量%)的比例(质量%)(甲醇可溶组分率)。
[0074]
[树脂组合物]
[0075]
本实施方式的偏振分束器用树脂制棱镜,优选由包含甲基丙烯酸系树脂的树脂组合物构成。
[0076]
[甲基丙烯酸系树脂]
[0077]
本实施方式的偏振分束器用树脂制棱镜所包含的甲基丙烯酸系树脂,优选包含具有在主链上具有环结构的结构单元x和源自甲基丙烯酸酯单体的结构单元的甲基丙烯酸系树脂。通过包含甲基丙烯酸系树脂、特别是包含具有在主链上具有环结构的结构单元x和甲基丙烯酸酯单体单元的甲基丙烯酸系树脂,由此能够获得棱镜的相位差充分小且光弹性系数也充分小的树脂制棱镜。
[0078]
以下,对各结构单元进行说明。
[0079]-源自甲基丙烯酸酯单体的结构单元-[0080]
作为源自甲基丙烯酸酯单体的结构单元,例如,可以举出源自选自以下所示的甲基丙烯酸酯类中的单体的结构单元。作为甲基丙烯酸酯,例如,可以举出甲基丙烯酸甲酯、甲基丙烯酸乙酯、甲基丙烯酸正丙酯、甲基丙烯酸异丙酯、甲基丙烯酸正丁酯、甲基丙烯酸
异丁酯、甲基丙烯酸叔丁酯、甲基丙烯酸2-乙基己酯、甲基丙烯酸环戊酯、甲基丙烯酸环己酯、甲基丙烯酸环辛酯、甲基丙烯酸三环癸酯、甲基丙烯酸二环辛酯、甲基丙烯酸三环十二烷酯、甲基丙烯酸异冰片酯、甲基丙烯酸苯酯、甲基丙烯酸苄酯、甲基丙烯酸1-苯基乙酯、甲基丙烯酸2-苯氧基乙酯、甲基丙烯酸3-苯基丙酯、甲基丙烯酸2,4,6-三溴苯酯等。
[0081]
这些单体,有时单独使用,也有时合用两种以上。
[0082]
作为源自上述甲基丙烯酸酯单体的结构单元,从所获得的甲基丙烯酸系树脂的透明性和耐候性优异的方面来看,优选为源自甲基丙烯酸甲酯和甲基丙烯酸苄酯的结构单元。
[0083]
源自甲基丙烯酸酯单体的结构单元,既可以含有一种,又可以含有两种以上。
[0084]
本实施方式的偏振分束器用树脂制棱镜,通过适当地调节主链上具有环结构的结构单元x与源自甲基丙烯酸酯单体的结构单元的比率,降低因成型时的取向、残余应力而产生的双折射,能够获得相位差的绝对值的平均值为10nm以下的树脂制棱镜。另外,通过适当地调节上述比率,能够对甲基丙烯酸系树脂充分地赋予耐热性。从这些观点出发,作为源自甲基丙烯酸酯单体的结构单元的含量,以甲基丙烯酸系树脂为100质量%计,优选为50~97质量%,更优选为55~97质量%,进一步优选为55~95质量%,进一步更优选为60~93质量%,特别优选为60~90质量%。
[0085]
需要说明的是,源自甲基丙烯酸酯单体的结构单元的含量,可以通过1h-nmr测定和
13
c-nmr测定来求出。例如,可以作为测定溶剂使用cdcl3或dmso-d6,在测定温度40℃进行1h-nmr测定和
13
c-nmr测定。
[0086]
以下,对主链上具有环结构的结构单元x进行说明。
[0087]
作为主链上具有环结构的结构单元x,优选包含选自由源自n-取代马来酰亚胺单体的结构单元、戊二酰亚胺系结构单元以及内酯环结构单元构成的组中至少一种结构单元,更优选为仅由选自由源自n-取代马来酰亚胺单体的结构单元、戊二酰亚胺系结构单元以及内酯环结构单元构成的组中的至少一种结构单元构成。主链上具有环结构的结构单元x,既可以是一种,又可以组合复数种以上。
[0088]-源自n-取代马来酰亚胺单体的结构单元-[0089]
接着,对源自n-取代马来酰亚胺单体的结构单元进行说明。
[0090]
源自n-取代马来酰亚胺单体的结构单元,可以是选自由下述式(1)表示的结构单元和下述式(2)表示的结构单元构成的组中的至少一种结构单元,优选的是,由下述式(1)和下述式(2)表示的结构单元的两种结构单元形成。
[0091]
[化学式1]
[0092][0093]
式(1)中,r1表示碳原子数7~14的芳基烷基、碳原子数6~14的芳基的任一种,r2和r3分别独立地表示氢原子、氧原子、硫原子、碳原子数为1~12的烷基或碳原子数6~14的芳基的任一种。
[0094]
另外,在r2或r3为芳基的情况下,r2或r3可以作为取代基包含卤素原子。
[0095]
另外,r1可以被卤素原子、碳原子数为1~6的烷基、碳原子数为1~6的烷氧基、硝基、苄基等取代基取代。
[0096]
[化学式2]
[0097][0098]
式(2)中,r4表示氢原子、碳原子数3~12的环烷基、碳原子数为1~12的烷基的任一种,r5和r6分别独立地表示氢原子、氧原子、硫原子、碳原子数为1~12的烷基或碳原子数6~14的芳基的任一种。
[0099]
以下,示出具体的例子。
[0100]
作为形成由式(1)表示的结构单元的单体(n-芳基马来酰亚胺类、n-芳香族取代马来酰亚胺等),例如,可以举出n-苯基马来酰亚胺、n-苄基马来酰亚胺、n-(2-氯苯基)马来酰亚胺、n-(4-氯苯基)马来酰亚胺、n-(4-溴苯基)马来酰亚胺、n-(2-甲基苯基)马来酰亚胺、n-(2,6-二甲基苯基)马来酰亚胺、n-(2-乙基苯基)马来酰亚胺、n-(2-甲氧基苯基)马来酰亚胺、n-(2-硝基苯基)马来酰亚胺、n-(2,4,6-三甲基苯基)马来酰亚胺、n-(4-苄基苯基)马来酰亚胺、n-(2,4,6-三溴苯基)马来酰亚胺、n-萘基马来酰亚胺、n-蒽基马来酰亚胺、3-甲基-1-苯基-1h-吡咯-2,5-二酮、3,4-二甲基-1-苯基-1h-吡咯-2,5-二酮、1,3-二苯基-1h-吡咯-2,5-二酮、1,3,4-三苯基-1h-吡咯-2,5-二酮等。
[0101]
在这些单体中,从所获得的甲基丙烯酸系树脂的耐热性和双折射等光学特性优异的方面出发,优选为n-苯基马来酰亚胺和n-苄基马来酰亚胺。
[0102]
这些单体,有时单独使用,也有时合用两种以上来使用。
[0103]
作为形成由式(2)表示的结构单元的单体,例如,可以举出n-甲基马来酰亚胺、n-乙基马来酰亚胺、n-正丙基马来酰亚胺、n-异丙基马来酰亚胺、n-正丁基马来酰亚胺、n-异丁基马来酰亚胺、n-仲丁基马来酰亚胺、n-叔丁基马来酰亚胺、n-正戊基马来酰亚胺、n-正己基马来酰亚胺、n-正庚基马来酰亚胺、n-正辛基马来酰亚胺、n-月桂基马来酰亚胺、n-环戊基马来酰亚胺、n-环己基马来酰亚胺、1-环己基-3-甲基-1h-吡咯-2,5-二酮、1-环己基-3,4-二甲基-1h-吡咯-2,5-二酮、1-环己基-3-苯基-1h-吡咯-2,5-二酮、1-环己基-3,4-二苯基-1h-吡咯-2,5-二酮等。
[0104]
在这些单体中,从甲基丙烯酸系树脂的耐候性优异的方面出发,优选为n-甲基马来酰亚胺、n-乙基马来酰亚胺、n-异丙基马来酰亚胺、n-环己基马来酰亚胺,从近年来光学材料所要求的低吸湿性优异的方面出发,特别优选为n-环己基马来酰亚胺。
[0105]
这些单体,有时单独使用,也可以合用两种以上来使用。
[0106]
在本实施方式的甲基丙烯酸系树脂中,在能够表现高度控制的双折射特性的方面,特别优选合用由式(1)表示的结构单元与由式(2)表示的结构单元后使用。
[0107]
由式(1)表示的结构单元的含量(x1)相对于由式(2)表示的结构单元的含量(x2)
[0124]
作为主链上具有戊二酰亚胺系结构单元的甲基丙烯酸系树脂,例如,可以举出在日本特开2006-249202号公报、日本特开2007-009182号公报、日本特开2007-009191号公报、日本特开2011-186482号公报、日本再公表专利2012/114718号公报等中记载的具有戊二酰亚胺系结构单元的甲基丙烯酸系树脂,可以通过该公报中记载的方法来形成。
[0125]
本实施方式的构成甲基丙烯酸系树脂的戊二酰亚胺系结构单元,可以在树脂聚合后形成。
[0126]
具体而言,戊二酰亚胺系结构单元,可以设为由下述通式(3)表示的物质。
[0127]
[化学式3]
[0128][0129]
在上述通式(3)中,优选的是,r7和r8分别独立地为氢原子或甲基,r9为氢原子、甲基、丁基、环己基的任一种,更优选的是,r7为甲基,r8为氢原子,r9为甲基。
[0130]
戊二酰亚胺系结构单元,既可以仅包含单一种类,又可以包含复数种种类。
[0131]
在具有戊二酰亚胺系结构单元的甲基丙烯酸系树脂中,以甲基丙烯酸系树脂为100质量%计,戊二酰亚胺系结构单元的含量优选为3~70质量%的范围,更优选为3~60质量%的范围。
[0132]
当戊二酰亚胺系结构单元的含量在上述范围时,能够获得成型加工性、耐热性以及光学特性良好的树脂,因此优选。
[0133]
另外,通过在此范围内适当地调节戊二酰亚胺系结构单元的含量,降低因成型时的取向、残余应力而产生的双折射,能够获得相位差的绝对值的平均值为10nm以下的偏振分束器用树脂制棱镜。最合适的戊二酰亚胺系结构单元的含量因通式(3)的r7~r9的取代基的种类的不同而不同,例如,在r7和r8为氢原子、r9为甲基的情况下,当戊二酰亚胺系结构单元的含量为3~10质量%的范围时,降低因成型时的取向、残余应力而产生的双折射,能够获得相位差的绝对值的平均值为10nm以下的偏振分束器用树脂制棱镜。
[0134]
需要说明的是,甲基丙烯酸系树脂中的戊二酰亚胺系结构单元的含量,可以使用前述的专利文献记载的方法来确定。
[0135]
根据需要,具有戊二酰亚胺系结构单元的甲基丙烯酸系树脂,可以进一步包含芳香族乙烯基单体单元。
[0136]
作为芳香族乙烯基单体,没有特别限定,可以举出苯乙烯、α-甲基苯乙烯,优选为苯乙烯。
[0137]
作为具有戊二酰亚胺系结构单元的甲基丙烯酸系树脂中的芳香族乙烯基单元的含量,没有特别限定,以具有戊二酰亚胺系结构单元的甲基丙烯酸系树脂为100质量%计,优选为0~20质量%。
[0138]
当芳香族乙烯基单元的含量在上述范围时,能够兼具耐热性和优异的光弹性特性,因此优选。
[0139]
例如,在对作为甲基丙烯酸酯单体使用甲基丙烯酸甲酯、作为芳香族乙烯基单体使用苯乙烯进行共聚而获得的甲基丙烯酸甲酯-苯乙烯共聚物进行戊二酰亚胺化而获得树脂的情况下,通过在源自甲基丙烯酸甲酯的结构单元65~90质量%、源自苯乙烯的结构单元5~15质量%、戊二酰亚胺系结构单元5~20质量%的范围内进行调节,降低因成型时的取向和残余应力而产生的双折射,能够获得相位差的绝对值的平均值为10nm以下的偏振分束器用树脂制棱镜。
[0140]-内酯环结构单元-[0141]
主链上具有内酯环结构单元的甲基丙烯酸系树脂,例如,能够通过日本特开2001-151814号公报、日本特开2004-168882号公报、日本特开2005-146084号公报、日本特开2006-96960号公报、日本特开2006-171464号公报、日本特开2007-63541号公报、日本特开2007-297620号公报、日本特开2010-180305号公报等所记载的方法来形成。
[0142]
本实施方式的构成甲基丙烯酸系树脂的内酯环结构单元,可以在树脂聚合后形成。
[0143]
作为本实施方式中的内酯环结构单元,从环结构的稳定性优异的方面出发,优选为六元环。
[0144]
作为六元环的内酯环结构单元,例如,特别优选为由下述通式(4)表示的结构。
[0145]
[化学式4]
[0146][0147]
在上述通式(4)中,r
10
、r
11
和r
12
相互独立地为氢原子或碳原子数为1~20的有机残基。
[0148]
作为有机残基,例如,可以举出甲基、乙基、丙基等碳原子数为1~20的饱和脂肪族烃基(烷基等);乙烯基、丙烯基等碳原子数2~20的不饱和脂肪族烃基(链烯基等);苯基、萘基等碳原子数6~20的芳香族烃基(芳基等);这些饱和脂肪族烃基、不饱和脂肪族烃基、芳香族烃基中的一个以上的氢原子被选自由羟基、羧基、醚基、酯基构成的组中的至少一种基团取代而成的基团;等。
[0149]
例如,通过使具有羟基的丙烯酸系单体与甲基丙烯酸甲酯等甲基丙烯酸酯单体共聚而在分子链上导入羟基与酯基或羧基之后,在这些羟基与酯基或羧基之间产生脱醇(酯化)或脱水缩合(以下,也称为“环化缩合反应”),从而能够形成内酯环结构单元。
[0150]
作为使用于聚合中的具有羟基的丙烯酸系单体,例如,可以举出2-(羟甲基)丙烯酸、2-(羟乙基)丙烯酸、2-(羟甲基)丙烯酸烷基酯(例如,2-(羟甲基)丙烯酸甲酯、2-(羟甲基)丙烯酸乙酯、2-(羟甲基)丙烯酸异丙酯、2-(羟甲基)丙烯酸正丁酯、2-(羟甲基)丙烯酸叔丁酯)、2-(羟乙基)丙烯酸烷基酯等,优选为具有羟烷基部位的单体、即2-(羟甲基)丙烯酸和2-(羟甲基)丙烯酸烷基酯,特别优选为2-(羟甲基)丙烯酸甲酯、2-(羟甲基)丙烯酸乙酯。
[0151]
主链上具有内酯环结构单元的甲基丙烯酸系树脂中的内酯环结构单元的含量,相
对于甲基丙烯酸系树脂100质量%,优选为5~40质量%,更优选为5~35质量%。
[0152]
当内酯环结构单元的含量在此范围时,能够保持成型加工性的同时表现出耐溶剂性提高和表面硬度提高等的环结构导入效果。另外,通过在此范围内适当地调节内酯环结构单元的含量,降低因成型时的取向、残余应力而产生的双折射,能够获得相位差的绝对值的平均值为10nm以下的偏振分束器用树脂制棱镜。
[0153]
需要说明的是,甲基丙烯酸系树脂中的内酯环结构的含量,可以使用前述的专利文献记载的方法来确定。
[0154]
主链上具有内酯环结构单元的甲基丙烯酸系树脂,可以具有源自可与上述甲基丙烯酸酯单体和具有羟基的丙烯酸系单体共聚的其他单体的结构单元。
[0155]
作为这样的可共聚的其他单体,例如,可以举出苯乙烯、乙烯基甲苯、α-甲基苯乙烯、α-羟甲基苯乙烯、α-羟乙基苯乙烯、丙烯腈、甲基丙烯腈、甲基烯丙醇、乙烯、丙烯、4-甲基-1-戊烯、乙酸乙烯酯、2-羟甲基-1-丁烯、甲基乙烯基酮、n-乙烯基吡咯烷酮、n-乙烯基咔唑等的具有聚合性双键的单体等。
[0156]
这些其他单体(构成单元),既可以仅具有一种,又可以具有两种以上。
[0157]
作为这些源自可共聚的其他单体的结构单元的含量,相对于甲基丙烯酸系树脂100质量%,优选为0~20质量%,从耐候性的观点出发,更优选小于10质量%,进一步优选小于7质量%。
[0158]
本实施方式中的甲基丙烯酸系树脂,既可以仅具有一种上述的源自可共聚的其他单体的结构单元,又可以具有两种以上的上述的源自可共聚的其他单体的结构单元。
[0159]
本实施方式中的甲基丙烯酸系树脂,优选具有选自由源自n-取代马来酰亚胺单体的结构单元、戊二酰亚胺系结构单元、内酯环结构单元构成的组中的至少一种结构单元,其中,特别是从容易高超地控制光弹性系数等光学特性而不混合其他热塑性树脂的方面出发,特别优选具有源自n-取代马来酰亚胺单体的结构单元。另外,特别是从获得高强度的树脂制棱镜的观点出发,特别优选具有戊二酰亚胺系结构单元。
[0160]
[甲基丙烯酸系树脂的制造方法]
[0161]
以下,对本实施方式的甲基丙烯酸系树脂的制造方法进行说明。
[0162]-包含源自n-取代马来酰亚胺单体的结构单元的甲基丙烯酸系树脂的制造方法-[0163]
作为主链上具有源自n-取代马来酰亚胺单体的结构单元的甲基丙烯酸系树脂(以下,有时记为”马来酰亚胺共聚物”)的制造方法,可以举出本体聚合法、溶液聚合法、悬浮聚合法、沉淀聚合法、乳液聚合法的任一种聚合方法,从能够减少树脂制棱镜中所包含残留单体量和杂质量的观点出发,优选为悬浮聚合、本体聚合、溶液聚合法,更优选为溶液聚合法。
[0164]
在本实施方式中的制造方法中,作为聚合方式,例如,可以使用间歇聚合法、半间歇法、连续聚合法的任一种。
[0165]
在本实施方式中,从能够降低聚合终止时的残留马来酰亚胺量且减小荧光强度(减小荧光发光性物质的含量)的观点出发,优选使用所谓的半间歇聚合法,所述半间歇聚合法为将单体的一部分在聚合开始前装入到反应器内,通过添加聚合引发剂来开始聚合,然后供给单体的剩余部分的方法。
[0166]
在具有源自n-取代马来酰亚胺单体的结构单元的甲基丙烯酸系树脂中,作为以使获得的树脂制棱镜表示规定的荧光强度(荧光发光性物质的含量成为规定的范围)的方式
进行控制的方法,可以举出进行选自(1)~(3)等中的至少一种:(1)作为原料,使用控制了特定杂质的含量的n-取代马来酰亚胺单体;(2)适用减少在聚合结束后残留的未反应的n-取代马来酰亚胺量的聚合方法;(3)适用降低剪切的脱挥方法。其中,优选选择组合(1)和(3)的制造方法或者选择组合三种的的制造方法。
[0167]
(1)n-取代马来酰亚胺中的杂质的控制
[0168]
作为对于甲基丙烯酸系树脂提供荧光发光性的n-取代马来酰亚胺中的杂质的一种,可以举出n-取代马来酰亚胺与伯胺反应而产生的2-氨基-n-取代琥珀酰亚胺。作为2-氨基-n-取代琥珀酰亚胺,可以举出2-环己基氨基-n-环己基琥珀酰亚胺、2-苯胺基-n-苯基琥珀酰亚胺等。2-氨基-n-取代琥珀酰亚胺不但其本身具有荧光发光性,而且通过发明人们的研究来明确了虽然详细的改性机理不清楚,但如果将此在300℃以上加热或者供给到具有剪切的脱挥装置,则产生具有荧光发光性的热改性物。即,为了控制甲基丙烯酸系树脂和所获得的树脂制棱镜的荧光强度,优选降低n-取代马来酰亚胺中所包含的2-氨基-n-取代琥珀酰亚胺以及在甲基丙烯酸系树脂的制造中使用不具有旋转部的脱挥装置并在300℃以下进行脱挥。
[0169]
