本发明属于有机化学合成领域,具体涉及一种通过连续电化学微反应器装置制备1,3-苯并噁嗪衍生物的方法。
背景技术:
近年来,含氮杂环化合物有着特殊的药物活性,毒性弱,内吸性好,常被用于农药和医药的组成结构单元。1,3-苯并噁嗪是一类含有氮氧原子的双环杂环化合物,在有机和药物化学中是一类极其重要和有用的分子结构,具有广泛的生物活性,比如抗癌、抗肿瘤、消炎、镇痛、杀虫、杀菌、植物生长调节等生物活性。此外,1,3-苯并噁嗪类化合物还是苯并噁嗪树脂的重要中间体和新型的光色化合物,1,3-苯并噁嗪树脂是近年来发展的一类新型热固性树脂,结构与酚醛树脂类似。该类树脂因具有良好的电学性能、耐热性能、耐磨性能、低表面活化能、抗吸湿性和优良的尺寸稳定性,在航空航天、电子封装材料、轨道交通等领域有广阔的应用前景。螺苯并噁嗪类化合物具有化学性质稳定,光响应快,抗疲劳性好等优点,在光信息记录、非银感成像、非线性光电器件、光过滤器、防伪以及装饰等领域具有很大的应用潜力。因此,研究1,3-苯并噁嗪衍生物的合成和应用日益受到广泛关注,近年来也取得了不少成果,使用较多的传统合成方法是利用邻氨基苯甲醇与醛或酮在乙酸或对甲苯磺酸的催化作用下经缩合反应来制备的。然而,这些传统的合成方法仍具有很多局限性,如2-氨基苯甲醇与羰基化合物的缩合反应局限于制备二氢-1,3-苯并噁嗪类化合物,且催化剂种类不多,而其它一些方法则往往针对某些特殊的反应物,普适性不广,还有繁琐的程序,苛刻的反应条件、有限的底物范围和贵重金属催化剂、氧化剂的参与及不利于环保等缺点。因此,仍然需要寻找其他步骤和原子经济性的方法来构建苯并噁嗪类衍生物。
随着社会的发展和资源的枯竭,绿色化学的发展已经成为科学家的使命之一。苯并噁嗪衍生物的研究也在积极响应“绿色化学”的号召,为此,本发明提供了一种通过连续电化学微反应器装置制备1,3-苯并噁嗪衍生物的方法。
技术实现要素:
发明目的:本发明所要解决的技术问题是针对现有技术的不足,提供一种通过连续电化学微反应器装置制备1,3-苯并噁嗪衍生物的方法。
为了解决上述技术问题,本发明提供了一种通过连续电化学微反应器装置制备1,3-苯并噁嗪衍生物的方法,如图2a所示,将式1所示的n-(2-苯乙烯基)苯甲酰胺类化合物、式2所示的二硒醚类化合物、电解质和溶剂的混合均相溶液泵入设有电极的微通道反应装置中进行连续电解质反应,得到式3所示的1,3-苯并噁嗪衍生物;
其中,
r1选自自苯、4-乙基苯、4-碘苯、4-氯苯、4-溴苯、4-甲氧基苯、4-硝基苯或萘环;优选地,r1选自4-乙基苯、4-碘苯、4-氯苯、4-溴苯、4-甲氧基苯、4-硝基苯、苯基;进一步优选地,r1选自苯、4-碘苯、4-氯苯或4-溴苯。
r2选自氢、2-甲基、3-甲基、4-甲基、4-氟、4-氯、4-溴、4-甲氧基、4-乙基、4-硝基、苯基、环戊烷、呋喃、3-氯或3-硝基;优选地,r2选自氢、4-甲基、3-氯;进一步优选地,r2选自氢、4-氯。
r选自甲基、乙基、苯基、4-氟苯基、4-氯苯基、4-硝基苯基、噻吩或4-甲氧基苯基;优选地,r选自苯基、乙基或4-甲氧基苯基;进一步优选地,r选自苯基。
其中,所述电解质为四丁基六氟磷酸铵、四丁基四氟硼酸铵、四丁基溴化铵、四丁基碘化铵和碘化钠中的任意一种或几种组合;优选地,所述电解质为四丁基四氟硼酸铵。
其中,所述溶剂为乙腈、甲醇、二氯甲烷、乙腈和水中任意一种或几种组合;优选地,所述溶剂为乙腈。
其中,所述混合均相溶液中,式1所示的n-(2-苯乙烯基)苯甲酰胺类化合物的浓度为0.01-0.05mmol/ml;优选地,所述混合均相溶液中,式1所示的n-(2-苯乙烯基)苯甲酰胺类化合物的浓度为0.02mmol/ml。
其中,所述混合均相溶液中,式2所示的二硒醚类化合物的浓度为0.01-0.03mmol/ml;优选地,所述混合均相溶液中,式2所示的二硒醚类化合物的浓度为0.01mmol/ml。
其中,所述混合均相溶液中,电解质的浓度为0.01-0.10mmol/ml;优选地,所述混合均相溶液中,电解质的浓度为0.04mmol/ml。
其中,所述设有电极的微通道反应装置包括进料泵、微反应器、阴极片、阳极片和接收器;其中,所述微反应器的两侧分别设有阴极片和阳极片;其中,所述进料泵、微反应器和接收器依次串联连接;所述连接为通过管道连接。