作为控制n-取代马来酰亚胺中的杂质的方法,可以举出设置对n-取代马来酰亚胺进行水洗(水洗工序)和/或脱水(脱水工序)的前处理工序等。
[0170]
上述前处理工序既可以仅是水洗,又可以是水洗与脱水的组合。另外,水洗和脱水既可以各进行一次,又可以进行两次以上。在上述前处理工序中,可以进一步设置对由脱水工序获得的n-取代马来酰亚胺溶液进行浓度调整的浓度调整工序。
[0171]
在本实施方式中,例如,通过以下所示的水洗工序来去除n-取代马来酰亚胺中的2-氨基-n-取代琥珀酰亚胺,接着在脱水工序中去除水分,由此能够获得n-取代马来酰亚胺溶液,所述n-取代马来酰亚胺溶液适合于制备控制荧光强度且具有良好的色调的甲基丙烯酸系树脂。
[0172]
在水洗工序中,例如,可以采用如下方法:将n-取代马来酰亚胺溶解于非水溶性有机溶剂,分离成有机层和水层,将此有机层利用酸性水溶液、水、碱性水溶液中的一种以上的液体,通过分次式、连续式或这两种方式来进行混合、清洗,此后分离有机层和水层。
[0173]
在将有机层中的n-取代马来酰亚胺量设为100质量%的情况下,水洗工序后的有机层中的2-氨基-n-取代琥珀酰亚胺量,优选为5质量ppm以下,更优选为0.1质量ppm以上1质量ppm以下。当有机层中的2-氨基-n-取代琥珀酰亚胺量在此范围时,能够将甲基丙烯酸系树脂和所获得的树脂制棱镜中的荧光发光性物质的浓度控制在上述的范围内,能够得到能够获得清晰的影像的偏振分束器用树脂制棱镜,因此优选。另外,从清洗中的成本方面出发也优选。在没有设置脱水工序的情况下,可以将水洗工序中所获得的包含n-取代马来酰亚胺的有机层,作为n-取代马来酰亚胺溶液使用于聚合工序。
[0174]
需要说明的是,关于有机层中的2-氨基-n-取代琥珀酰亚胺量,例如,可以将苯甲酸异丙酯作为内部标准物质,通过气相色谱仪、液相色谱仪等来测定,具体而言,可以通过后述的实施例记载的方法来测定。
[0175]
所使用的非水溶性有机溶剂,只要是溶解n-取代马来酰亚胺和2-氨基-n-取代琥珀酰亚胺、与水进行相分离且具有与水的共沸点的溶剂,就没有特别限制。具体而言,例如,可以使用甲苯、二甲苯等芳香族烃;正己烷、环己烷等脂肪族烃;三氯甲烷、二氯乙烷等卤代
烃;等。
[0176]
所使用的水,可以使用污水、纯水、自来水的任一种。
[0177]
另外,酸性水溶液、碱性水溶液的酸度没有特别限制。
[0178]
水洗工序前的有机层中的n-取代马来酰亚胺的浓度,优选为0.5质量%以上30质量%以下,更优选为10质量%以上25质量%以下,进一步优选为20质量%以上25质量%以下。
[0179]
对有机层和水层进行混合、清洗时的温度,只要是40℃以上即可,优选为40℃以上80℃以下,更优选为50℃以上60℃以下。
[0180]
在将有机层的质量设为100质量%的情况下,水层相对于有机层的质量比例,优选为5质量%以上300质量%以下,更优选为10质量%以上200质量%,进一步优选为30质量%以上100质量%以下。
[0181]
当有机层中的n-取代马来酰亚胺的浓度、清洗时的液温以及水层的质量比例在这些范围时,n-取代马来酰亚胺与水的反应难以进行,并且2-氨基-n-取代琥珀酰亚胺容易向水层侧萃取,因此优选。
[0182]
在以分次式进行清洗的情况下,所使用的反应槽既可以是不锈钢制、搪玻璃制的任一种,又可以使用除了这些以外的反应槽。另外,关于搅拌叶片的形状,没有特别限制。作为具体的搅拌叶片,例如,可以使用三片后掠叶片、四片桨式叶片、四片倾斜桨式叶片、六片涡轮叶片、锚式叶片,另外,也可以使用神钢环境解决方案公司(神钢环境
ソリューション
)制的双星(twin star,
ツインスター
)和全区(full zone,
フルゾーン
)。
[0183]
搅拌时间优选为10分钟以上120分钟以下,更优选为30分钟以上60分钟以下。另外,关于搅拌速度,适当地选择混合液成为紊流状态且不乳化的速度。当搅拌时间、搅拌速度分别在此范围时,能够提高搅拌效率和2-氨基-n-取代琥珀酰亚胺向水层中的萃取效率,能够更加降低n-取代马来酰亚胺中的2-氨基-n-取代琥珀酰亚胺,因此优选。
[0184]
在以连续式进行清洗的情况下,可以使用空塔、填充塔或板式塔,也可以使用静态混合器等静止型混合器、动态混合器等旋转式混合器。
[0185]
有机层与水层的接触时间,优选设定为1秒以上60分钟以下,更优选为30秒以上10分钟以下。当接触时间在此范围时,n-取代马来酰亚胺与水的反应难以进行,并且2-氨基-n-取代琥珀酰亚胺容易向水层侧萃取,因此优选。
[0186]
在脱水工序中,在反应槽中供给有机层,通过在减压下进行加热来去除水分。压力和温度,只要是所使用的溶剂和水形成共沸组分的条件,就没有特别限制。
[0187]
脱水工序后的有机层中的水分含量,在将有机层的质量设为100质量%的情况下,优选为100质量ppm以下。当脱水工序后的水分含量在此范围时,能够抑制甲基丙烯酸系树脂聚合时因水而引起的色调变差,并且能够减少与水一起蒸馏除去的溶剂量,因此从成本方面出发也优选。
[0188]
可以将由脱水工序获得的包含n-取代马来酰亚胺的有机层,作为n-取代马来酰亚胺溶液使用于聚合工序,此外也可以将在浓度调整工序中调整了浓度的n-取代马来酰亚胺溶液使用于聚合工序。
[0189]
在浓度调整工序中,例如,可以将由脱水工序等获得的上述n-取代马来酰亚胺的有机层通过上述非水溶性有机溶剂来稀释。浓度调整工序中所使用的非水溶性有机溶剂,
优选与水洗工序中所使用的非水溶性有机溶剂相同。
[0190]
由上述前处理工序获得的n-取代马来酰亚胺溶液,优选为上述非水溶性有机溶剂的溶液(有机层)。
[0191]
由上述前处理工序所获得的n-取代马来酰亚胺溶液中,相对于上述n-取代马来酰亚胺溶液中所包含的n-取代马来酰亚胺100质量%,上述n-取代马来酰亚胺溶液中所包含的2-氨基-n-取代琥珀酰亚胺的质量比例,优选为5质量ppm以下,更优选为0.1质量ppm以上1质量ppm以下。
[0192]
由上述前处理工序获得的n-取代马来酰亚胺溶液中所包含的水分含量,相对于n-取代马来酰亚胺溶液100质量%,优选为200质量ppm以下,更优选为100~200质量ppm。
[0193]
由上述前处理工序获得的n-取代马来酰亚胺溶液中包含的n-取代马来酰亚胺的质量比例,相对于n-取代马来酰亚胺溶液100质量%,优选为5~30质量%,更优选为5~25质量%。在此范围的情况下,n-取代马来酰亚胺难以析出,能够以均匀的溶液方式转运,因此优选。
[0194]
在聚合工序中使用复数种n-取代马来酰亚胺溶液的情况下,优选复数种n-取代马来酰亚胺溶液的混合液满足上述2-氨基-n-取代琥珀酰亚胺的质量比例、上述水分含量和/或上述n-取代马来酰亚胺的质量比例,更优选各n-取代马来酰亚胺溶液满足上述2-氨基-n-取代琥珀酰亚胺的质量比例、上述水分含量和/或上述n-取代马来酰亚胺的质量比例。
[0195]
通过采用如上所述的n-取代马来酰亚胺的水洗工序和脱水工序,能够减少具有荧光发光性的2-氨基-n-取代琥珀酰亚胺,以及能够去除成为甲基丙烯酸系树脂的色调变差的原因的水分,能够获得即使在光程长度长的棱镜中也具有良好色调的甲基丙烯酸系树脂和该树脂的组合物,因此优选。
[0196]
在聚合工序中,可以在由上述前处理工序获得的n-取代马来酰亚胺溶液中混合甲基丙烯酸酯单体、任意的其他单体、聚合引发剂、聚合溶剂、链转移剂等,制成单体混合液之后,使用于聚合中。
[0197]
(2)聚合结束时的未反应n-取代马来酰亚胺的减少
[0198]
本发明人发现,虽然详细的机理不清楚,但如果甲基丙烯酸系树脂的聚合结束时存在未反应n-取代马来酰亚胺,则在加热的脱挥装置内等中生成作为其结构单元包含n-取代马来酰亚胺的低分子量且具有荧光发光性的反应副产物。
[0199]
为了将甲基丙烯酸系树脂和所获得的树脂制棱镜中的荧光发光性物质的含量控制在上述的范围内,优选将聚合结束后残留的未反应n-取代马来酰亚胺的总质量,相对于聚合结束时的聚合溶液100质量%为1000质量ppm以下,更优选为10质量ppm以上500质量ppm以下。
[0200]
另外,在作为n-取代马来酰亚胺使用n-苯基马来酰亚胺等n-芳基马来酰亚胺类时,聚合结束后残留的未反应n-芳基马来酰亚胺类的总质量,相对于聚合结束时的聚合溶液100质量%,优选为500质量ppm以下,更优选为10质量ppm以上500质量ppm以下,进一步优选为10质量ppm以上50质量ppm以下。
[0201]
在这些范围时,能够将甲基丙烯酸系树脂和所获得的树脂制棱镜中的荧光性物质的含量控制在上述的范围内,因此优选。另外,为了将未反应n-取代马来酰亚胺量设为小于10质量ppm,需要提高聚合温度,或者需要增加聚合引发剂的量,因此马来酰亚胺热改性物、
活性自由基会增加,成为甲基丙烯酸系树脂的色调变差的原因,因此不优选。
[0202]
作为将聚合结束后的未反应n-取代马来酰亚胺量控制上述的范围的手段,可以举出半间歇聚合法。在半间歇聚合法中,优选在聚合工序中从开始添加聚合引发剂至30分钟之后,以供给于聚合的全部单体(例如,甲基丙烯酸酯、n-取代马来酰亚胺以及任意的其他单体)的总质量为100质量%计,补充添加5~35质量%的甲基丙烯酸酯单体。换言之,优选在供给于聚合的全部单体的总质量100质量%中将65~95质量%在聚合引发剂添加前装入反应器内,从开始添加聚合引发剂至30分钟以后补充添加5~35质量%的甲基丙烯酸酯单体的剩余部分。补充添加的甲基丙烯酸酯单体量,以供给于聚合的全部单体的总质量为100质量%计,更优选为10~30质量%。当补充添加的甲基丙烯酸酯单体量在上述范围时,未反应n-取代马来酰亚胺与补充添加的甲基丙烯酸酯单体反应,能够将聚合结束后的未反应n-取代马来酰亚胺量控制在上述的范围内,因此优选。
[0203]
单体的补充添加的开始时刻、补充添加的速度等,只要根据聚合转化率适当地选择即可。另外,在不阻碍本发明的效果、不阻碍减少未反应n-取代马来酰亚胺量的范围,除甲基丙烯酸酯单体之外,可以补充添加包含n-取代马来酰亚胺单体、其他单体的单体混合物。
[0204]
通过采用如上所述的半间歇聚合法,能够减少聚合后半时刻中的未反应的n-取代马来酰亚胺单体量,使脱挥工序中的荧光发光性物质的生成处于最小限度,能够获得即使在光程长度长的棱镜中也具有良好色调的甲基丙烯酸系树脂和该树脂的组合物,因此优选。
[0205]
以下,作为具有源自n-取代马来酰亚胺单体的结构单元的甲基丙烯酸系树脂的制造方法的一例,对使用溶液聚合法以半间歇式由自由基聚合进行制造的情况,进行具体说明。
[0206]
在半间歇聚合法中,优选从开始添加聚合引发剂至30分钟以后,以供给于聚合的全部单体(甲基丙烯酸酯、n-取代马来酰亚胺以及任意的其他单体)的总质量为100质量%计,补充添加5~35质量%的甲基丙烯酸酯单体。换言之,优选在供给于聚合的全部单体的总质量100质量%中将65~95质量%在聚合开始前装入反应器内,从开始添加聚合引发剂至30分钟以后补充添加5~35质量%的甲基丙烯酸酯单体的剩余部分。
[0207]
补充添加的甲基丙烯酸酯单体量,以供给于聚合的全部单体的总质量为100质量%计,更优选为10~30质量%。
[0208]
单体的补充添加的开始时刻、补充添加的速度等,只要根据聚合转化率适当地选择即可。
[0209]
另外,在不阻碍本发明的效果、不阻碍提高n-取代马来酰亚胺单体的转化率的范围,除甲基丙烯酸酯单体以外,可以补充添加包含n-取代马来酰亚胺单体和其他单体的单体混合物。
[0210]
通过采用如上所述的半间歇聚合法,能够提高聚合后半时刻中的n-取代马来酰亚胺单体的转化率,减少荧光发光性物质的含量,能够获得光程长度长的棱镜的透光率优异、容易控制所获得的聚合物的分子量分布、具有特别适合于注射成型的流动性的树脂以及该树脂的组合物,因此优选。
[0211]
作为所使用的聚合溶剂,从提高通过聚合而获得的马来酰亚胺共聚物的溶解度、
防止凝胶化等目的出发,只要是适当地保持反应液的粘度的溶剂,就没有特别限制。
[0212]
作为具体的聚合溶剂,例如,可以使用甲苯、二甲苯、乙基苯、异丙苯等芳香族烃;甲基异丁基酮、丁基溶纤剂、甲基乙基酮、环己酮等酮;二甲基甲酰胺、2-甲基吡咯烷酮等极性溶剂。
[0213]
这些既可以单独使用又可以合用两种以上来使用。
[0214]
另外,在不阻碍聚合时的聚合产物的溶解的范围,可以作为聚合溶剂合用甲醇、乙醇、异丙醇等醇。
[0215]
作为聚合时的溶剂量,只要是使聚合进行、在生产时不发生共聚物和使用单体的析出等且能够容易去除的量,就没有特别限制,例如,在将配合的单体的总量设为100质量%的情况下,优选设为10~200质量%。更优选为25~150质量%,进一步优选为40~100质量%,进一步更优选为50~100质量%。
[0216]
在本实施方式中,也优选使用在将配合的单体的总量设为100质量%的情况下,聚合时的溶剂量在100质量%以下的范围内且在聚合中一边适当地改变溶剂浓度一边进行聚合的方法。
[0217]
更具体而言,可以例示出在聚合初期配合40~60质量%,在聚合途中配合剩余的的60~40质量%,最终在将配合的单体的总量设为100质量%的情况下,使溶剂量成为100质量%以下的范围的方法等。
[0218]
通过采用此方法,能够提高聚合转化率,此外能够控制分子量分布,能够获得注射成型性优异、减少荧光发光性物质的含量、即使在制作光程长度长的棱镜时也获得良好色调的树脂和树脂组合物,因此优选。
[0219]
在溶液聚合中,重要的是尽量减少聚合溶液中的溶解氧浓度,例如,溶解氧浓度,优选为10ppm以下的浓度。溶解氧浓度,例如,可以使用溶解氧计do计b-505(饭岛电子工业株式会社制)进行测定。作为降低溶解氧浓度的方法,可以适当地选择在聚合溶液中鼓泡非活性气体的方法、在聚合前反复进行对包含聚合溶液的容器中以非活性气体加压到0.2mpa左右之后进行放压的操作的方法、在包含聚合溶液的容器中通入非活性气体的方法等的方法。
[0220]
作为聚合温度,只要是使聚合进行的温度,就没有特别限制,优选为70~180℃,更优选为80~160℃,进一步优选为90~150℃,此外更优选为100~150℃。从生产率的观点出发,优选设为70℃以上,为了抑制聚合时的副反应、获得所期望的分子量、品质的聚合物,优选设为180℃以下。
[0221]
另外,关于聚合时间,只要是以需要的转化率能够获得需要的聚合度的时间就没有特别限定,从生产率等的观点出发,优选为2~15小时,更优选为3~12小时,进一步优选为4~10小时。
[0222]
作为聚合引发剂,可以使用通常用于自由基聚合的任意的引发剂,例如,异丙苯过氧化氢、二异丙苯过氧化氢、二叔丁基过氧化物、过氧化月桂酰、过氧化苯甲酰、叔丁基过氧化异丙基碳酸酯、叔戊基过氧化-2-乙基己酸酯、叔戊基过氧化异壬酸酯、1,1-二(叔丁基过氧化)环己烷等有机过氧化物;2,2
’-
偶氮双(异丁腈)、1,1
’-
偶氮双(环己烷甲腈)、2,2
’-
偶氮双(2,4-二甲基戊腈)、二甲基-2,2
’-
偶氮双异丁酸酯等偶氮化合物;等。
[0223]
这些既可以单独使用又可以合用两种以上来使用。
[0224]
作为聚合引发剂的添加量,在将使用于聚合中的单体的总量设为100质量%的情况下,可以是0.01~1质量%,优选为0.05~0.5质量%的范围。
[0225]
作为聚合引发剂的添加方法,只要不是恒定添加速度而是根据在聚合溶液内残留的单体浓度的可变的添加,就没有特别限制,既可以连续地加入又可以间歇地添加。在间歇地添加聚合引发剂的情况下,未添加的时间不考虑每单位时间的添加量。
[0226]
在本实施方式中,优选适当地选择聚合引发剂的种类和添加量、以及聚合温度等,以使因聚合引发剂而产生的自由基总量相对于在反应体系内残留的未反应的单体总量的比例始终成为恒定值以下。
[0227]
通过采用这些方法,也能抑制聚合后期中的低聚物、低分子量物质的生成量,或者也能抑制聚合时的过热而谋求聚合的稳定性。
[0228]
在聚合反应时,可以根据需要添加链转移剂而进行聚合。
[0229]
作为链转移剂,可以使用在通常的自由基聚合中所使用的链转移剂,例如,可以举出正丁基硫醇、正辛基硫醇、正癸基硫醇、正十二烷基硫醇、巯基乙酸2-乙基己酯等硫醇化合物;四氯化碳、二氯甲烷、三溴甲烷等卤素化合物;α-甲基苯乙烯二聚物、α-萜品烯、二戊烯、萜品油烯等不饱和烃化合物;等。
[0230]
这些既可以单独使用又可以合用两种以上来使用。
[0231]
这些链转移剂,只要在聚合反应进行中,就可以在任意阶段添加,没有特别限定。
[0232]
作为链转移剂的添加量,在将使用于聚合中的单体的总量设为100质量%的情况下,可以设为0.01~1质量%,优选为0.05~0.5质量%。
[0233]
作为从由溶液聚合获得的聚合液回收聚合物的方法,可以举出经由称作脱挥工序的工序,分离聚合溶剂和未反应的单体,回收聚合产物的方法。在此,脱挥工序是指,在加热、减压条件下,去除聚合溶剂、残留单体、反应副产物等的挥发成分的工序。
[0234]
在本实施方式中,优选将供给到脱挥工序的包含甲基丙烯酸系树脂的聚合溶液中所包含的未反应的n-取代马来酰亚胺单体的含量控制在规定浓度以下。详细内容如上述的“(2)聚合结束时的未反应n-取代马来酰亚胺的减少”所述,除上述的方法以外,例如,也可以使用:为了提高单体的转化率,尽量延长聚合时间,或者根据聚合溶液中的未反应的单体浓度来变化聚合引发剂的添加速度,从而进行聚合的方法;在聚合中一边适当地改变溶剂浓度一边进行聚合的方法;在聚合后半期补充添加与残留的n-取代马来酰亚胺单体的反应性高的其他单体的方法;在聚合结束时添加α-萜品烯等与n-取代马来酰亚胺的反应性高的化合物的方法等。
[0235]-n-取代马来酰亚胺单体的残留量-[0236]
作为求出在包含甲基丙烯酸系树脂的聚合溶液中残留的n-取代马来酰亚胺单体的残留量的方法,例如,可以采集并称量聚合溶液的一部分,将该试样溶解于三氯甲烷,制备5质量%的溶液,作为内部标准物质添加正癸烷,使用气相色谱仪(岛津制作所制,gc-2010),测定试样中残留的n-取代马来酰亚胺单体的浓度,由此求出n-取代马来酰亚胺单体的残留量。作为更具体的测定条件,可以使用后述的实施例记载的条件。
[0237]
(3)减少剪切的脱挥方法
[0238]
作为使用于上述脱挥工序的装置,优选使用以热交换器和减压容器为主要构成、作为其结构不具有旋转部的脱挥装置。通过不设置旋转部,能够减少脱挥中的剪切,能够获
得荧光发光性更低的树脂。
[0239]
具体而言,可以采用由脱挥槽和排出装置构成的脱挥装置,所述脱挥槽的构成是在其上部配置有热交换器且在具有能脱挥的尺寸的减压容器中附带减压单元,所述排出装置是用于排出脱挥后的聚合物的齿轮泵等。
[0240]
关于上述脱挥装置,将聚合溶液供给到配置于减压容器的上部并被加热的热交换器,例如,多管式热交换器、散热片式热交换器、具有平板型流路和加热器的平板式热交换器等进行预热之后,供给到处于加热、减压下的脱挥槽,分离并去除聚合溶剂、未反应原料混合物、聚合副产物等与聚合物。通过使用如上所述不具有旋转部的脱挥装置,能够抑制源自未反应的n-取代马来酰亚胺的低分子量且具有荧光发光性的反应副产物,能够将甲基丙烯酸系树脂和所获得的树脂制棱镜中的荧光强度(荧光发光性物质的含量)控制在规定的范围内,因此优选。另外,能够获得具有良好色调的甲基丙烯酸系树脂,因此优选。
[0241]
作为上述具有旋转部的装置,可以举出神钢环境解决方案公司(神钢环境
ソリューション
社)制的wiprene(
ワイプレン
)和exeva(
エクセバ
)、日立制作所制的kontro(
コントラ
)和倾斜叶片kontro(
コントラ
)等薄膜蒸发器;附带通气孔的挤出机等。
[0242]
在本实施方式中的脱挥工序中,优选将施加于聚合溶液的剪切速度设为20s-1
以下,更优选设为10s-1
以下,进一步优选以0.1s-1
以上10s-1
以下的方式实施。通过将剪切速度设为0.1s-1
以上,熔融树脂的流动不会过慢,能够抑制因滞留时间的增加而引起的色调变差。另外,通过设为20s-1
以下,能够抑制因剪切而引起的具有荧光发光性的反应副生物的生成。
[0243]
在此,例如,挤出机中的剪切速度γ由下述式计算。
[0244]
γ=(π
×
d
×
n)/h
[0245]
(式中,d表示螺杆直径(m),n表示每1秒的螺杆转速、h表示螺杆沟槽深度(m)。)
[0246]
另外,在平板型流路的情况下,剪切速度γ由下述式计算。
[0247]
γ=(6
×
q)/(w
×
h2)
[0248]
(式中,q表示通过平板型流路的体积流量(m3/s),w表示平板型流路的宽度(m),h表示平板之间的距离(m)。)
[0249]
在本实施方式中,作为配置于减压容器上部的热交换器,优选使用具有平板型流路和加热器的平板式热交换器。更优选为具有平板型流路和加热器的平板式热交换器,该平板型流路具有层叠结构,该层叠结构在同一平面上具有截面为矩形的复数个狭缝状流路。
[0250]
供给到脱挥装置的聚合溶液,从该热交换器的中央部传送到该狭缝状流路而被加热。被加热的聚合溶液从狭缝状流路供给到与热交换器一体化的减压下的减压容器内,而被闪蒸。
[0251]
这样的脱挥方法,有时称作急骤脱挥,在本发明中,以后也记作急骤脱挥。
[0252]
还可以采用串联设置两台以上的上述脱挥装置而以两阶段以上进行脱挥的方法。
[0253]
作为利用附带脱挥装置的热交换器进行加热的温度的范围,可以设为100℃以上300℃以下,优选为甲基丙烯酸系树脂的玻璃化转变温度(tg) 100℃~tg 160℃的温度、更优选为tg 110℃~tg 150℃的温度。作为被加热、保温的脱挥槽的温度的范围,可以设为100℃以上300℃以下,优选为tg 100℃~tg 160℃的温度、更优选为tg 110℃~tg 150℃
的温度。当热交换器和脱挥槽的温度在此范围时,能够抑制残留的2-氨基-n-取代琥珀酰亚胺的热改性,能够抑制荧光发光性物质的生成,因此优选。另外,有效地防止残留挥发成分变多,提高所获得的甲基丙烯酸系树脂的热稳定性、产品品质,因此优选。
[0254]
作为脱挥槽内的真空度,可以设为5~300torr的范围,其中,优选为10~200torr的范围。当此真空度为300torr以下时,能够有效地分离去除未反应单体或未反应单体与聚合溶剂的混合物,所获得的热塑性共聚物的热稳定性、品质不会降低。当真空度为5torr以上时,工业实施更容易。
[0255]
作为脱挥槽内的平均滞留时间,可以是5~60分钟,优选为5~45分钟。当平均滞留时间在此范围时,能够有效地脱挥的同时,能够抑制因聚合物的热改性而引起的着色、分解,因此优选。
[0256]
经过脱挥工序而回收的聚合物,利用称作造粒工序的工序来被加工成颗粒状。
[0257]
在造粒工序中,将熔融状态的树脂,利用选自具有多孔模作为附带设备的齿轮泵、单轴挤出机以及双轴挤出机等中的至少一种搬运造粒装置,以股线状挤出,利用冷切方式、空气热切方式、水中线束切割方式以及水下切割方式加工成颗粒状。从控制因剪切而引起的具有荧光发光性的反应副生物的生成的观点出发,优选选择剪切速度小的搬运装置而不使用挤出机。
[0258]
在本实施方式中,为了获得高度控制的树脂组合物,优选采用使在高温下处于熔融状态的树脂组合物尽量不与空气接触且能迅速冷却固化的造粒方式。
[0259]
在此情况下,更优选在可能的范围降低熔融树脂温度,且极力减少从多孔模出口至冷却水面为止的滞留时间,冷却水的温度也在可能的范围的较高的温度,在能实施的条件下进行造粒。
[0260]
例如,作为熔融树脂温度,优选为220~280℃,更优选为230~270℃,从多孔模出口至冷却水面为止的滞留时间优选为5秒以内,更优选为3秒以内,作为冷却水的温度,优选为30~80℃,更优选为40~60℃的范围。
[0261]
通过在这些熔融树脂温度以及冷却水温度的范围进行实施,能够获得着色更少、所含有的水分率低的甲基丙烯酸系树脂及其组合物,因此优选。
[0262]
从热稳定性和产品品质的观点出发,脱挥工序后的甲基丙烯酸系树脂中残留的单体的含量越少越优选。具体而言,作为甲基丙烯酸酯单体的含量,优选为3000质量ppm以下,更优选为2000质量ppm以下。作为n-取代马来酰亚胺单体的含量,以总量计优选为200质量ppm以下,更优选为100质量ppm以下。
[0263]
另外,作为残留的聚合溶剂的含量,优选为500质量ppm以下,更优选为300质量ppm以下。
[0264]-包含戊二酰亚胺系结构单元的甲基丙烯酸系树脂的制造方法-[0265]
作为主链上具有戊二酰亚胺系结构单元的甲基丙烯酸系树脂的制造方法,可以举出本体聚合法、溶液聚合法、悬浮聚合法、沉淀聚合法、乳液聚合聚合法的任一种聚合方法,优选为悬浮聚合、本体聚合、溶液聚合法,进一步优选为溶液聚合法。
[0266]
在本实施方式中的制造方法中,作为聚合方式,例如,可以使用间歇聚合法、半间歇法、连续聚合法的任一种。
[0267]
在本实施方式中的制造方法中,优选为通过自由基聚合来使单体进行聚合的方
法。
[0268]
主链上具有戊二酰亚胺系结构单元的甲基丙烯酸系树脂,例如,可以是日本特开2006-249202号公报、日本特开2007-009182号公报、日本特开2007-009191号公报、日本特开2011-186482号公报、国际公开第2012/114718号等中记载的具有戊二酰亚胺系结构单元的甲基丙烯酸系树脂,可以通过该公报中记载的方法来形成。
[0269]
以下,对作为具有戊二酰亚胺系结构单元的甲基丙烯酸系树脂的制造方法的一例而使用溶液聚合法以间歇式自由基聚合进行制造的情况,进行具体说明。
[0270]
首先,通过使甲基丙烯酸甲酯等(甲基)丙烯酸酯进行聚合,制造(甲基)丙烯酸酯聚合物。在具有戊二酰亚胺系结构单元的甲基丙烯酸系树脂中包含芳香族乙烯基单元的情况下,使(甲基)丙烯酸酯和芳香族乙烯基(例如,苯乙烯)进行共聚,从而制造(甲基)丙烯酸酯-芳香族乙烯基共聚物。
[0271]
作为使用于聚合中的溶剂,例如,可以举出甲苯、二甲苯、乙基苯等芳香族烃;甲基乙基酮、甲基异丁基酮等酮类;等。
[0272]
这些溶剂,既可以可以单独使用又可以合用两种以上。
[0273]
作为聚合时的溶剂量,只要是使聚合进行、在生产时不引起共聚物和使用单体的析出等且能够容易去除的量,就没有特别限制,例如,在将配合的单体的总量设为100质量%的情况下,优选为10~200质量%。更优选为25~200质量%,进一步优选为50~200质量%,进一步更优选为50~150质量%。
[0274]
作为聚合温度,只要是使聚合进行的温度就没有特别限制,优选为50~200℃,更优选为80~200℃。进一步优选为90~150℃,进一步更优选为100~140℃,更进一步优选为100~130℃。从生产率的观点出发,优选设为70℃以上,为了抑制聚合时的副反应而获得所期望的分子量、品质的聚合物,优选设为180℃以下。
[0275]
作为聚合时间,只要满足目的转化率,就不受特别限制,但从生产率等的观点出发,优选为0.5~15小时,更优选为2~12小时,进一步优选为4~10小时。
[0276]
在聚合反应时,可以根据需要添加聚合引发剂、链转移剂来进行聚合。
[0277]
作为聚合引发剂,没有特别限定,例如,可以利用在具有源自上述n-取代马来酰亚胺单体的结构单元的甲基丙烯酸系树脂的制备方法中公开的聚合引发剂等。
[0278]
这些聚合引发剂,既可以单独使用又可以合用两种以上。
[0279]
这些聚合引发剂,只要在聚合反应进行中,就可以在任意阶段添加。
[0280]
聚合引发剂的添加量,只要根据单体的组合和反应条件等适当地设定即可,没有特别限定,在将使用于聚合中的单体的总量设为100质量%的情况下,可以设为0.01~1质量%,优选为0.05~0.5质量%。
[0281]
作为链转移剂,可以使用在通常的自由基聚合中使用的链转移剂,例如,可以利用在具有源自上述n-取代马来酰亚胺单体的结构单元的甲基丙烯酸系树脂的制备方法中公开的链转移剂等。
[0282]
这些既可以单独使用又可以合用两种以上来使用。
[0283]
这些链转移剂只要在聚合反应进行中,就可以在任意阶段中添加,没有特别限定。
[0284]
关于链转移剂的添加量,只要在能够在使用的聚合条件下获得所期望的聚合度的范围,就没有特别限定,优选的是在将使用于聚合中的单体的总量设为100质量%的情况
下,可以设为0.01~1质量%,优选为0.05~0.5质量%。
[0285]
聚合工序中的合适的聚合引发剂和链转移剂的添加方法,例如,可以设为在具有源自上述n-取代马来酰亚胺单体的结构单元的甲基丙烯酸系树脂的制备方法中记载的方法。
[0286]
聚合溶液中的溶解氧浓度,例如,可以是在具有源自上述n-取代马来酰亚胺单体的结构单元的甲基丙烯酸系树脂的制备方法中公开的值。
[0287]
接着,通过使上述(甲基)丙烯酸酯聚合物或上述甲基丙烯酸酯-芳香族乙烯基共聚物与酰亚胺化剂反应,进行酰亚胺化反应(酰亚胺化工序)。由此,能够制造具有戊二酰亚胺系结构单元的甲基丙烯酸系树脂。
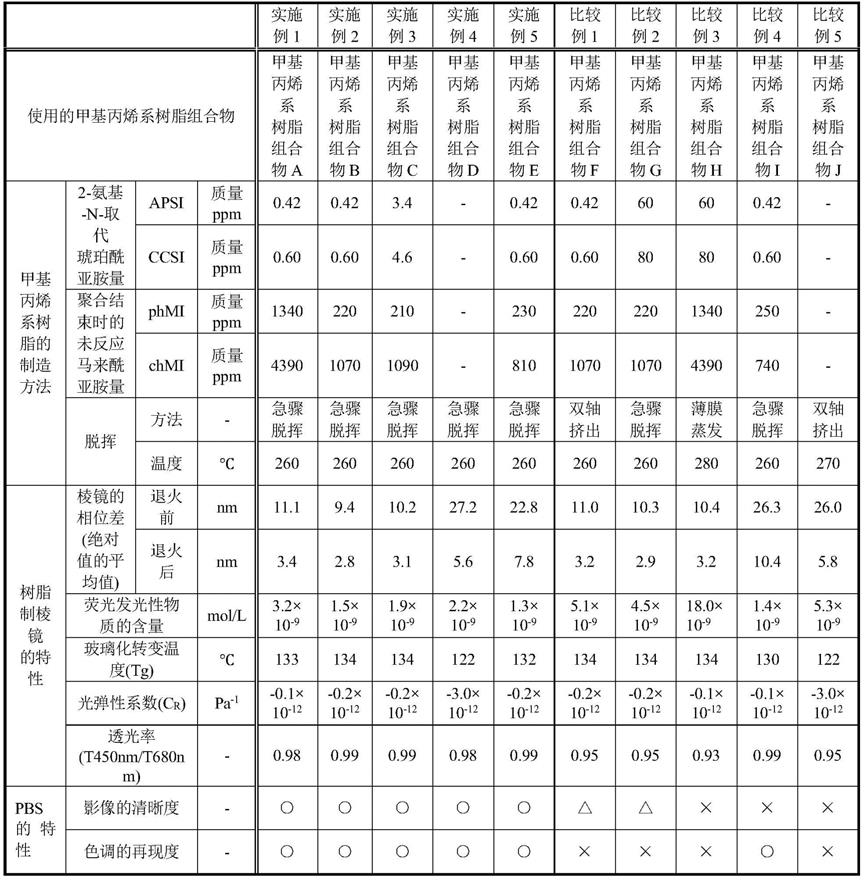
[0288]
作为上述酰亚胺化剂,没有特别限定,只要是能够生成由上述通式(3)表示的戊二酰亚胺系结构单元的酰亚胺化剂即可。
[0289]
作为酰亚胺化剂,具体而言,可以使用氨或伯胺。作为上述伯胺,例如,可以举出甲胺、乙胺、正丙胺、异丙胺、正丁胺、异丁胺、叔丁胺、正己胺等含脂肪族烃基的伯胺;环己胺等含脂环式烃基的伯胺;等。
[0290]
在上述酰亚胺化剂中,从成本、物性的方面出发,优选使用氨、甲胺、环己胺,特别优选使用甲胺。
[0291]
在此酰亚胺化工序中,通过调整上述酰亚胺化剂的添加比例,能够调整所获得的具有戊二酰亚胺系结构单元的甲基丙烯酸系树脂中的戊二酰亚胺系结构单元的含量。
[0292]
为了实施上述酰亚胺化反应的方法,没有特别限定,可以使用以往公知的方法,例如,可以通过使用挤出机或间歇式反应槽来进行酰亚胺化反应。
[0293]
作为上述挤出机,没有特别限定,例如,可以使用单轴挤出机、双轴挤出机、多轴挤出机等。
[0294]
其中,优选使用双轴挤出机。根据双轴挤出机,能够促进原料聚合物与酰亚胺化剂的混合。
[0295]
作为双轴挤出机,例如,可以举出非啮合型同向旋转式、啮合型同向旋转式、非啮合型异向旋转式、啮合型异向旋转式等。
[0296]
上述例示的挤出机,既可以单独使用,又可以串联连接复数个后使用。
[0297]
另外,在使用的挤出机上安装能减压到大气压以下的通气口,这样能够去除反应的酰亚胺化剂、甲醇等副生物或单体类,因此特别优选。
[0298]
在制造具有戊二酰亚胺系结构单元的甲基丙烯酸系树脂时,除上述酰亚胺化的工序之外,可以包括利用二甲基碳酸酯等酯化剂来处理树脂的羧基的酯化工序。此时,可以合用三甲胺、三乙胺、三丁胺等催化剂而进行处理。
[0299]
与上述酰亚胺化工序同样地,例如,可以通过使用挤出机或间歇式反应槽来进行酯化工序。
[0300]
另外,以去除过剩的酯化剂、甲醇等副生物或单体类的目的,优选在使用的装置上安装能减压到大气压以下的通气口。
[0301]
已经过酰亚胺化工序和根据需要而进行的酯化工序的甲基丙烯酸系树脂,从附带多孔模的挤出机以股线状熔融并挤出,利用冷切方式、空气热切方式、水中线束切割方式、水下切割方式等被加工成颗粒状。
[0302]
另外,为了减少树脂的异物数量,也优选使用将甲基丙烯酸系树脂溶解于甲苯、甲基乙基酮、二氯甲烷等有机溶剂,过滤所获得的甲基丙烯酸系树脂溶液,此后对有机溶剂进行脱挥的方法。
[0303]
从减少荧光强度(荧光发光性物质的含量)的观点出发,优选使聚合结束后的聚合溶液在间歇式反应槽中酰亚胺化且不使用会受到剪切力的双轴挤出机。
[0304]
酰亚胺化反应优选在130~250℃实施,更优选在150~230℃实施,进一步优选在170~190℃实施。另外,反应时间,优选为10分钟~5小时,更优选为30分钟~2小时。
[0305]
从减少荧光强度的观点出发,优选在酰亚胺化工序后,根据需要经过酯化工序之后,通过具有源自上述n-取代马来酰亚胺单体的结构单元的甲基丙烯酸系树脂的制备方法中记载的(3)减少剪切的脱挥方法来进行脱挥之后,进行造粒(
ペレタイズ
)。
[0306]-包含内酯环结构单元的甲基丙烯酸系树脂的制造方法-[0307]
作为主链上具有内酯环结构单元的甲基丙烯酸系树脂的制造方法,可以使用在聚合后通过环化反应来形成内酯环结构的方法,优选在促进环化反应的基础上,以使用溶剂的溶液聚合法,通过自由基聚合来对单体进行聚合。
[0308]
在本实施方式中的制造方法中,作为聚合方式,例如,可以使用间歇聚合法、半间歇法、连续聚合法的任一种。
[0309]
主链上具有内酯环结构单元的甲基丙烯酸系树脂,例如,可以通过日本特开2001-151814号公报、日本特开2004-168882号公报、日本特开2005-146084号公报、日本特开2006-96960号公报、日本特开2006-171464号公报、日本特开2007-63541号公报、日本特开2007-297620号公报、日本特开2010-180305号公报等中记载的方法来形成。
[0310]
以下,作为具有内酯环结构单元的甲基丙烯酸系树脂的制造方法的一例,对使用溶液聚合法以间歇式由自由基聚合进行制造的情况,进行具体说明。
[0311]
作为具有内酯环结构单元的甲基丙烯酸系树脂的制造方法,可以使用在聚合后通过环化反应来形成内酯环结构的方法,优选为在促进环化反应的基础上使用溶剂的溶液聚合。
[0312]
作为使用于聚合中的溶剂,例如,可以举出甲苯、二甲苯、乙基苯等芳香族烃;甲基乙基酮、甲基异丁基酮等酮类;等。
[0313]
这些溶剂,既可以单独使用又可以合用两种以上。
[0314]
作为聚合时的溶剂量,只要是使聚合进行、能抑制凝胶化的条件就没有特别限制,例如,在将配合的单体的总量设为100质量%的情况下,优选设为50~200质量%,更优选为100~200质量%。
[0315]
为了充分地抑制聚合液的凝胶化、促进聚合后的环化反应,优选以聚合后所获得的反应混合物中生成的聚合物的浓度成为50质量%以下的方式进行聚合。另外,优选在反应混合物适当地添加聚合溶剂并以上述浓度成为50质量%以下的方式进行控制。
[0316]