其中,所述阳极片为石墨碳电极或铂片电极;优选地,所述阳极片为石墨碳电极。
其中,所述阴极片为石墨碳电极或铂片电极;优选地,所述阴极片为铂片电极。
优选地,所述为微反应器的厂家为syrrisltd,名称为theasiafluxmodule,型号为modelno.2200554。
其中,所述微通道反应装置中管道等装置的材料为聚四氟乙烯。
其中,所述反应的温度为20-30℃;优选地,所述反应的温度为室温。
其中,所述反应的电流强度为5-15ma;优选地,所述反应的电流强度为10ma。
其中,所述反应的停留时间为0.25-5min;优选地,所述反应的停留时间为4min。
其中,当所述微反应器的体积为225μl时,所述混合均相溶液的流速为45-900μl/min;优选地,当所述微反应器的体积为225μl时,所述混合均相溶液的流速为56.25μl/min。
其中,所述反应结束后,收集为反应器的流出液,用乙酸乙酯稀释五倍后,经水洗、干燥、过滤后,用乙酸乙酯/石油醚(1:30)的混合溶剂淋洗下分离即得式3所示的1,3-苯并噁嗪衍生物。
其中,所述柱层析为在流动相为乙酸乙酯/石油醚的混合溶剂(体积比为1:10-1:30)淋洗下得到目标产物。
有益效果:与现有技术相比,本发明具有如下优势:
本发明所提供的方法无需添加贵重有机催化剂或金属催化剂,操作简单,安全性高,更加经济环保、绿色实用。可以有效克服传统合成路径的缺点,如反应时间长、反应温度高、原子效率低、成本高昂、不利环保等,解决该传统反应过程中步骤繁杂、反应时间长,需要昂贵催化剂,过量的强氧化剂,高反应温度和低原子效率等问题,并能够提高反应效率,适于工业化生产。
附图说明
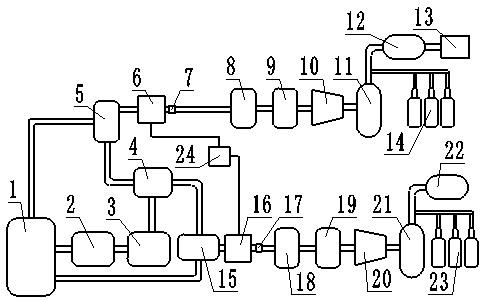
图1是本发明微通道电合成反应装置图。
图2为本发明的反应路径图。
图3为3a的氢谱和碳谱图。
图4为3b的氢谱和碳谱图。
图5为3c的氢谱和碳谱图。
图6为3d的氢谱和碳谱图。
图7为3e的氢谱和碳谱图。
图8为3f的氢谱和碳谱图。
图9为3g的氢谱和碳谱图。
图10为3h的氢谱和碳谱图。
图11为3i的氢谱和碳谱图。
图12为3j的氢谱和碳谱图。
图13为3k的氢谱和碳谱图。
图14为3l的氢谱和碳谱图。
具体实施方式
根据下述实施例,可以更好地理解本发明。然而,本领域的技术人员容易理解,实施例所描述的内容仅用于说明本发明,而不应当也不会限制权利要求书中所详细描述的本发明。
以下实施例所述的微通道反应装置,如图1所示,包括注射泵、微反应器、阴极片、阳极片和接收器;其中,所述微反应器的两侧分别设有阴极片(铂片)和阳极片(石墨碳);其中,所述进料泵、微反应器和接收器依次串联连接;所述连接为通过管道连接;所述微反应器的厂家为syrrisltd,名称为theasiafluxmodule,型号为modelno.2200554。
以下实施例中按照下述步骤:(1)将按比例配置好的混合均相溶液添加到注射泵中;(2)通过注射泵按照一定比例注入到微通道反应装置中进行混合和反应;(3)调节所需电流;(4)收集流出反应液,以过柱称重方法计算产物收率;通过高效液相测得产物收率,再经柱层析分离得到目标产物。
以下实施例中,若无特殊说明,所述反应的温度为室温。
其中,如表1所示的1,3-苯并噁嗪衍生物,均为通过本发明方法合成得到的产物,且经过核磁表征确证,如图3-图14所示。
表1
实施例1化合物3a的合成:
(1)将0.2mmol(0.045g)化合物1an-(2-苯乙烯基)苯甲酰胺,0.12mmol(0.037g)二苯基二硒醚2a和0.4mmol(0.132g)四丁基四氟硼酸铵溶于乙腈(10ml)溶剂中,得到均相溶液,添加到注射泵中;注射泵的注射流速为56.25μl/min;施加电流为10ma;微反应器反应体积v=225μl,反应时间4min;在微反应器反应历经一个周期后,收集反应液体,以hplc的方法计算产物收率为78%。反应液体用乙酸乙酯稀释五倍后,经水洗、干燥、过滤后,用乙酸乙酯/石油醚(1:30)的混合溶剂淋洗下分离得到产物3a。