作为在反应混合物适当地添加聚合溶剂的方法,没有特别限定,例如,既可以连续地添加聚合溶剂,又可以间歇地添加聚合溶剂。
[0317]
添加的聚合溶剂,既可以是仅一种的单一溶剂又可以是两种以上的混合溶剂。
[0318]
作为聚合温度,只要是使聚合进行的温度就没有特别限制,从生产率的观点出发,优选为50~200℃,更优选为80~180℃。
[0319]
作为聚合时间,只要满足目的转化率,就不受特别限制,从生产率等的观点出发,优选为0.5~10小时,更优选为1~8小时。
[0320]
在聚合反应时,可以根据需要添加聚合引发剂、链转移剂来进行聚合。
[0321]
作为聚合引发剂,没有特别限定,例如,可以利用在具有源自上述n-取代马来酰亚胺单体的结构单元的甲基丙烯酸系树脂的制备方法中公开的聚合引发剂等。
[0322]
这些聚合引发剂,既可以单独使用又可以合用两种以上。
[0323]
这些聚合引发剂,只要在聚合反应进行中,就可以在任意阶段添加。
[0324]
聚合引发剂的添加量,只要根据单体的组合、反应条件等适当地设定即可,没有特别限定,在将使用于聚合中的单体的总量设为100质量%的情况下,可以设为0.05~1质量%。
[0325]
作为链转移剂,可以使用在通常的自由基聚合中使用的链转移剂,例如,可以利用在具有源自上述n-取代马来酰亚胺单体的结构单元的甲基丙烯酸系树脂的制备方法中公开的链转移剂等。
[0326]
这些既可以单独使用又可以合用两种以上来使用。
[0327]
这些链转移剂只要在聚合反应进行中,就可以在任意阶段添加,没有特别限定。
[0328]
作为链转移剂的添加量,只要在能够在使用的聚合条件下获得所期望的聚合度的范围,就没有特别限定,优选的是在将使用于聚合中的单体的总量设为100质量%的情况下,可以设为0.05~1质量%。
[0329]
聚合工序中的合适的聚合引发剂和链转移剂的添加方法,例如,可以是在具有源自上述n-取代马来酰亚胺单体的结构单元的甲基丙烯酸系树脂的制备方法中记载的方法。
[0330]
聚合溶液中的溶解氧浓度,例如,可以是在具有源自上述n-取代马来酰亚胺单体的结构单元的甲基丙烯酸系树脂的制备方法中公开的值。
[0331]
本实施方式中的具有内酯环结构单元的甲基丙烯酸系树脂,可以在上述聚合反应结束后,通过进行环化反应来获得。因此,优选以包含溶剂的状态,供给到内酯环化反应而不从聚合反应液中去除聚合溶剂。
[0332]
通过聚合而获得的共聚物,通过加热处理来引起在共聚物的分子链中存在的羟基(氢氧基)与酯基之间的环化缩合反应,从而形成内酯环结构。
[0333]
在形成内酯环结构的加热处理时,也可以使用具备有用于去除因环化缩合而可能副产的醇的真空装置或脱挥装置的反应装置、具备有脱挥装置的挤出机等。
[0334]
在形成内酯环结构时,为了促进环化缩合反应,可以根据需要使用环化缩合催化剂进行加热处理。
[0335]
作为环化缩合催化剂的具体的例子,例如,可举出亚磷酸甲酯、亚磷酸乙酯、亚磷酸苯酯、亚磷酸二甲酯、亚磷酸二乙酯、亚磷酸二苯酯、亚磷酸三甲酯、亚磷酸三乙酯等亚磷酸单烷基酯、二烷基酯或三酯;磷酸甲酯、磷酸乙酯、磷酸2-乙基己酯、磷酸辛酯、磷酸异癸酯、磷酸月桂酯、磷酸硬脂酯、磷酸异硬脂酯、磷酸二甲酯、磷酸二乙酯、磷酸二-2-乙基己酯、磷酸二异癸酯、磷酸二月桂酯、磷酸二硬脂酯、磷酸二异硬脂酯、磷酸三甲酯、磷酸三乙酯、磷酸三异癸酯、磷酸三月桂酯、磷酸三硬脂酯、磷酸三异硬脂酯等磷酸单烷基酯、二烷基酯或三烷基酯;乙酸锌、丙酸锌、辛基锌等有机锌化合物;等。
[0336]
这些既可以单独使用又可以合用两种以上。
[0337]
作为环化缩合催化剂的使用量,没有特别限定,例如,相对于甲基丙烯酸系树脂100质量%,优选为0.01~3质量%,更优选为0.05~1质量%。
[0338]
当催化剂的使用量为0.01质量%以上时,有效地提高环化缩合反应的反应率,当催化剂的使用量为3质量%以下时,有效地防止所获得的聚合物的着色、聚合物交联而难以熔融成型。
[0339]
作为环化缩合催化剂的添加时期,没有特别限定,例如,既可以在环化缩合反应初期添加,又可以在反应途中添加,还可以在这两个时期添加。
[0340]
在溶剂的存在下进行环化缩合反应时,也可以同时进行脱挥。
[0341]
关于在同时进行环化缩合反应和脱挥工序的情况下使用的装置,没有特别限定,可以是由热交换器和脱挥槽构成的脱挥装置、附带通气孔的挤出机,另外,优选为串联配置脱挥装置和挤出机的装置,更优选为附带通气孔的双轴挤出机。
[0342]
作为使用的附带通气孔的双轴挤出机,优选为具有复数个通气孔口的附带通气孔的挤出机。
[0343]
在使用附带通气孔的挤出机的情况下的反应处理温度,优选为150~350℃,更优选为200~300℃。当反应处理温度小于150℃时,环化缩合反应会变得不充分而会使残留挥发成分变多。相反,当反应处理温度大于350℃时,会引起所获得的聚合物的着色、分解。
[0344]
作为在使用附带通气孔的挤出机的情况下的真空度,优选为10~500torr,更优选为10~300torr。当真空度大于500torr时,有时挥发成分会容易残留。相反,当真空度小于10torr时,工业实施有时会变得困难。
[0345]
在进行上述的环化缩合反应时,也优选以使残留的环化缩合催化剂失活的目的,在造粒时添加有机酸的碱土类金属和/或两性金属盐。
[0346]
作为有机酸的碱土类金属和/或两性金属盐,例如,可以使用乙酰乙酸钙、硬脂酸钙、乙酸锌、辛酸锌、2-乙基己酸锌等。
[0347]
在经过环化缩合反应工序之后,甲基丙烯酸系树脂从附带多孔模的挤出机以股线状熔融并挤出,利用冷切方式、空气热切方式、水中线束切割方式以及水下切割方式加工成颗粒状。
[0348]
需要说明的是,上述的用于形成内酯环结构单元的内酯化,既可以在树脂的制造后树脂组合物的制造(后述)前进行,又可以在树脂组合物的制造中,与树脂与除了树脂以外的成分的熔融混炼一起进行。
[0349]
从减少荧光强度(荧光发光性物质的含量)的观点出发,优选使聚合结束后的聚合溶液在间歇式反应槽中内酯环化且不使用受到剪切力的双轴挤出机。从减少荧光强度的观点出发,优选在内酯环化工序后,通过具有源自上述n-取代马来酰亚胺单体的结构单元的甲基丙烯酸系树脂的制备方法中记载的(3)减少剪切的脱挥方法来进行脱挥之后,进行造粒。
[0350]
[添加剂]
[0351]
本实施方式的构成偏振分束器用树脂制棱镜的树脂组合物,在上述的不显著损害本发明的效果的范围内,可以含有各种添加剂。
[0352]
作为添加剂,没有特别限制,例如,可以举出抗氧化剂、受阻胺系光稳定剂等光稳定剂、紫外线吸收剂、脱模剂、其他热塑性树脂、链烷烃系加工油、环烷烃系加工油、芳香族
系加工油、链烷烃、有机聚硅氧烷、矿物油等软化剂/增塑剂、阻燃剂、抗静电剂、有机纤维、氧化铁等颜料等无机填充剂、玻璃纤维、碳纤维、金属晶须等增强剂、着色剂;亚磷酸酯类、亚膦酸酯类、磷酸酯类等有机磷化合物、其他添加剂或者这些的混合物等。
[0353]-抗氧化剂-[0354]
本实施方式的构成偏振分束器用树脂制棱镜的树脂组合物,优选含有抑制在成型加工时或者使用中的劣化和着色的抗氧化剂。
[0355]
作为上述抗氧化剂,例如,可以举出受阻酚系抗氧化剂、磷系抗氧化剂、硫系抗氧化剂等,但不限定于这些抗氧化剂。
[0356]
这些抗氧化剂,可以使用一种或合用两种以上。
[0357]
另外,从提高热稳定性和抑制成型不良的观点出发,优选合用复数种热稳定剂,例如,优选合用选自磷系抗氧化剂和硫系抗氧化剂中的至少一种和受阻酚系抗氧化剂。
[0358]
作为受阻酚系抗氧化剂,例如,可以举出季戊四醇四[3-(3,5-二叔丁基-4-羟基苯基)丙酸酯]、硫代二乙撑双[3-(3,5-二叔丁基-4-羟基苯基)丙酸酯]、十八烷基-3-(3,5-二叔丁基-4-羟基苯基)丙酸酯、3,3’,3”,5,5’,5
”-
六叔丁基-a,a’,a
”-
(均三甲苯-2,4,6-三基)三对甲酚、4,6-双(辛基硫代甲基)-邻甲酚、4,6-双(十二烷基硫代甲基)-邻甲酚、乙撑双(氧基乙撑)双[3-(5-叔丁基-4-羟基间甲苯基)丙酸酯]、六亚甲基双[3-(3,5-二叔丁基-4-羟基苯基)丙酸酯]、1,3,5-三(3,5-二叔丁基-4-羟基苄基)-1,3,5-三嗪-2,4,6(1h,3h,5h)-三酮、1,3,5-三[(4-叔丁基-3-羟基-2,6-二甲苯基)甲基]-1,3,5-三嗪-2,4,6(1h,3h,5h)-三酮、2,6-二叔丁基-4-(4,6-双(辛基硫基)-1,3,5-三嗪-2-基氨基)苯酚、丙烯酸2-[1-(2-羟基-3,5-二叔戊基苯基)乙基]-4,6-二叔戊基苯酯、丙烯酸2-叔丁基-4-甲基-6-(2-羟基-3-叔丁基-5-甲基苄基)苯酯等,但不限定于这些受阻酚系抗氧化剂。
[0359]
特别优选为季戊四醇四[3-(3,5-二叔丁基-4-羟基苯基)丙酸酯]、十八烷基-3-(3,5-二叔丁基-4-羟基苯基)丙酸酯、丙烯酸2-[1-(2-羟基-3,5-二叔戊基苯基)乙基]-4,6-二叔戊基苯酯。
[0360]
另外,作为上述抗氧化剂的受阻酚系抗氧化剂,可以使用市售的苯酚系抗氧化剂,作为这样的市售的苯酚系抗氧化剂,例如,可以举出irganox 1010(
イルガノックス
1010:季戊四醇四[3-(3,5-二叔丁基-4-羟基苯基)丙酸酯]、巴斯夫公司制)、irganox 1076(
イルガノックス
1076:十八烷基-3-(3,5-二叔丁基-4-羟基苯基)丙酸酯、巴斯夫公司制)、irganox 1330(
イルガノックス
1330:3,3’,3”,5,5’,5
”-
六叔丁基-a,a’,a
”-
(均三甲苯-2,4,6-三基)三对甲酚、巴斯夫公司制)、irganox 3114(
イルガノックス
3114:1,3,5-三(3,5-二叔丁基-4-羟基苄基)-1,3,5-三嗪-2,4,6(1h,3h,5h)-三酮、巴斯夫公司制)、irganox 3125(
イルガノックス
3125、巴斯夫公司制)、adk stab ao-60(
アデカスタブ
ao-60)(季戊四醇四[3-(3,5-二叔丁基-4-羟基苯基)丙酸酯]、adeka公司制)、adk stab ao-80(
アデカスタブ
ao-80)(3,9-双{2-[3-(3-叔丁基-4-羟基-5-甲基苯基)丙酰氧基]-1,1-二甲基乙基}-2,4,8,10-四氧杂螺[5.5]十一烷、adeka公司制)、sumilizer bht(
スミライザー
bht、住友化学制)、cyanox 1790(
シアノックス
1790、新特公司(sitech,
サイテック
)制)、sumilizer ga-80(
スミライザー
ga-80、住友化学制)、sumilizer gs(
スミライザー
gs:丙烯酸2-[1-(2-羟基-3,5-二叔戊基苯基)乙基]-4,6-二叔戊基苯酯、住友化学制)、sumilizer gm(
スミライザー
gm:丙烯酸2-叔丁基-4-甲基-6-(2-羟基-3-叔丁基-5-甲基苄基)苯酯、住友化学制)、维生素e(卫
材公司(eisai,
エーザイ
)制)等,但不限定于这些。
[0361]
在这些市售的苯酚系抗氧化剂中,从赋予该树脂中的热稳定性的效果的观点出发,优选为irganox 1010(
イルガノックス
1010)、adk stab ao-60(
アデカスタブ
ao-60)、adk stab ao-80(
アデカスタブ
ao-80)、irganox 1076(
イルガノックス
1076)、sumilizer gs(
スミライザー
gs)等。
[0362]
这些既可以仅单独使用一种,又可以合用两种以上。
[0363]
另外,作为上述抗氧化剂的磷系抗氧化剂,例如,可以举出三(2,4-二叔丁基苯基)亚磷酸酯、亚磷酸双(2,4-双(1,1-二甲基乙基)-6-甲基苯基)乙酯、四(2,4-二叔丁基苯基)(1,1-联苯基)-4,4
’-
二基二亚膦酸酯、双(2,4-二叔丁基苯基)季戊四醇二亚磷酸酯、双(2,6-二叔丁基-4-甲基苯基)季戊四醇二亚磷酸酯、双(2,4-二枯基苯基)季戊四醇-二亚磷酸酯、四(2,4-叔丁基苯基)(1,1-联苯基)-4,4
’-
二基二亚膦酸酯、二叔丁基间甲苯基亚膦酸酯、4-[3-[(2,4,8,10-四叔丁基二苯并[d,f][1,3,2]二氧杂膦环庚)-6-基氧基]丙基]-2-甲基-6-叔丁基苯酚等,但不限定于这些。
[0364]
此外,作为磷系抗氧化剂可以使用市售的磷系抗氧化剂,作为这样的市售的磷系抗氧化剂,例如,可以举出irgafos 168(
イルガフォス
168:三(2,4-二叔丁基苯基)亚磷酸酯、巴斯夫制)、irgafos 12(
イルガフォス
12:三[2-[[2,4,8,10-四叔丁基二苯并[d,f][1,3,2]二氧磷杂环庚-6-基]氧基]乙基]胺、巴斯夫制)、irgafos 38(
イルガフォス
38:亚磷酸双(2,4-双(1,1-二甲基乙基)-6-甲基苯基)乙酯、巴斯夫制)、adk stab-229k(
アデカスタブ
329k、adeka制)、adk stab pep-36(
アデカスタブ
pep-36、adeka制)、adk stab pep-36a(
アデカスタブ
pep-36a、adeka制)、adk stab pep-8(
アデカスタブ
pep-8、adeka制)、adk stab hp-10(
アデカスタブ
hp-10、adeka制)、adk stab 2112(
アデカスタブ
2112、adeka公司制)、adk stab 1178(
アデカスタブ
1178、adeka制)、adk stab 1500(
アデカスタブ
1500、adeka制)、sandstab p-epq(科莱恩公司(clariant,
クラリアント
)制)、weston 618(
ウェストン
618、ge制)、weston 619g(
ウェストン
619g、ge制)、ultranox 626(
ウルトラノックス
626、ge制)、sumilizer gp(
スミライザー
gp:4-[3-[(2,4,8,10-四叔丁基二苯并[d,f][1,3,2]二氧杂膦环庚)-6-基氧基]丙基]-2-甲基-6-叔丁基苯酚、住友化学制)、hca(9,10-二氢-9-氧杂-10-膦菲-10-氧化物、三光株式会社制)等,但不限定于这些。
[0365]
在这些市售的磷系抗氧化剂中,从赋予该树脂中的热稳定性的效果、与多种抗氧化剂的合用效果的观点出发,优选为irgafos 168(
イルガフォス
168)、adk stab pep-36(
アデカスタブ
pep-36)、adk stab pep-36a(
アデカスタブ
pep-36a)、adk stab hp-10(
アデカスタブ
hp-10)、adk stab 1178(
アデカスタブ
1178),特别优选为adk stab pep-36a(
アデカスタブ
pep-36a)、adk stab pep-36(
アデカスタブ
pep-36)。
[0366]
这些磷系抗氧化剂,既可以仅单独使用一种,又可以合用两种以上。
[0367]
另外,作为上述抗氧化剂的硫系抗氧化剂,例如,可以举出2,4-双(十二烷基硫代甲基)-6-甲基苯酚(irganox 1726(
イルガノックス
1726)、巴斯夫公司制)、2,4-双(辛基硫代甲基)-6-甲基苯酚(irganox 1520l(
イルガノックス
1520l)、巴斯夫公司制)、2,2-双{[3-(十二烷基硫基)-1-氧代丙氧基]甲基}丙烷-1,3-二基双[3-十二烷基硫基]丙酸酯](adk stab ao-412s(
アデカスタブ
ao-412s)、adeka公司制)、2,2-双{[3-(十二烷基硫基)-1-氧代丙氧基]甲基}丙烷-1,3-二基双[3-十二烷基硫基]丙酸酯](keminoxpls(
ケミノックス
pls)、chemipro化成株式会社(
ケミプロ
化成株式会社)制)、二(十三烷基)3,3
’-
硫代二丙酸酯(ao-503、adeka公司制)等,但不限定于这些。
[0368]
在这些市售的硫抗氧化剂中,从赋予该树脂中的热稳定性的效果、与多种抗氧化剂的合用效果的观点、处理性的观点出发,优选为adk stab ao-412s(
アデカスタブ
ao-412s)、keminox pls(
ケミノックス
pls)。
[0369]
这些硫系抗氧化剂,既可以仅单独使用一种,又可以合用两种以上。
[0370]
抗氧化剂的含量,只要是能够获得提高热稳定性的效果的量即可,在含量过剩的情况下,有时在加工时发生渗出等问题,因此相对于甲基丙烯酸系树脂100质量%,优选为5质量%以下,更优选为3质量%以下,进一步优选为1质量%以下,进一步更优选为0.8质量%以下,更进一步优选为0.01~0.8质量%,特别优选为0.01~0.5质量%。
[0371]
关于添加抗氧化剂的时机,没有特别限定,可以举出:添加于聚合前的单体溶液之后开始聚合的方法;添加于聚合后的聚合物溶液并混合之后供给到脱挥工序的方法;添加于脱挥后的溶融状态的聚合物并混合之后进行造粒的方法;在对脱挥、造粒后的颗粒再次进行熔融挤出时添加并混合的方法等。在这些中,从防止脱挥工序中热劣化、着色的观点出发,优选为添加于聚合后的聚合物溶液并混合之后在脱挥工序之前添加抗氧化剂,然后供给到脱挥工序的方法。
[0372]-受阻胺系光稳定剂-[0373]
本实施方式的构成偏振分束器用树脂制棱镜的树脂组合物,可以含有受阻胺系光稳定剂。
[0374]
受阻胺系光稳定剂没有特别限定,优选为包含三个以上的环结构的化合物。在此,环结构优选为选自由芳香族环、脂肪族环、芳香族杂环和非芳香族杂环构成的组中的至少一种,在一个化合物中具有两个以上的环结构的情况下,这些环结构彼此既可以相同又可以不同。
[0375]
作为受阻胺系光稳定剂,例如,具体而言,可以举出双(1,2,2,6,6-五甲基-4-哌啶基)[[3,5-双(1,1-二甲基乙基)-4-羟基苯基]甲基]丁基丙二酸酯、双(1,2,2,6,6-五甲基-4-哌啶基)癸二酸酯与甲基1,2,2,6,6-五甲基-4-哌啶基癸二酸酯的混合物、双(2,2,6,6-四甲基-4-哌啶基)癸二酸酯、n,n
’-
双(2,2,6,6-四甲基-4-哌啶基)-n,n
’-
二甲酰六亚甲基二胺、二丁胺
·
1,3,5-三嗪
·
n,n
’-
双(2,2,6,6-四甲基-4-哌啶基)-1,6-六亚甲基二胺与n-(2,2,6,6-四甲基-4-哌啶基)丁胺的缩聚物、聚[{6-(1,1,3,3-四甲基丁基)氨基-1,3,5-三嗪-2,4-二基}{(2,2,6,6-四甲基-4-哌啶基)亚氨基}六亚甲基{(2,2,6,6-四甲基-4-哌啶基)亚氨基}]、四(1,2,2,6,6-五甲基-4-哌啶基)丁烷-1,2,3,4-四羧酸酯、四(2,2,6,6-四甲基-4-哌啶基)丁烷-1,2,3,4-四羧酸酯、1,2,2,6,6-五甲基-4-哌啶醇与β,β,β’,β
’-
四甲基-2,4,8,10-四氧杂螺[5.5]十一烷-3,9-二乙醇的反应产物、2,2,6,6-四甲基-4-哌啶醇与β,β,β’,β
’-
四甲基-2,4,8,10-四氧杂螺[5.5]十一烷-3,9-二乙醇的反应产物、双(1-十一烷氧基-2,2,6,6-四甲基哌啶-4-基)碳酸酯、1,2,2,6,6-五甲基-4-哌啶基甲基丙烯酸酯、2,2,6,6-四甲基-4-哌啶基甲基丙烯酸酯等,但不限定于这些。
[0376]
其中,优选为包含三个以上的环结构的双(1,2,2,6,6-五甲基-4-哌啶基)[[3,5-双(1,1-二甲基乙基)-4-羟基苯基]甲基]丁基丙二酸酯、二丁胺
·
1,3,5-三嗪
·
n,n
’-
双(2,2,6,6-四甲基-4-哌啶基)-1,6-六亚甲基二胺与n-(2,2,6,6-四甲基-4-哌啶基)丁胺的
缩聚物、聚[{6-(1,1,3,3-四甲基丁基)氨基-1,3,5-三嗪-2,4-二基}{(2,2,6,6-四甲基-4-哌啶基)亚氨基}六亚甲基{(2,2,6,6-四甲基-4-哌啶基)亚氨基}]、1,2,2,6,6-五甲基-4-哌啶醇与β,β,β’,β
’-
四甲基-2,4,8,10-四氧杂螺[5.5]十一烷-3,9-二乙醇的反应产物、2,2,6,6-四甲基-4-哌啶醇与β,β,β’,β
’-
四甲基-2,4,8,10-四氧杂螺[5.5]十一烷-3,9-二乙醇的反应产物。
[0377]
受阻胺系光稳定剂的含量,只要是能够获得提高光稳定性的效果的量即可,在含量过剩的情况下,有时在加工时发生渗出等问题,因此相对于甲基丙烯酸系树脂100质量%,优选为5质量%以下,更优选为3质量%以下,进一步优选为1质量%以下,进一步更优选为0.8质量%以下,更进一步优选为0.01~0.8质量%,特别优选为0.01~0.5质量%。
[0378]
--
紫外线吸收剂
--
[0379]
本实施方式的构成偏振分束器用树脂制棱镜的树脂组合物,可以含有紫外线吸收剂。
[0380]
作为紫外线吸收剂,没有特别限定,优选为在280~380nm具有其最大吸收波长的紫外线吸收剂,例如,可以举出苯并三唑系化合物、苯并三嗪系化合物、二苯甲酮系化合物、氧代二苯甲酮系化合物、苯甲酸酯系化合物、酚系化合物、恶唑系化合物、氰基丙烯酸酯系化合物、苯并恶嗪酮系化合物等。
[0381]
作为苯并三唑系化合物,可以举出2,2
’-
亚甲基双[4-(1,1,3,3-四甲基丁基)-6-(2h-苯并三唑-2-基)苯酚]、2-(3,5-二叔丁基-2-羟基苯基)-5-氯苯并三唑、2-(2h-苯并三唑-2-基)-对甲酚、2-(2h-苯并三唑-2-基)-4,6-双(1-甲基-1-苯基乙基)苯酚、2-苯并三唑-2-基-4,6-二叔丁基苯酚、2-[5-氯(2h)-苯并三唑-2-基]-4-甲基-6-叔丁基苯酚、2-(2h-苯并三唑-2-基)-4,6-二叔丁基苯酚、2-(2h-苯并三唑-2-基)-4-(1,1,3,3-四甲基丁基)苯酚、2-(2h-苯并三唑-2-基)-4-甲基-6-(3,4,5,6-四氢邻苯二甲酰亚胺基甲基)苯酚、3-(3-(2h-苯并三唑-2-基)-5-叔丁基-4-羟基苯基)丙酸甲酯与聚乙二醇300的反应产物、2-(2h-苯并三唑-2-基)-6-(直链和支链十二烷基)-4-甲基苯酚、2-(5-甲基-2-羟基苯基)苯并三唑、2-[2-羟基-3,5-双(α,α-二甲基苄基)苯基]-2h-苯并三唑、3-(2h-苯并三唑-2-基)-5-(1,1-二甲基乙基)-4-羟基-c7-9支链和直链烷基酯。
[0382]
这些中,优选为分子量为400以上的苯并三唑系化合物,例如,在市售品的情况下,可以举出kemisorb(注册商标)2792(chemipro化成(
ケミプロ
化成)制)、adk stab(
アデカスタブ
)(注册商标)la31(株式会社adeka制)、tinuvin(
チヌビン
)(注册商标)234(巴斯夫公司制)等。
[0383]
作为苯并三嗪系化合物,可以举出2-单(羟基苯基)-1,3,5-三嗪化合物、2,4-双(羟基苯基)-1,3,5-三嗪化合物、2,4,6-三(羟基苯基)-1,3,5-三嗪化合物,具体而言,可以举出2,4-二苯基-6-(2-羟基-4-甲氧基苯基)-1,3,5-三嗪、2,4-二苯基-6-(2-羟基-4-乙氧基苯基)-1,3,5-三嗪、2,4-二苯基-(2-羟基-4-丙氧基苯基)-1,3,5-三嗪、2,4-二苯基-(2-羟基-4-丁氧基苯基)-1,3,5-三嗪、2,4-二苯基-6-(2-羟基-4-丁氧基苯基)-1,3,5-三嗪、2,4-二苯基-6-(2-羟基-4-己氧基苯基)-1,3,5-三嗪、2,4-二苯基-6-(2-羟基-4-辛氧基苯基)-1,3,5-三嗪、2,4-二苯基-6-(2-羟基-4-十二烷氧基苯基)-1,3,5-三嗪、2,4-二苯基-6-(2-羟基-4-苄氧基苯基)-1,3,5-三嗪、2,4-二苯基-6-(2-羟基-4-丁氧基乙氧基)-1,3,5-三嗪、2,4-双(2-羟基-4-丁氧基苯基)-6-(2,4-二丁氧基苯基)-1,3-5-三嗪、2,4,6-三
(2-羟基-4-甲氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-4-乙氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-4-丙氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-4-丁氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-4-丁氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-4-己氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-4-辛氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-4-十二烷氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-4-苄氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-4-乙氧基乙氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-4-丁氧基乙氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-4-丙氧基乙氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-4-甲氧基羰基丙氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-4-乙氧基羰基乙氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-4-(1-(2-乙氧基己氧基)-1-氧代丙烷-2-基氧基)苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-3-甲基-4-甲氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-3-甲基-4-乙氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-3-甲基-4-丙氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-3-甲基-4-丁氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-3-甲基-4-丁氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-3-甲基-4-己氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-3-甲基-4-辛氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-3-甲基-4-十二烷氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-3-甲基-4-苄氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-3-甲基-4-乙氧基乙氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-3-甲基-4-丁氧基乙氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-3-甲基-4-丙氧基乙氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-3-甲基-4-甲氧基羰基丙氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-3-甲基-4-乙氧基羰基乙氧基苯基)-1,3,5-三嗪、2,4,6-三(2-羟基-3-甲基-4-(1-(2-乙氧基己氧基)-1-氧代丙烷-2-基氧基)苯基)-1,3,5-三嗪等。
[0384]
作为苯并三嗪系化合物,可以使用市售品,例如可以使用kemisorb102(
ケミプロ
化成公司制)、la-f70(株式会社adeka制)、la-46(株式会社adeka制)、tinuvin405(
チヌビン
405)(巴斯夫公司制)、tinuvin460(
チヌビン
460)(巴斯夫公司制)、tinuvin479(
チヌビン
479)(巴斯夫公司制)、tinuvin1577ff(
チヌビン
1577ff)(巴斯夫公司制)等。
[0385]
其中,从与丙烯酸系树脂的相溶性高、紫外线吸收特性优异的方面出发,进一步优选使用具有2,4-双(2,4-二甲基苯基)-6-[2-羟基-4-(3-烷氧基-2-羟基丙氧基)-5-α-枯基苯基]-s-三嗪骨架的紫外线吸收剂(“烷氧基”,是指辛氧基、壬氧基、癸氧基等长链烷氧基)。
[0386]
作为紫外线吸收剂,特别从与树脂的相溶性、加热时的挥发性的观点出发,优选为分子量400以上的苯并三唑系化合物、苯并三嗪系化合物,另外,从紫外线吸收剂本身的挤出加工时抑制因加热而引起的分解的观点出发,特别优选为苯并三嗪系化合物。
[0387]
另外,上述紫外线吸收剂的熔点(tm),优选为80℃以上,更优选为100℃以上,进一步优选为130℃以上,进一步更优选为160℃以上。
[0388]
上述紫外线吸收剂,在从23℃至260℃以20℃/分钟的速度进行升温的情况下,重量减少率优选为50%以下,更优选为30%以下,进一步优选为15%以下,进一步更优选为10%以下,更进一步优选为5%以下。
[0389]
这些紫外线吸收剂,既可以仅单独使用一种,又可以合用两种以上。通过合用两种结构不同的紫外线吸收剂,能够吸收宽波长区域的紫外线。
[0390]
上述紫外线吸收剂的含量,只要是不阻碍耐热性、耐湿热性、热稳定性以及成型加
工性且发挥本发明的效果的量就没有特别限制,相对于甲基丙烯酸系树脂100质量%,优选为0.1~5质量%,优选为0.2~4质量%以下,更优选为0.25~3质量%,进一步更优选为0.3~3质量%。当在此范围时,紫外线吸收性能、成型性等的平衡优异。
[0391]
--
脱模剂
--
[0392]
本实施方式的构成偏振分束器用树脂制棱镜的树脂组合物,可以含有脱模剂。作为上述脱模剂,例如,可以举出脂肪酸酯、脂肪酸酰胺、脂肪酸金属盐、烃系滑剂、醇系滑剂、聚亚烷基二醇类、羧酸酯类、烃类的链烷烃系矿物油等,但不限定于这些。
[0393]
作为能够用作上述脱模剂的脂肪酸酯,没有特别限制,可以使用以往公知的脂肪酸酯。
[0394]
作为脂肪酸酯,例如,可以使用月桂酸、棕榈酸、十七烷酸、硬脂酸、油酸、花生酸、山萮酸等碳原子数为12~32的脂肪酸与棕榈醇、硬脂醇、山萮醇等一价脂肪族醇或甘油、季戊四醇、二季戊四醇、山梨糖醇酐等多价脂肪族醇的酯化合物;脂肪酸和多价有机酸与一价脂肪族醇或多价脂肪族醇的复合酯化合物等。
[0395]
作为这样的脂肪酸酯,例如,可以举出棕榈酸十六烷基酯、硬脂酸丁酯、硬脂酸硬脂酯、柠檬酸硬脂酯、甘油单辛酸酯、甘油单癸酸酯、甘油单月桂酸酯、甘油单棕榈酸酯、甘油二棕榈酸酯、甘油单硬脂酸酯、甘油二硬脂酸酯、甘油三硬脂酸酯、甘油单油酸酯、甘油二油酸酯、甘油三油酸酯、甘油单亚油酸酯、甘油单山嵛酸酯、甘油单12-羟基硬脂酸酯、甘油二12-羟基硬脂酸酯、甘油三12-羟基硬脂酸酯、甘油二乙酰单硬脂酸酯、甘油柠檬酸脂肪酸酯、季戊四醇己二酸硬脂酸酯、褐煤酸部分皂化酯、季戊四醇四硬脂酸酯、二季戊四醇六硬脂酸酯、山梨糖醇酐三硬脂酸酯等。
[0396]
这些脂肪酸酯,既可以单独使用一种或者又可以组合两种以上后使用。
[0397]
作为市售品,例如,可以举出理研维他公司制rikemal(
リケマール
)系列、poem(
ポエム
)系列、rikestar(
リケスター
)系列、rikemaster(
リケマスター
)系列、花王公司制excel(
エキセル
)系列、rheodol(
レオドール
)系列、exceparl(
エキセパール
)系列、coconad(
ココナード
)系列,更具体而言,可以举出rikemal s-100(
リケマール
s-100)、rikemal h-100(
リケマール
h-100)、poem v-100(
ポエム
v-100)、rikemal b-100(
リケマール
b-100)、rikemal hc-100(
リケマール
hc-100)、rikemal s-200(
リケマール
s-200)、poem b-200(
ポエム
b-200)、rikestar ew-200(
リケスター
ew-200)、rikestar ew-400(
リケスター
ew-400)、excel s-95(
エキセル
s-95)、rheodol ms-50(
レオドール
ms-50)等。
[0398]
关于脂肪酸酰胺,也没有特别限制,可以使用以往公知的脂肪酸酰胺。
[0399]
作为脂肪酸酰胺,例如,可以举出月桂酸酰胺、棕榈酸酰胺、硬脂酸酰胺、山萮酸酰胺、羟基硬脂酸酰胺等饱和脂肪酸酰胺;油酸酰胺、芥酸酰胺、蓖麻油酸酰胺等不饱和脂肪酸酰胺;n-硬脂基硬脂酸酰胺、n-油基油酸酰胺、n-硬脂基油酸酰胺、n-油基硬脂酸酰胺、n-硬脂基芥酸酰胺、n-油基棕榈酸酰胺等取代酰胺;羟甲基硬脂酸酰胺、羟甲基山萮酸酰胺等羟甲基酰胺;亚甲基双硬脂酸酰胺、乙撑双癸酸酰胺、乙撑双月桂酸酰胺、乙撑双硬脂酸酰胺(乙撑双硬脂基酰胺)、乙撑双异硬脂酸酰胺、乙撑双羟基硬脂酸酰胺、乙撑双山萮酸酰胺、六亚甲基双硬脂酸酰胺、六亚甲基双山萮酸酰胺、六亚甲基双羟基硬脂酸酰胺、n,n
’-
二硬脂基己二酸酰胺、n,n
’-
二硬脂基癸二酸酰胺等饱和脂肪酸双酰胺;乙撑双油酸酰胺、六亚甲基双油酸酰胺、n,n
’-
二油基己二酸酰胺、n,n
’-
二油基癸二酸酰胺等不饱和脂肪酸双
酰胺;间亚二甲苯基双硬脂酸酰胺、n,n
’-
二硬脂基异邻苯二甲酸酰胺等芳香族系双酰胺等。
[0400]