1hnmr(400mhz,chloroform-d)δ8.21–8.11(m,2h),7.52–7.40(m,5h),7.33–7.28(m,2h),7.24–7.20(m,3h),7.18–7.12(m,1h),7.06–7.00(m,1h),5.57(dd,j=8.2,5.0hz,1h),3.45–3.37(m,1h),3.28–3.20(m,1h).13cnmr(101mhz,chloroform-d)δ156.20,138.92,133.01,132.38,131.46,129.45,129.28,129.20,128.23,128.17,127.31,126.43,125.09,124.71,124.48,75.29,33.46.
(2)将0.2mmol(0.045g)化合物1an-(2-苯乙烯基)苯甲酰胺,0.12mmol(0.037g)二苯基二硒醚2a和0.4mmol(0.132g)四丁基四氟硼酸铵溶于乙腈(10ml)溶剂中,得到均相溶液,添加到注射泵中;注射泵的注射流速为56.25μl/min;施加电流为5ma;微反应器反应体积v=225μl,反应时间4min;在微反应器反应历经一个周期后,收集反应液体,以hplc的方法计算产物收率为58%。反应液体用乙酸乙酯稀释五倍后,经水洗、干燥、过滤后,用乙酸乙酯/石油醚(1:30)的混合溶剂淋洗下分离得到产物3a。
(3)将0.2mmol(0.045g)化合物1an-(2-苯乙烯基)苯甲酰胺,0.12mmol(0.037g)二苯基二硒醚2a和0.4mmol(0.132g)四丁基四氟硼酸铵溶于乙腈(10ml)溶剂中,得到均相溶液,添加到注射泵中;注射泵的注射流速为56.25μl/min;施加电流为15ma;微反应器反应体积v=225μl,反应时间4min;在微反应器反应历经一个周期后,收集反应液体,以hplc的方法计算产物收率为42%。反应液体用乙酸乙酯稀释五倍后,经水洗、干燥、过滤后,用乙酸乙酯/石油醚(1:30)的混合溶剂淋洗下分离得到产物3a。
(4)将0.2mmol(0.045g)化合物1an-(2-苯乙烯基)苯甲酰胺,0.12mmol(0.037g)二苯基二硒醚2a和0.4mmol(0.129g)四丁基溴化铵溶于乙腈(10ml)溶剂中,得到均相溶液,添加到注射泵中;注射泵a的注射流速为56.25μl/min;施加电流为10ma;微反应器反应体积v=225μl,反应时间4min;在微反应器反应历经一个周期后,收集反应液体,以hplc的方法计算产物收率为23%。反应液体用乙酸乙酯稀释五倍后,经水洗、干燥、过滤后,用乙酸乙酯/石油醚(1:30)的混合溶剂淋洗下分离得到产物3a。
(5)将0.2mmol(0.045g)化合物1an-(2-苯乙烯基)苯甲酰胺,0.12mmol(0.037g)二苯基二硒醚2a和0.4mmol(0.148g)四丁基碘化铵溶于乙腈(10ml)溶剂中,得到均相溶液,添加到注射泵中;注射泵的注射流速为56.25μl/min;施加电流为10ma;微反应器反应体积v=225μl,反应时间4min;在微反应器反应历经一个周期后,收集反应液体,以hplc的方法计算产物收率为28%。反应液体用乙酸乙酯稀释五倍后,经水洗、干燥、过滤后,用乙酸乙酯/石油醚(1:30)的混合溶剂淋洗下分离得到产物3a。
(6)将0.2mmol(0.045g)化合物1an-(2-苯乙烯基)苯甲酰胺,0.12mmol(0.037g)二苯基二硒醚2a和0.4mmol(0.155g)四丁基六氟磷酸铵溶于乙腈(10ml)溶剂中,得到均相溶液,添加到注射泵中;注射泵的注射流速为56.25μl/min;施加电流为10ma;微反应器反应体积v=225μl,反应时间4min;在微反应器反应历经一个周期后,收集反应液体,以hplc的方法计算产物收率为40%。反应液体用乙酸乙酯稀释五倍后,经水洗、干燥、过滤后,用乙酸乙酯/石油醚(1:30)的混合溶剂淋洗下分离得到产物3a。
实施例2化合物3b的合成:
将0.2mmol(0.050g)化合物1b4-乙基-n-(2-乙烯基苯基)苯甲酰胺,0.12mmol(0.037g)二苯基二硒醚2a和0.4mmol(0.132g)四丁基四氟硼酸铵溶于乙腈(10ml)溶剂中,得到均相溶液,添加到注射泵中;注射泵的注射流速为56.25μl/min;施加电流为10ma;微反应器反应体积v=225μl,反应时间4min;在微反应器反应历经一个周期后,收集反应液体,以hplc的方法计算产物收率为76%。反应液体用乙酸乙酯稀释五倍后,经水洗、干燥、过滤后,用乙酸乙酯/石油醚(1:30)的混合溶剂淋洗下分离得到产物3b。1hnmr(400mhz,chloroform-d)δ8.13–8.01(m,2h),7.55–7.45(m,2h),7.34–7.29(m,2h),7.28–7.20(m,5h),7.16–7.11(m,1h),7.05–7.00(m,1h),5.55(dd,j=8.2,5.0hz,1h),3.45–3.37(m,1h),3.26–3.19(m,1h),2.70(q,j=7.6hz,2h),1.26(t,j=7.6hz,3h).13cnmr(101mhz,chloroform-d)δ156.39,148.21,139.41,133.02,129.88,129.54,129.25,129.21,128.30,127.81,127.29,126.20,125.00,124.78,124.48,75.22,33.41,28.92,15.34.
实施例3化合物3c的合成:
将0.2mmol(0.070g)化合物1c4-碘-n-(2-乙烯基苯基)苯甲酰胺,0.12mmol(0.037g)二苯基二硒醚2a和0.4mmol(0.132g)四丁基四氟硼酸铵溶于乙腈(10ml)溶剂中,得到均相溶液,添加到注射泵中;注射泵的注射流速为56.25μl/min;施加电流为10ma;微反应器反应体积v=225μl,反应时间4min;在微反应器反应历经一个周期后,收集反应液体,以hplc的方法计算产物收率为82%。反应液体用乙酸乙酯稀释五倍后,经水洗、干燥、过滤后,用乙酸乙酯/石油醚(1:30)的混合溶剂淋洗下分离得到产物3c。1hnmr(400mhz,chloroform-d)δ7.86–7.81(m,2h),7.77–7.73(m,2h),7.48(ddt,j=6.9,4.7,2.1hz,2h),7.34–7.28(m,2h),7.24–7.19(m,3h),7.18–7.14(m,1h),7.03–6.99(m,1h),5.57(dd,j=8.3,4.8hz,1h),3.42–3.36(m,1h),3.25–3.20(m,1h).13cnmr(101mhz,chloroform-d)δ155.48,138.67,137.44,132.99,131.92,129.65,129.35,129.22,127.35,126.70,125.18,124.65,124.45,98.62,75.44,33.51.
实施例4化合物3d的合成:
将0.2mmol(0.052g)化合物1d4-氯-n-(2-乙烯基苯基)苯甲酰胺,0.12mmol(0.037g)二苯基二硒醚2a和0.4mmol(0.132g)四丁基四氟硼酸铵溶于乙腈(10ml)溶剂中,得到均相溶液,添加到注射泵中;注射泵的注射流速为56.25μl/min;施加电流为10ma;微反应器反应体积v=225μl,反应时间4min;在微反应器反应历经一个周期后,收集反应液体,以hplc的方法计算产物收率为86%。反应液体用乙酸乙酯稀释五倍后,经水洗、干燥、过滤后,用乙酸乙酯/石油醚(1:30)的混合溶剂淋洗下分离得到产物3d。1hnmr(400mhz,chloroform-d)δ8.09–8.02(m,2h),7.52–7.46(m,2h),7.40–7.36(m,2h),7.34–7.28(m,2h),7.24–7.19(m,3h),7.19–7.14(m,1h),7.05–7.01(m,1h),5.58(dd,j=8.3,4.8hz,1h),3.44–3.36(m,1h),3.27–3.20(m,1h).13cnmr(101mhz,chloroform-d)δ155.26,138.73,137.63,133.03,130.90,129.48,129.37,129.24,128.49,127.37,126.68,125.17,124.64,124.46,75.49,33.53.