这些脂肪酸酰胺,既可以单独使用一种或者又可以组合两种以上后使用。
[0401]
作为市售品,例如,可以举出diamido(
ダイヤミッド
)系列(日本化成公司制)、酰胺系列(日本化成公司制)、nikka amide(
ニッカアマイド
)系列(日本化成公司制)、羟甲基酰胺系列、双酰胺系列、suripakkusu(
スリパックス
)系列(日本化成公司制)、花王蜡系列(花王公司制)、脂肪酸酰胺系列(花王公司制)、乙撑双硬脂酸酰胺类(大日化学工业公司制)等。
[0402]
脂肪酸金属盐,是指高级脂肪酸的金属盐,例如,可以举出硬脂酸锂、硬脂酸镁、硬脂酸钙、月桂酸钙、蓖麻油酸钙、硬脂酸锶、硬脂酸钡、月桂酸钡、蓖麻油酸钡、硬脂酸锌、月桂酸锌、蓖麻油酸锌、2-乙基己酸锌、硬脂酸铅、二元硬脂酸铅、环烷酸铅、12-羟基硬脂酸钙、12-羟基硬脂酸锂等,其中,从使所获得的透明树脂组合物的加工性优异、透明性极其优异的方面出发,特别优选为硬脂酸钙、硬脂酸镁、硬脂酸锌。
[0403]
作为市售品,当举出一例时,可以举出堺化学工业公司制sz系列、sc系列、sm系列、sa系列等。
[0404]
在使用上述脂肪酸金属盐的情况下的的含量,从保持透明性的观点出发,相对于树脂组合物100质量%,优选为0.2质量%以下。
[0405]
上述脱模剂,既可以单独使用一种,又可以合用两种以上后使用。
[0406]
作为供使用的脱模剂,优选为分解开始温度为200℃以上的脱模剂。在此,分解开始温度可以通过基于tga的减少1%质量的温度来测定。
[0407]
脱模剂的含量,只要是能够获得作为脱模剂的效果的量即可,在含量过剩的情况下,有时可能在加工时发生渗出或因螺杆的滑动而引起的挤出不良等问题,因此相对于甲基丙烯酸系树脂100质量%,优选为5质量%以下,更优选为3质量%以下,进一步优选为1质量%以下,进一步更优选为0.8质量%以下,更进一步优选为0.01~0.8质量%,特别优选为0.01~0.5质量%。当以上述范围的量添加时,在抑制因脱模剂添加而引起的透明性的降低的基础上,存在注射成型时的脱模不良得到抑制的倾向,因此优选上述情形。
[0408]-其他热塑性树脂-[0409]
本实施方式的构成偏振分束器用树脂制棱镜的树脂组合物,只要不损害本发明的目的,可以以调整双折射率和提高挠性的目的含有除了甲基丙烯酸系树脂以外的其他热塑性树脂。
[0410]
作为其他热塑性树脂,例如,可以举出聚丙烯酸丁酯等聚丙烯酸酯类;聚苯乙烯、苯乙烯-甲基丙烯酸甲酯共聚物、苯乙烯-丙烯酸丁酯共聚物、苯乙烯-丙烯腈共聚物、丙烯腈-丁二烯-苯乙烯嵌段共聚物等苯乙烯系聚合物;此外,例如,可以举出日本特开昭59-202213号公报、日本特开昭63-27516号公报、日本特开昭51-129449号公报、日本特开昭52-56150号公报等中记载的3~4层结构的丙烯酸系橡胶粒子;日本特公昭60-17406号公报、日本特开平8-245854公报中公开的橡胶质聚合物;国际公开第2014-002491号中记载的由多段聚合获得的含甲基丙烯酸系橡胶的接枝共聚物粒子;等。
[0411]
其中,从获得良好的光学特性和机械特性的观点出发,优选为苯乙烯-丙烯腈共聚物、表面层具有接枝部的含橡胶的接枝共聚物粒子,所述接枝部由能够与含有主链上具有环结构的结构单元x的甲基丙烯酸系树脂相溶的组成来构成。
[0412]
作为前述的丙烯酸系橡胶粒子、含甲基丙烯酸系橡胶的接枝共聚物粒子以及橡胶质聚合物的平均粒径,从提高由本实施方式的组合物获得的膜的冲击强度和光学特性等观点出发,优选为0.03~1μm,更优选为0.05~0.5μm。
[0413]
作为其他热塑性树脂的含量,在将甲基丙烯酸系树脂设为100质量%的情况下,优选为0~50质量%,更优选为0~25质量。
[0414]
[树脂组合物的制造方法]
[0415]
作为制造本实施方式的构成偏振分束器用树脂制棱镜的树脂组合物的方法,只要能够获得满足本发明的条件的组合物,就没有特别限定。例如,可以举出使用挤出机、加热辊、捏合机、辊式混合器、班伯里混合器等混炼机进行混炼的方法。其中,从生产率的方面出发,优选为基于挤出机的混炼。混炼温度,只要根据构成甲基丙烯酸系树脂的聚合物和混合的其他树脂的优选加工温度即可,作为基准可以为140~300℃的范围,优选为180~280℃的范围。另外,以减少挥发成分的目的,优选在挤出机设置通气孔口。
[0416]
在此,在本实施方式的构成偏振分束器用树脂制棱镜的树脂组合物中,残留的溶剂量(残留溶剂量)优选小于1000质量ppm,更优选小于800质量ppm,进一步优选小于700质量ppm。
[0417]
在此,残留的溶剂,是指在聚合时所使用的聚合溶剂(其中,醇类除外)以及在将由聚合而获得的树脂再次溶解而溶液化时所使用的溶剂,具体而言,作为聚合溶剂,可以例示甲苯、二甲苯、乙基苯、异丙苯等芳香族烃;甲基异丁基酮、丁基溶纤剂、甲基乙基酮、环己酮等酮类;二甲基甲酰胺、2-甲基吡咯烷酮等极性溶剂;等,作为再溶解所使用的溶剂,可以例示甲苯、甲基乙基酮、二氯甲烷等。
[0418]
关于本实施方式的构成偏振分束器用树脂制棱镜的树脂组合物,残留的醇量(残留醇量)优选小于为500质量ppm,更优选小于400质量ppm,进一步优选小于350质量ppm。
[0419]
在此,残留的醇,是指由环化缩合反应而副产的醇,具体而言,可以例示甲醇、乙醇、异丙醇等脂肪族醇等。
[0420]
上述残留溶剂量和上述残留醇量,可以通过气相色谱仪来测定。
[0421]
在选择任意方法的情况下,优选在尽量减少氧和水的基础上,制备组合物。
[0422]
例如,作为在溶液聚合中的聚合溶液中的溶解氧浓度,在聚合工序中,优选小于300ppm,另外,在利用挤出机等的制备法中,作为挤出机内的氧浓度,优选设为小于1容量%,进一步优选设为小于0.8容量%。作为甲基丙烯酸系树脂的水分含量,优选调整为1000质量ppm以下,更优选调整为500质量ppm以下。
[0423]
只要在这些范围内,就比较容易地制备满足本发明的必要条件的组合物,因此有利。
[0424]
[偏振分束器用树脂制棱镜的制造方法]
[0425]
本实施方式的偏振分束器用树脂制棱镜,对上述树脂组合物进行成型而成。作为本实施方式的偏振分束器用树脂制棱镜的制造方法,可以使用注射成型、压缩成型、挤出成型等成型方法。其中,从生产率的观点出发,优选为注射成型。
[0426]
通常,注射成型法由如下工序构成:(1)注射工序,对树脂进行熔融,在温度控制的模具的型腔填充熔融树脂;(2)保压工序,直到浇口密封为止在型腔内施加压力,注入相当于由注射工序填充的熔融树脂接触模具而冷却并收缩的量的树脂;(3)冷却工序,开放保压
之后,直到树脂被冷却为止保持成型品;(4)打开模具而取出被冷却的成型品的工序。
[0427]
此时,作为成型温度,以树脂组合物的玻璃化转变温度(tg)为基准,可以是tg 100℃~tg 160℃的范围,优选为tg 110℃~tg 150℃的范围。在此,成型温度,是指在注射喷嘴上缠绕的带加热器的控制温度。成型温度越高能够获得相位差越低的树脂制棱镜,但在高温下促进在成型机内滞留时的因热劣化而引起的着色,因此需要适当地选择成型温度。
[0428]
另外,作为模具温度,以树脂组合物的玻璃化转变温度(tg)为基准,优选为tg-70℃~tg的范围,更优选为tg-50℃~tg-20℃的范围。
[0429]
另外,作为注射速度,可以根据所要获得的树脂制棱镜的厚度和尺寸,适当地选择,例如,可以在10~1000mm/秒的范围适当地选择。
[0430]
另外,作为用于保压的压力,可以根据所要获得的树脂制棱镜的形状,适当地选择,例如可以在30~120mpa的范围适当地选择。
[0431]
在此,用于保压的压力,是指在填充熔融树脂之后,通过用于从浇口进一步送出熔融树脂的螺杆来保持的压力。
[0432]
另外,为了缓解因注射成型而产生的残余应力、减少树脂制棱镜的相位差,可以经过退火工序。关于退火时的温度,以树脂组合物的玻璃化转变温度(tg)为基准,优选为tg-50℃~tg的范围,更优选为tg-30℃~tg-10℃的范围。
[0433]
对本实施方式的偏振分束器用树脂制棱镜的表面,例如,也可以进一步进行硬涂布处理、抗反射处理、透明导电处理、电磁波屏蔽处理、气体阻隔处理等表面功能化处理。这些功能层的厚度,没有特别限制,通常为0.01~10μm的范围。
[0434]
作为赋予在其表面的硬涂布层,例如,将在有机溶剂中溶解或分散硅酮系固化性树脂、含有机聚合物复合无机微粒的固化性树脂、氨基甲酸酯丙烯酸酯、环氧丙烯酸酯、多官能丙烯酸酯等丙烯酸酯和光聚合引发剂而成的涂布液,利用以往公知的涂布方法,涂布于由本实施方式的树脂组合物获得的膜或板上,进行干燥,并进行光固化来形成。
[0435]
另外,为了改良粘接性,例如,也可以在涂布硬涂布层之前预先设置在其组分中包含无机微粒的易粘接层、底涂层、锚固层等之后形成硬涂布层的方法。
[0436]
作为赋予其表面的防眩层,对二氧化硅、三聚氰胺树脂、丙烯酸酯树脂等微粒进行墨(ink)化,利用以往公知的涂布方法涂布于其他功能层上,进行热固化或光固化来形成。
[0437]
作为赋予在其表面的抗反射层,可以例示由金属氧化物、氟化物、硅化物、硼化物、氮化物、硫化物等无机物的薄膜构成的抗反射层;丙烯酸酯树脂、氟树脂等折射率不同的树脂单层或层叠成多层的抗反射层等,另外,也可以利用层叠包含无机系化合物和有机系化合物的复合微粒的薄层而成的抗反射层。
[0438]
[实施例]
[0439]
以下,举出实施例和比较例对本发明的内容进行具体说明。需要说明的是,本发明不限定于下述实施例。
[0440]
<1.n-环己基马来酰亚胺中的2-环己基氨基-n-环己基琥珀酰亚胺量的测定>
[0441]
采集并称量n-环己基马来酰亚胺、或n-环己基马来酰亚胺/间二甲苯溶液,制备25质量%的n-环己基马来酰亚胺的间二甲苯溶液,作为内部标准物质添加苯甲酸异丙酯,使用气相色谱仪(岛津制作所制,gc-2014),在以下的条件下进行测定而确定。
[0442]
检测器:fid。
[0443]
使用色谱柱:hp-5ms。
[0444]
测定条件:在80℃保持5分钟之后,以10℃/分钟的升温速度升温至300℃,此后保持5分钟。
[0445]
<2.n-苯基马来酰亚胺中的2-苯胺基-n-苯基琥珀酰亚胺量的测定>
[0446]
采集并称量n-苯基马来酰亚胺、或n-苯基马来酰亚胺/间二甲苯溶液,制备10质量%的n-苯基马来酰亚胺的间二甲苯溶液,作为内部标准物质添加苯甲酸异丙酯,使用液相色谱仪(waters株式会社制,uplc h-class),在以下的条件下进行测定而确定。
[0447]
检测器:pda(检测波长:210nm~300nm)。
[0448]
使用色谱柱:acquity uplc hss t3。
[0449]
色谱柱温度:40℃。
[0450]
流动相:含有0.1%甲酸的50%乙腈水溶液。
[0451]
流速:0.4ml/min。
[0452]
<3.n-取代马来酰亚胺单体的残留量的确定>
[0453]
采集并称量分析对象(聚合后的聚合溶液或甲基丙烯酸系树脂颗粒)的一部分,将此试样溶解于三氯甲烷,制备5质量%的溶液,作为内部标准物质添加正癸烷,使用气相色谱仪(岛津制作所制,gc-2010),在以下的条件下进行测定而确定。
[0454]
检测器:fid。
[0455]
使用色谱柱:zb-1。
[0456]
测定条件:在45℃保持5分钟之后,以20℃/分钟的升温速度升温至300℃,此后保持15分钟。
[0457]
<4.结构单元的解析>
[0458]
在后述的制造实施例和比较例中制造的甲基丙烯酸系树脂中的各结构单元,除非另有说明,通过1h-nmr测定和
13
c-nmr测定来鉴定甲基丙烯酸系树脂和甲基丙烯酸系树脂组合物的各结构单元,计算其存在量。1h-nmr测定和
13
c-nmr测定的测定条件,如下所述。
[0459]
·
测定设备:日本电子株式会社制jnm-ecz400s。
[0460]
·
测定溶剂:cdcl3或d
6-dmso。
[0461]
·
测定温度:40℃。
[0462]
<5.分子量和分子量分布>
[0463]
在后述的制造实施例和制造比较例中制造的甲基丙烯酸系树脂组合物的重均分子量(mw)、数均分子量(mn)、z均分子量(mz),利用下述的装置和条件进行测定。
[0464]
·
测定装置:东曹株式会社制凝胶渗透色谱(hlc-8320gpc)。
[0465]
·
测定条件:
[0466]
色谱柱:依次串联一根tskguardcolumn superh-h、两根tskgel superhm-m、一根tskgel superh2500来使用。
[0467]
色谱柱温度:40℃。
[0468]
展开溶剂:四氢呋喃、流速:0.6ml/min、作为内部标准,以0.1g/l添加2,6-二叔丁基-4-甲基苯酚(bht)。
[0469]
检测器:ri(差示折射)检测器。
[0470]
检测灵敏度:3.0mv/分钟。
[0471]
样品:0.02g的甲基丙烯酸系树脂组合物的四氢呋喃20ml溶液。
[0472]
进样量:10μl。
[0473]
检量线用标准样品:使用单分散的重量峰分子量已知且分子量不同的以下十种聚甲基丙烯酸甲酯(聚合物实验室(polymerlaboratories)制;pmma calibration kit m-m-10)。
[0474]
重量峰分子量(mp)
[0475]
标准试样11,916,000标准试样2625,500标准试样3298,900标准试样4138,600标准试样560,150标准试样627,600标准试样710,290标准试样85,000标准试样92,810标准试样10850
[0476]
在上述的条件下,测定相对于甲基丙烯酸系树脂组合物的洗脱时间的ri检出强度。
[0477]
基于通过上述检量线用标准样品的测定来获得的各检量线,求出甲基丙烯酸系树脂组合物的重均分子量(mw)、数均分子量(mn)以及z均分子量(mz),并使用其值而确定分子量分布(mw/mn)和(mz/mw)。
[0478]
<6.树脂制棱镜的相位差>
[0479]
将由实施例和比较例获得的一对直角棱镜的斜边彼此相合而构成立方体,使用光子晶格公司(photonic lattice,inc,
フォトニックラティス
社)制双折射评价系统pa-200-l,以波长520nm使光透过入射面和射出面而测定棱镜的相位差的面分布,将棱镜内进行区域指定,求出相位差的绝对值的平均值(nm)。
[0480]
<7.树脂制棱镜中的荧光发光性物质的含量>
[0481]
细切割由实施例和比较例获得的树脂制棱镜,称量到玻璃制的样品瓶并加入三氯甲烷,利用振荡机以每分钟800次的速度振荡30分钟,由此制备2.0质量%的树脂制棱镜的三氯甲烷溶液,使用荧光分光光度计(堀场乔宾伊冯公司(堀场jobin yvon社)制、fluorolog3-22)进行荧光强度的测定。
[0482]
关于测定条件,使用氙灯作为光源,使用pmt作为检测器,激发波长为436nm,将狭缝宽度在激发侧、观测侧同时设为2nm,时间常数为0.2s,测定模式为以各波长的激发光强度对发光强度进行标准化的sc/rc,使用光程长度1cm的石英比色皿进行90
°
观测来测定。
[0483]
对所获得的波长530nm中的荧光强度,使用荧光素/乙醇溶液的荧光强度进行标准化。具体而言,如下所述。
[0484]
求出分别对乙醇和5
×
10-8
mol/l和1
×
10-6
mol/l的荧光素/乙醇溶液以激发波长436nm进行分光分析时波长530nm中的荧光强度,减去乙醇的空白之后,制作浓度-强度换算式。测定三氯甲烷的荧光发光光谱,从2.0质量%的树脂制棱镜的三氯甲烷溶液的波长
530nm的发光强度减去三氯甲烷的波长530nm发光强度而获得的发光强度,使用先前求出的荧光素/乙醇溶液的浓度-强度换算式换算浓度,求出荧光发光性物质的含量(mol/l)。
[0485]
<8.树脂制棱镜的玻璃化转变温度>
[0486]
依据jis-k7121,测定树脂制棱镜的玻璃化转变温度(tg)(℃)。
[0487]
首先,作为试验片,在标准状态(23℃、50%rh)下从已状态调节(在23℃放置1周)的树脂制棱镜分别在四点(四处)切出约10mg。
[0488]
接着,在氮气流量25ml/分钟的条件下使用示差扫描热量计(珀金埃尔默日本有限公司(
パーキンエルマージャパン
(株))制,diamond dsc),在此,以10℃/分钟从室温(23℃)升温至200℃(一次升温),在200℃保持5分钟,使试样完全熔解之后,以10℃/分钟从200℃降温至40℃,在40℃保持5分钟,进一步在上述升温条件下再次升温(二次升温),在此期间描绘的dsc曲线中,测定二次升温时的台阶状变化部分曲线与在纵轴方向上从各基线延长线处于等距离的直线的交点(中间点玻璃化转变温度)作为玻璃化转变温度(tg)(℃)。对每一试样进行四点的测定,将四点的算术平均值(小数点以下四舍五入)作为测定值。
[0489]
<9.树脂制棱镜的光弹性系数>
[0490]
细切割由实施例和比较例获得的树脂制棱镜之后,使用真空压缩成型机来制成压膜,由此作为测定用试样。
[0491]
作为具体的试样制备条件,使用真空压缩成型机(神藤金属工业所制,sfv-30型),在260℃、减压下(约10kpa)预热10分钟之后,将树脂制棱镜在260℃、约10mpa压缩5分钟,解除减压和压缩压之后,移至冷却用压缩成型机而进行冷却固化。将所获得的压膜,在调整为23℃、湿度60%的恒温恒湿室内熟化24小时以上,在此基础上,切出测定用试验片(厚度约150μm、宽度6mm)。
[0492]
使用在《polymer engineering and science》(1999,39,2349-2357)中有详细记载的双折射测定装置,测定光弹性系数c
r
(pa-1
)。
[0493]
将膜状试验片配置于同样设置于恒温恒湿室的膜拉伸装置(井元制作所制)以使夹头间成为50mm。接下来,以使双折射测定装置(大塚电子制,rets-100)的激光束经路位于膜的中心部的方式配置装置,以变形速度50%/分钟(夹头间:50mm、夹头移动速度:5mm/分钟)施加拉伸应力的同时,测定试验片的双折射。
[0494]
从测定而获得的双折射(δn)与拉伸应力(σ
r
)的关系,通过最小二乗近似来求出其直线的斜率,计算光弹性系数(c
r
)(pa-1
)。在计算中,使用拉伸应力为2.5mpa≤σ
r
≤10mpa之间的数据。
[0495]
c
r
=δn/σ
r
[0496]
在此,双折射(δn)为如下所示的值。
[0497]
δn=nx-ny
[0498]
(nx:拉伸方向的折射率、ny:在面内与拉伸方向相垂直的方向的折射率)
[0499]
<10.树脂制棱镜的透光率>
[0500]
对于由实施例和比较例获得的树脂制棱镜,将一对直角棱镜的斜边彼此相合而制成立方体,使光透过入射面和射出面,使用分光色彩计(日本电色工业株式会社制,sd-5000),以d65光源10
°
视野在波长380~780nm的范围每5nm测定透过率(%)。使用测定值,求出波长450nm的透过率(t450)相对于波长680nm的透过率(t680)的比率(t450/t680)。
[0501]
<11.影像的清晰度>
[0502]
使用由实施例和比较例获得的树脂制棱镜,在一对直角棱镜的斜边和斜边之间夹入并粘合反射型偏光膜,从而制作了立方体型偏振分束器。使用此偏振分束器,制作了图1所示的评价系统。
[0503]
该评价系统是将蓝色led(峰波长:约465nm)1、准直透镜2、偏振分束器3、反射型液晶面板(lcos:liquid crystal on silicon)4、投影透镜5、屏幕6进行组合而形成。偏振分束器3是在由实施例和比较例获得的一对树脂制直角棱镜31、32的斜边和斜边之间夹入并粘合反射型偏光膜33而制作。来自蓝色led 1的光通过准直透镜2来成为平行光之后,入射到偏振分束器3,通过树脂制棱镜31之后,通过反射型偏光膜33来被分割成p偏振光和s偏振光。s偏振光通过反射型偏光膜33来折回90
°
,并入射到反射型液晶面板4。在反射型液晶面板4中,入射的s偏振光在开启信号的像素中被转换为p偏振光而被反射,在关闭信号的像素中以s偏振光的状态被反射,入射到树脂制棱镜31中并到达反射型偏光膜33。p偏振光的光透过反射型偏光膜33,从而通过树脂制棱镜32,利用投影透镜5被放大并投影到屏幕6。
[0504]
通过上述模拟装置观察在屏幕6上显示的静止图像,并根据以下评价基准对影像的清晰度进行评价。
[0505]
[评价标准]
[0506]
影像的清晰度
[0507]
〇:在影像中观察不到渗色和模糊。
[0508]
△
:由于渗色和模糊,影像变得稍微不清晰,但能够辨别出放映什么影像。
[0509]
×
:由于渗色和模糊,影像变得不清晰,因此无法辨别出放映什么影像。
[0510]
色调的再现度
[0511]
〇:清楚地再现蓝色。
[0512]
×
:蓝色的再现度变差。
[0513]
[原料]
[0514]
对后述的实施例和比较例中使用的原料,如下所示。
[0515]
[[单体]]
[0516]
·
甲基丙烯酸甲酯(mma):旭化成株式会社制。
[0517]
·
n-环己基马来酰亚胺(chmi):株式会社日本触媒制。
[0518]
(相对于chmi质量的2-环己基氨基-n-环己基琥珀酰亚胺(ccsi)的质量比例为80质量ppm)
[0519]
·
n-苯基马来酰亚胺(phmi):株式会社日本触媒制。
[0520]
(相对于phmi质量的2-苯胺基-n-苯基琥珀酰亚胺(apsi)的质量比例为60质量ppm)
[0521]-n-取代马来酰亚胺(chmi、phmi)的前处理(水洗工序和脱水工序)-[0522]
对上述n-环己基马来酰亚胺(chmi)和n-苯基马来酰亚胺(phmi),通过下述的水洗和脱水工序,进行2-氨基-n-取代琥珀酰亚胺(ccsi、apsi)的去除和含有水分的去除。
[0523]
--
n-环己基马来酰亚胺(chmi)的水洗和脱水
--
[0524]
---
将n-环己基马来酰亚胺中的ccsi减少为5质量ppm以下
---
[0525]
量取250.0kg的chmi、750.0kg的间二甲苯(以下记作”mxy”),加入到具备基于护套
的温度调节装置和作为搅拌叶片的三片后掠叶片的2.0m3搪玻璃制反应器,在护套内吹入蒸汽而将反应器内的溶液温度提升至56℃,进行搅拌,获得有机层。接下来,量取2质量%的硫酸水350.0kg并加入到反应器,将溶液温度保持在56℃,以100rpm搅拌10分钟间。停止搅拌,静置10分钟,将水层抽取到圆桶罐。
[0526]
将同样的操作进一步重复两次,将有机层利用硫酸水清洗的操作进行合计三次。利用气相色谱仪定量有机层中的ccsi量,其结果相对于有机层中的chmi为4.9质量ppm。
[0527]
此后,将离子交换水350.0kg加入到反应器,将溶液温度保持在55℃,以100rpm搅拌10分钟。停止搅拌,静置20分钟,将水层抽取到圆桶罐。
[0528]
将同样的操作再次重复一次,将有机层利用离子交换水清洗的操作进行合计两次。气相色谱仪定量有机层中的ccsi量,其结果相对于有机层中的chmi为4.6质量ppm。
[0529]
接着,将溶液温度保持在50℃,一边以100rpm搅拌一边使反应器内逐渐减压,使反应器内的压力成为5kpa。此后,将溶液温度提升至60℃,进行共沸脱水操作。蒸馏除去131.6kg的水/mxy混合液,通过卡尔费休水分测定仪定量有机层中的水分浓度,其结果相对于有机层的质量为102质量ppm。
[0530]
在有机层中加入浓度调整用mxy,获得chmi为24.0质量%、水分浓度为191质量ppm、ccsi相对于chmi的质量为4.6质量ppm的有机层1034.3kg。
[0531]
---
将n-环己基马来酰亚胺中的ccsi减少为1质量ppm以下
---
[0532]
量取80.0kg的chmi、240.0kg的mxy,加入到具备基于护套的温度调节装置和作为搅拌叶片的神钢环境解决方案(神钢环境
ソリューション
)制全区域(full zone,
フルゾーン
)的0.50m3搪玻璃制反应器,在护套内吹入蒸汽而将反应器内的溶液温度提升至55℃,进行搅拌,获得有机层。接下来,量取2质量%的硫酸水112.0kg并加入到反应器,将溶液温度保持在55℃,以100rpm搅拌30分钟。停止搅拌,静置10分钟,将水层抽取到圆桶罐。
[0533]
将同样的操作进一步重复三次,将有机层利用硫酸水清洗的操作进行合计4次。利用气相色谱仪定量有机层中的ccsi量,其结果相对于有机层中的chmi为0.57质量ppm。
[0534]
此后,将离子交换水112.0kg加入到反应器,将溶液温度保持在55℃,以100rpm搅拌30分钟。停止搅拌,静置10分钟,将水层抽取圆桶罐。
[0535]
将溶液温度保持在50℃,一边以100rpm搅拌一边使反应器内逐渐减压,使反应器内的压力成为5kpa。此后,将溶液温度提升至60℃,进行共沸脱水操作。蒸馏除去54kg的水/mxy混合液,通过卡尔费休水分测定仪定量有机层中的水分浓度,其结果相对于有机层的质量为46质量ppm。
[0536]
在有机层中加入浓度调整用mxy,获得chmi为20.3质量%、水分浓度为125质量ppm、ccsi相对于chmi的质量为0.60质量ppm的有机层384.0kg。
[0537]
--
n-苯基马来酰亚胺(phmi)的水洗和脱水
--
[0538]
---
将n-苯基马来酰亚胺中的apsi减少为5质量ppm以下
---
[0539]
量取150.0kg的phmi、720.0kg的mxy,加入到具备基于护套的温度调节装置和作为搅拌叶片的三张后掠叶片的2.0m3搪玻璃制反应器,在护套内吹入蒸汽而反应器内的溶液温度提升至55℃,进行搅拌,获得有机层。接下来,量取7质量%的碳酸氢钠水336.0kg并加入到反应器,将溶液温度保持在55℃,以100rpm搅拌10分钟。停止搅拌,静置10分钟,将水层抽取到圆桶罐。
[0540]
接下来,将离子交换水336.0kg加入到反应器,将溶液温度保持在55℃,以100rpm搅拌10分钟。停止搅拌,静置10分钟,将水层抽取到圆桶罐。
[0541]
此后,量取2质量%的硫酸水336.0kg并加入到反应器,将溶液温度保持在55℃,以100rpm搅拌10分钟。停止搅拌,静置10分钟,将水层抽取到圆桶罐。
[0542]
与上述同样地,将利用离子交换水清洗的操作进一步进行两次。
[0543]
利用液相色谱仪定量有机层中的apsi量,其结果相对于有机层中的phmi为3.6质量ppm。
[0544]
接着,将溶液温度保持在50℃,一边以100rpm搅拌一边使反应器内逐渐减压,使反应器内的压力成为5kpa。此后,将溶液温度提升至55℃,进行共沸脱水操作。蒸馏除去190kg的水/mxy混合液,通过卡尔费休水分测定仪定量有机层中的水分浓度,其结果相对于有机层的质量为47质量ppm。
[0545]
在有机层中加入浓度调整用mxy,获得phmi为10.8质量%、水分浓度为170质量ppm、apsi相对于phmi的质量为3.4质量ppm的有机层1340.7kg。
[0546]
---
将n-苯基马来酰亚胺中的apsi减少为1质量ppm以下
---
[0547]
量取50.0kg的phmi、240.0kg的mxy,加入到具备基于护套的温度调节装置和作为搅拌叶片的神钢环境解决方案(神钢环境
ソリューション
)制的全区域(full zone,
フルゾーン
)的0.50m3搪玻璃制反应器,在护套内吹入蒸汽而将反应器内的溶液温度提升至55℃,进行搅拌,获得有机层。接下来,量取7质量%的碳酸氢钠水112.0kg并加入到反应器,将溶液温度保持在55℃,以100rpm搅拌15分钟。停止搅拌,静置10分钟,将水层抽取到圆桶罐。将同样的操作再次重复一次,将利用碳酸氢钠水清洗有机层的操作进行合计两次。
[0548]
接下来,将离子交换水112.0kg加入到反应器,将溶液温度保持在55℃,以100rpm搅拌30分钟。停止搅拌,静置10分钟,将水层抽取到圆桶罐。
[0549]
此后,量取2质量%硫酸水112.0kg并加入到反应器,将溶液温度保持在55℃,以100rpm搅拌30分钟。停止搅拌,静置10分钟,将水层抽取到圆桶罐。将同样的操作进一步重复两次,将利用硫酸水清洗有机层的操作进行合计三次。
[0550]
与上述同样地,将利用离子交换水清洗的操作进一步进行两次。
[0551]
利用液相色谱仪定量有机层中的apsi量,其结果相对于有机层中的phmi为0.37质量ppm。
[0552]
将溶液温度保持在50℃,一边以100rpm搅拌一边使反应器内逐渐减压,使反应器内的压力成为5kpa。此后,将溶液温度提升至55℃,进行共沸脱水操作。蒸馏除去72kg的水/mxy混合液,通过卡尔费休水分测定仪定量有机层中的水分浓度,其结果相对于有机层的质量为60质量ppm。
[0553]
在有机层中加入浓度调整用mxy,获得phmi为10.3质量%、水分浓度为180质量ppm、apsi相对于phmi的质量为0.42质量ppm的有机层432.8kg。
[0554]
[[聚合引发剂]]
[0555]
·
1,1-二(叔丁基过氧化)环己烷:日油株式会社制“perhexa c(
パーヘキサ
c)”。
[0556]
[[链转移剂]]
[0557]
·
正辛基硫醇:花王株式会社制。
[0558]
[[受阻酚系抗氧化剂]]
[0559]
·
irganox 1010(
イルガノックス
1010):巴斯夫公司制。
[0560]
[[磷系抗氧化剂]]
[0561]
·
irgafos 168(
イルガフォス
168)(熔点180~190℃):巴斯夫公司制。
[0562]
[甲基丙烯酸系树脂组合物]
[0563]
[制造实施例1]
[0564]
量取经过水洗工序和脱水工序的phmi为10.3质量%的mxy溶液(apsi相对于phmi的质量为0.42质量ppm)374.8kg和经过水洗工序和脱水工序的chmi为20.3质量%的mxy溶液(ccsi相对于chmi的质量为0.60质量ppm)323.6kg,加入到具备基于护套的温度调节装置和搅拌叶片的1.25m3反应器,在溶液温度60℃、反应器内压力5kpa条件下,一边搅拌一边在减压下蒸馏除去160.8kg的mxy。接下来,将反应釜恢复至常压,添加16.7kg的mxy,由此制备38.6kg的phmi、65.7kg的chmi、450.0kg的mxy的混合溶液。量取445.7kg的mma和作为链转移剂的正辛基硫醇0.413kg,投入并搅拌,获得单体混合溶液。
[0565]
对于反应器的内容液体,以30l/分钟的速度实施基于氮的鼓泡1小时,从而去除溶解氧。
[0566]
此后,在护套内吹入蒸汽而将反应器内的溶液温度提升至125℃,一边以50rpm搅拌,一边以0.5kg/小时的速度添加将1,1-二(叔丁基过氧化)环己烷0.23kg溶解于2.77kg的mxy的聚合引发剂溶液,从而开始聚合,在聚合开始6小时后停止添加。
[0567]
需要说明的是,在聚合中利用基于护套的温度调节来将反应器内的溶液温度控制在125
±
2℃。
[0568]
从聚合开始至经过8小时之后,获得包含主链上具有环结构的甲基丙烯酸系树脂的聚合溶液。
[0569]
评价所获得的聚合溶液中包含的n-取代马来酰亚胺量,其结果包含1340质量ppm的phmi、4390质量ppm的chmi。
[0570]
在此聚合溶液中,在搅拌下添加相对于溶液中包含的聚合物100质量%为0.1质量%的irganox 1010(
イルガノックス
1010)和0.05质量%的irgafos168(
イルガフォス
168)。
[0571]
将包含此抗氧化剂的聚合溶液通过由sus316l制金属纤维构成的过滤精度2μm的过滤器,由此,来进行过滤。
[0572]
为了从聚合溶液中回收聚合物,作为使用于脱挥工序的装置,使用由具有平板狭缝型流路和热介质流路的平板式热交换器与内容积约0.3m3的附带sus制热介质护套的减压容器(以下,记作脱挥槽)构成的脱挥装置。
[0573]
将包含通过聚合而获得的聚合物的溶液,以30升/小时的速度供给至设置于减压容器的上部的热交换器,在加热至260℃之后,供给至被加热、减压至内温260℃、真空度30torr的条件的脱挥槽,实施脱挥处理。根据装置形状、运转条件计算脱挥装置内的剪切速度,其为5.3s-1
。
[0574]
对脱挥后的聚合物从脱挥槽下部利用齿轮泵进行升压,从股线模头挤出、水冷之后,进行颗粒化而获得甲基丙烯酸系树脂组合物a。
[0575]
确认甲基丙烯酸系树脂组合物a的组成,其结果源自mma、phmi、chmi各单体的结构单元,分别为81.2质量%、7.1质量%、11.7质量%。另外,重均分子量mw为148,000,mw/mn为
2.12,mz/mw为1.63,玻璃化转变温度为133℃。
[0576]
[制造实施例2]
[0577]
量取经过水洗工序和脱水工序的phmi为10.3质量%的mxy溶液(apsi相对于phmi的质量为0.42质量ppm)352.4kg和经过水洗工序和脱水工序的chmi为20.3质量%的mxy溶液(ccsi相对于chmi的质量为0.60质量ppm)310.3kg,加入到具备基于护套的温度调节装置和搅拌叶片的1.25m3反应器,在溶液温度60℃、反应器内压力5kpa的条件下,一边搅拌一边在减压下蒸馏除去335.4kg的mxy。接下来,将反应釜恢复常压,添加8.9kg的mxy,由此制备36.3kg的phmi、63.0kg的chmi、236.9kg的mxy的混合溶液。量取340.7kg的mma和作为链转移剂的正辛基硫醇0.275kg,投入并搅拌,获得单体混合溶液。
[0578]
接下来,量取123.1kg的mxy,并加入到罐1。
[0579]
此外,在罐2中量取110.0kg的mma和90.0kg的mxy并搅拌,作为补充添加用单体溶液。
[0580]
对于反应器的内容液体,以30l/分钟的速度实施基于氮的鼓泡1小时,对于各自罐1、罐2,以10l/分钟的速度实施基于氮的鼓泡30分钟,去除溶解氧。
[0581]
此后,在护套内吹入蒸汽而将反应器内的溶液温度提升至128℃,一边以50rpm搅拌,一边以1kg/小时的速度添加将1,1-二(叔丁基过氧化)环己烷0.37kg溶解于3.005kg的mxy的聚合引发剂溶液,从而开始聚合。在聚合开始0.5小时后,将引发剂溶液的添加速度降低至0.25kg/小时的同时,从罐1以35.2kg/小时将mxy添加3.5小时。
[0582]
需要说明的是,在聚合中利用基于护套的温度调节来将反应器内的溶液温度控制在128
±
2℃。
[0583]
接下来,在聚合开始4小时之后将引发剂溶液的添加速度变化为0.75kg/小时的同时,从罐2以100.0kg/小时的速度将包含mma的单体溶液添加2小时。
[0584]
此外,在聚合开始6小时之后将引发剂溶液的添加速度降低至0.5kg/小时,在聚合开始7小时之后停止添加。将聚合进一步继续1小时,获得包含主链上具有环结构单元的甲基丙烯酸系树脂的聚合溶液。
[0585]
评价所获得的聚合溶液中包含的n-取代马来酰亚胺量,其结果包含220质量ppm的phmi、1070质量ppm的chmi。
[0586]
在此聚合溶液中,在搅拌下添加相对于溶液中包含的聚合物100质量%为0.1质量%的irganox 1010(
イルガノックス
1010)和0.05质量%的irgafos168(
イルガフォス
168)。
[0587]
将包含抗氧化剂的聚合溶液通过由sus316l制金属纤维构成的过滤精度2μm的过滤器,由此来进行过滤。
[0588]
为了从聚合溶液中回收聚合物,作为使用于脱挥工序的装置,使用由具有平板狭缝型流路和热介质流路的平板式热交换器与内容积约0.3m3的附带sus制热介质护套的减压容器(以下,记作脱挥槽)构成的脱挥装置,所述脱挥装置不具有旋转部。
[0589]
将包含通过聚合而获得的聚合物的溶液,以30升/小时的速度供给至设置于减压容器的上部的热交换器,在加热至260℃之后,供给至被加热、减压至内温260℃、真空度30torr的条件的脱挥槽,实施脱挥处理。根据装置形状、运转条件计算脱挥装置内的剪切速度,其为5.3s-1
。
[0590]
对脱挥后的聚合物从脱挥槽下部利用齿轮泵进行升压,从股线模头挤出、水冷之后,进行颗粒化而获得甲基丙烯酸系树脂组合物b。
[0591]
确认甲基丙烯酸系树脂组合物b的组成,其结果源自mma、phmi、chmi各单体的结构单元,分别为80.9质量%、7.0质量%、12.1质量%。另外,重均分子量mw为142,000,mw/mn为2.32,mz/mw为1.75,玻璃化转变温度为134℃。
[0592]
[制造实施例3]
[0593]
作为经过水洗工序和脱水工序的phmi使用10.8质量%的mxy溶液(apsi相对于phmi的质量为3.4质量ppm),以及作为经过水洗工序和脱水工序的chmi使用24.0质量%的mxy溶液(ccsi相对于chmi的质量为4.6质量ppm),并将反应器内制备的混合溶液的组成与制造实施例2一致,除此之外,与制造实施例2同样地进行,获得甲基丙烯酸系树脂组合物c。
[0594]
需要说明的是,评价所获得的聚合溶液中包含的n-取代马来酰亚胺量,其结果包含210质量ppm的phmi、1090质量ppm的chmi。
[0595]
确认甲基丙烯酸系树脂组合物c的组成,其结果源自mma、phmi、chmi各单体的结构单元,分别为80.8质量%、7.1质量%、12.1质量%。另外,重均分子量mw为141,000,mw/mn为2.31,mz/mw为1.75,玻璃化转变温度为134℃。
[0596]
[制造实施例4]
[0597]
在具备基于护套的温度调节装置和搅拌叶片的反应器,装入40质量份的mma和60质量份的mxy、作为链转移剂的正辛基硫醇0.08质量份,实施基于氮的鼓泡1小时,去除溶解氧。此后,在护套吹入蒸汽而将反应器内的溶液温度提升至120℃,一边以50rpm搅拌,一边以等速添加将1,1-二(叔丁基过氧化)环己烷0.02质量份溶解于0.10质量份的mxy的聚合引发剂溶液5小时,从而进行聚合,进一步在120℃熟化3小时,从聚合开始至8小时后结束聚合而获得pmma的mxy溶液。在聚合结束后将液温降低至50℃。
[0598]
接着,将由单甲基胺12质量份和甲醇12质量份构成的混合液在室温滴加至反应器内,将液温提高至170℃并在加压下搅拌1小时,从而进行戊二酰亚胺环化反应。将液温降低至120℃,使反应器内减压而蒸馏除去未反应的单甲基胺和甲醇以及mxy的一部分,获得戊二酰亚胺环化甲基丙烯酸系聚合物的约50质量%的mxy溶液。
[0599]
在此聚合溶液中,在搅拌下添加相对于溶液中包含的聚合物100质量%为0.1质量%的irganox 1010(
イルガノックス
1010)和0.05质量%的irgafos168(
イルガフォス
168)。
[0600]
将包含抗氧化剂的聚合溶液通过由sus316l制金属纤维构成的过滤精度2μm的过滤器,由此来进行过滤。
[0601]
为了从聚合溶液中回收聚合物,作为使用于脱挥工序的装置,使用由具有平板狭缝型流路和热介质流路的平板式热交换器与内容积约0.3m3的附带sus制热介质护套的减压容器(以下,记作脱挥槽)构成的脱挥装置,所述脱挥装置不具有旋转部。
[0602]
将包含通过聚合而获得的聚合物的溶液,以30升/时的速度供给至设置于减压容器的上部的热交换器,在加热至260℃之后,供给至被加热、减压至内温260℃、真空度30torr的条件的脱挥槽并实施脱挥处理。根据装置形状、运转条件计算脱挥装置内的剪切速度,其为5.3s-1
。
[0603]
对脱挥后的聚合物从脱挥槽下部利用齿轮泵进行升压,从股线模头挤出、水冷之
后,进行颗粒化而获得甲基丙烯酸系树脂组合物d。
[0604]
确认所获得的甲基丙烯酸系树脂组合物d的组成,其结果酰亚胺化率为3.2%,戊二酰亚胺结构单元量为5.1质量%。另外,重均分子量mw为93,000,mw/mn为1.79,mz/mw为1.51,玻璃化转变温度为122℃。
[0605]
[制造实施例5]
[0606]
除了在反应器内制备的溶液中的各单体量设为39.3kg的phmi、46.5kg的chmi、354.2kg的mma以外,与制造实施例2同样地获得甲基丙烯酸系树脂组合物e。
[0607]
确认甲基丙烯酸系树脂组合物e的组成,其结果源自mma、phmi、chmi各单体的结构单元,分别为83.2质量%、7.6质量%、9.2质量%。另外,重均分子量mw为143,000,mw/mn为2.35,mz/mw为1.81,玻璃化转变温度为132℃。
[0608]
[制造比较例1]
[0609]
量取经过水洗工序和脱水工序的phmi为10.3质量%的mxy溶液(apsi相对于phmi的质量为0.42质量ppm)352.4kg和经过水洗工序和脱水工序的chmi为20.3质量%的mxy溶液(ccsi相对于chmi的质量为0.60质量ppm)310.3kg,加入到具备基于护套的温度调节装置和搅拌叶片的1.25m3反应器,在溶液温度60℃、反应器内压力5kpa的条件下,一边搅拌一边在减压下蒸馏除去335.4kg的mxy。接下来,将反应釜恢复常压,添加8.9kg的mxy,因此制备36.3kg的phmi、63.0kg的chmi、236.9kg的mxy的混合溶液。量取340.7kg的mma和作为链转移剂的正辛基硫醇0.275kg,投入并搅拌,获得单体混合溶液。
[0610]
接下来,量取123.1kg的mxy,并加入到罐1。
[0611]
此外,在罐2中量取110.0kg的mma和90.0kg的mxy并搅拌,作为补充添加用单体溶液。
[0612]
对于反应器的内容液体,以30l/分钟的速度实施基于氮的鼓泡1小时,对于各自罐1、罐2,以10l/分钟的速度实施基于氮的鼓泡30分钟,去除溶解氧。
[0613]
此后,在护套内吹入蒸汽而将反应器内的溶液温度提升至128℃,一边以50rpm搅拌,一边以1kg/小时的速度添加将1,1-二(叔丁基过氧化)环己烷0.37kg溶解于3.005kg的mxy的聚合引发剂溶液,从而开始聚合。在聚合开始0.5小时后,将引发剂溶液的添加速度降低至0.25kg/小时的同时,从罐1以35.2kg/小时将mxy添加3.5小时。
[0614]
需要说明的是,在聚合中利用基于护套的温度调节将反应器内的溶液温度控制在128
±
2℃。
[0615]
接下来,在聚合开始4小时之后将引发剂溶液的添加速度变化为0.75kg/小时的同时,从罐2以100.0kg/小时的速度将包含mma的单体溶液添加2小时。
[0616]
此外,聚合开始6小时之后将引发剂溶液的添加速度降低至0.5kg/小时,在聚合开始7小时之后停止添加。将聚合进一步继续1小时,获得包含主链上具有环结构单元的甲基丙烯酸系树脂的聚合溶液。
[0617]
评价所获得的聚合溶液中包含的n-取代马来酰亚胺量,其结果包含220质量ppm的phmi、1070质量ppm的chmi。
[0618]
在此聚合溶液中,相对于溶液中包含的聚合物100质量%,在搅拌下添加0.1质量%的irganox 1010(
イルガノックス
1010)和0.05质量%的irgafos168(
イルガフォス
168)。
[0619]
将包含抗氧化剂的聚合溶液通过由sus316l制金属纤维构成的过滤精度2μm的过滤器,由此来进行过滤。
[0620]
接着,将所获得的聚合溶液导入到脱挥用的配备有复数个通气孔口的双轴挤出机,由此进行脱挥。在双轴挤出机中,以树脂换算计成为10kg/小时的方式,供给所获得的聚合溶液,设定为料筒温度260℃、螺杆转速150rpm、真空度10~40torr的条件。从股线模头挤出在双轴挤出机中脱挥的树脂,在水冷之后进行颗粒化,获得甲基丙烯酸系树脂组合物f。根据装置形状、运转条件计算脱挥装置内的剪切速度,其为80s-1
。
[0621]
确认甲基丙烯酸系树脂组合物f的组成,其结果源自mma、phmi、chmi各单体的结构单元,分别为80.9质量%、7.0质量%、12.1质量%。另外,重均分子量mw为136,000,mw/mn为2.35,mz/mw为1.81,玻璃化转变温度为134℃。
[0622]
[制造比较例2]
[0623]
除了作为phmi和chmi以未精制的状态直接使用市售品而制备36.3kg的phmi、63.0kg的chmi、236.9kg的mxy混合溶液以外,与制造实施例2同样地进行,获得甲基丙烯酸系树脂组合物g。根据装置形状、运转条件计算脱挥装置内的剪切速度,其为5.3s-1
。
[0624]
需要说明的是,评价所获得的聚合溶液中包含的n-取代马来酰亚胺量,其结果包含220质量ppm的phmi、1070质量ppm的chmi。
[0625]
确认所获得的颗粒状的聚合物的组成,其结果源自mma、phmi、chmi各单体的结构单元,分别为80.9质量%、7.1质量%、12.1质量%。另外,重均分子量mw为142,000,mw/mn为2.32,mz/mw为1.75,玻璃化转变温度为134℃。
[0626]
[制造比较例3]
[0627]
作为phmi和chmi以未精制的状态直接使用市售品,量取38.6kg的phmi、65.7kg的chmi、450.0kg的mxy、445.7kg的mma和作为链转移剂的正辛基硫醇0.413kg,投入到具备基于护套的温度调节装置和搅拌叶片的1.25m3反应器并搅拌,获得单体混合溶液。
[0628]
对于反应器的内容液体,以30l/分钟的速度实施基于氮的鼓泡1小时,去除溶解氧。
[0629]
此后,在护套内吹入蒸汽而将反应器内的溶液温度提升至125℃,一边以50rpm搅拌,一边以0.5kg/小时的速度添加将1,1-二(叔丁基过氧化)环己烷0.23kg溶解于2.77kg的mxy的聚合引发剂溶液,从而开始聚合,在聚合开始6小时之后停止添加。
[0630]
需要说明的是,在聚合中利用基于护套的温度调节来将反应器内的溶液温度控制在125
±
2℃。
[0631]
从聚合开始至经过8小时之后,获得包含主链上具有环结构的甲基丙烯酸系树脂的聚合溶液。
[0632]
评价所获得的聚合溶液中包含的n-取代马来酰亚胺量,其结果包含1340质量ppm的phmi、4390质量ppm的chmi。
[0633]
在此聚合溶液中,在搅拌下添加相对于溶液中包含的聚合物100质量%为0.1质量%的irganox 1010(
イルガノックス
1010)和0.05质量%的irgafos168(
イルガフォス
168)。
[0634]
将包含此抗氧化剂的聚合溶液通过由sus316l制金属纤维构成的过滤精度2μm的过滤器,由此来进行过滤。
[0635]
将添加有此抗氧化剂的聚合溶液供给到由预先被加热至170℃的管状热交换器和气化槽构成的浓缩装置,将溶液中包含的聚合物的浓度提高至70质量%,接着将所获得的聚合溶液供给至传热面积为0.2m2的具有旋转部的薄膜蒸发器,进行脱挥。
[0636]
此时在装置内温度为280℃、供给量30l/hr、转速400rpm、真空度30torr的条件下实施,对脱挥后的聚合物利用齿轮泵进行升压,从股线模头挤出、水冷之后,进行颗粒化而获得甲基丙烯酸系树脂组合物h。根据装置形状、运转条件计算具有旋转部的薄膜蒸发器内的剪切速度,其为3200s-1
。
[0637]
确认所获得的颗粒状的聚合物的组成,其结果源自mma、phmi、chmi各单体的结构单元,分别为81.0质量%、7.2质量%、11.8质量%。另外,重均分子量mw为136,000,mw/mn为2.21,mz/mw为1.71,玻璃化转变温度为134℃。
[0638]
[制造比较例4]
[0639]
除了在反应器内制备的溶液中的各单体量设为41.3kg的phmi、41.3kg的chmi、357.5kg的mma以外,与制造实施例2同样地进行,获得甲基丙烯酸系树脂组合物i。
[0640]
确认甲基丙烯酸系树脂组合物i的组成,其结果源自mma、phmi、chmi各单体的结构单元,分别为84.1质量%、8.0质量%、7.9质量%。另外,重均分子量mw为145,000,mw/mn为2.31,mz/mw为1.77,玻璃化转变温度为130℃。
[0641]
[制造比较例5]
[0642]
使用同向旋转式双轴挤出机,对聚甲基丙烯酸甲酯利用单甲基胺进行酰亚胺化,由此获得具有戊二酰亚胺系结构单元的甲基丙烯酸系树脂组合物j。
[0643]
具体而言,使用螺杆直径40mm的同向旋转式双轴挤出机,将挤出机料筒温度设定为270℃,将螺杆转速设定为150rpm,通过加料斗来以20kg/h供给重均分子量mw为10.8万的聚甲基丙烯酸甲酯的同时,将氮以200ml/min的流量流通到挤出机内。由捏合块使树脂熔融、充满之后,从喷嘴注入相对于原料树脂100质量份为1.8质量份的单甲基胺,进行酰亚胺化反应。在反应区域的末端(通气孔口的近前)放入反向螺纹(reverse flight)而使树脂充满。将通气孔口的压力减压至50torr而去除反应后的副产物和过剩的甲胺。将从设置于挤出机出口的模头以股线方式出来的树脂,在水槽中冷却之后,利用造粒机进行颗粒化,从而获得酰亚胺树脂。
[0644]
接下来,使用螺杆直径40mm的同向旋转式双轴挤出机,将挤出机料筒温度设定为250℃,将螺杆转速设定为150rpm,以20kg/hr供给所获得的酰亚胺树脂,由捏合块使树脂熔融、充满之后,从喷嘴作为酯化剂注入碳酸二甲酯和三乙胺的混合液,进行树脂中的羧酸基的减少。相对于酰亚胺树脂100质量份,碳酸二甲酯设为3.2质量份,三乙胺设为0.8质量份。在反应区域的末端放入反向螺纹而使树脂充满。将通气孔口的压力减压至50torr而去除反应后的副产物和过剩的碳酸二甲酯。将从设置于挤出机出口的模头以股线方式出来的树脂,在水槽中冷却之后,利用造粒机进行颗粒化,从而获得具有戊二酰亚胺结构的甲基丙烯酸系树脂组合物j。
[0645]
此树脂组合物的酰亚胺化率为3.3%,戊二酰亚胺系结构单元的含量为5.2质量%。根据装置形状、运转条件计算双轴挤出机内的剪切速度,其为约80s-1
。
[0646]
另外,重均分子量mw为96,000,mw/mn为1.82,mz/mw为1.48,玻璃化转变温度为122℃。
[0647]
[实施例1~5、比较例1~5]
[0648]
使用由制造实施例和制造比较例获得的甲基丙烯酸系树脂组合物a~j,利用以下的方法注射成型树脂制棱镜,进行诸特性的测定。将结果示于表1。
[0649]
〈树脂制棱镜的成型〉
[0650]
将甲基丙烯酸系树脂组合物a~j在强制送风循环式的干燥器中于100℃干燥4小时之后,使用以一边12.7mm的直角等边三角形为底面的高度12.7mm的三角柱(三角柱,其形状为将一边12.7mm的立方体以包含一对相对面的对角线的面进行切断而成)的模具,利用夹模力50t的注射成型机,以料筒温度270℃、模具温度110℃、注射速度10mm/s,在50mpa以10秒的保压、以冷却时间100秒进行直角棱镜的注射成型。
[0651]
进一步地,在设定为比甲基丙烯酸系树脂组合物的玻璃化转变温度低25℃的温度的烘箱中退火3小时。
[0652]
[表1]
[0653][0654]
本发明的偏振分束器用树脂制棱镜,在形成偏振分束器时能够获得清晰的影像,因此能够有助于头戴式显示器等的显示设备的轻量化。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。