实施例5化合物3e的合成:
将0.2mmol(0.060g)化合物1e4-溴-n-(2-乙烯基苯基)苯甲酰胺,0.12mmol(0.037g)二苯基二硒醚2a和0.4mmol(0.132g)四丁基四氟硼酸铵溶于乙腈(10ml)溶剂中,得到均相溶液,添加到注射泵中;注射泵的注射流速为56.25μl/min;施加电流为10ma;微反应器反应体积v=225μl,反应时间4min;在微反应器反应历经一个周期后,收集反应液体,以hplc的方法计算产物收率为80%。反应液体用乙酸乙酯稀释五倍后,经水洗、干燥、过滤后,用乙酸乙酯/石油醚(1:30)的混合溶剂淋洗下分离得到产物3e。1hnmr(400mhz,chloroform-d)δ8.02–7.95(m,2h),7.56–7.52(m,2h),7.51–7.46(m,2h),7.34–7.28(m,2h),7.24–7.20(m,3h),7.19–7.15(m,1h),7.05–7.01(m,1h),5.58(dd,j=8.3,4.8hz,1h),3.43–3.36(m,1h),3.26–3.20(m,1h).13cnmr(101mhz,chloroform-d)δ155.35,138.71,133.02,131.46,131.35,129.66,129.38,129.24,127.38,126.71,126.21,125.18,124.64,124.47,75.49,33.54.
实施例6化合物3f的合成:
将0.2mmol(0.051g)化合物1f4-甲氧基-n-(2-乙烯基苯基)苯甲酰胺,0.12mmol(0.037g)二苯基二硒醚2a和0.4mmol(0.132g)四丁基四氟硼酸铵溶于乙腈(10ml)溶剂中,得到均相溶液,添加到注射泵中;注射泵的注射流速为56.25μl/min;施加电流为10ma;微反应器反应体积v=225μl,反应时间4min;在微反应器反应历经一个周期后,收集反应液体,以hplc的方法计算产物收率为71%。反应液体用乙酸乙酯稀释五倍后,经水洗、干燥、过滤后,用乙酸乙酯/石油醚(1:20)的混合溶剂淋洗下分离得到产物3f。1hnmr(400mhz,chloroform-d)δ8.13–8.08(m,2h),7.53–7.48(m,2h),7.31–7.27(m,2h),7.24–7.20(m,3h),7.15–7.11(m,1h),7.04–7.00(m,1h),6.95–6.91(m,2h),5.54(dd,j=8.4,4.9hz,1h),3.85(s,3h),3.44–3.38(m,1h),3.26–3.20(m,1h).13cnmr(101mhz,chloroform-d)δ162.40,156.17,139.25,133.01,130.05,129.54,129.25,129.21,127.29,125.99,124.86,124.71,124.43,113.62,75.21,55.38,55.38,33.37.
实施例7化合物3g的合成:
将0.2mmol(0.054g)化合物1g4-硝基-n-(2-乙烯基苯基)苯甲酰胺,0.12mmol(0.037g)二苯基二硒醚2a和0.4mmol(0.132g)四丁基四氟硼酸铵溶于乙腈(10ml)溶剂中,得到均相溶液,添加到注射泵中;注射泵的注射流速为56.25μl/min;施加电流为10ma;微反应器反应体积v=225μl,反应时间4min;在微反应器反应历经一个周期后,收集反应液体,以hplc的方法计算产物收率为63%。反应液体用乙酸乙酯稀释五倍后,经水洗、干燥、过滤后,用乙酸乙酯/石油醚(1:20)的混合溶剂淋洗下分离得到产物3g。1hnmr(400mhz,chloroform-d)δ8.27–8.21(m,4h),7.52–7.46(m,2h),7.38–7.32(m,2h),7.25–7.19(m,4h),7.08–7.03(m,1h),5.67(dd,j=8.2,4.5hz,1h),3.45–3.39(m,1h),3.31–3.25(m,1h).13cnmr(101mhz,chloroform-d)δ154.92,149.43,138.28,138.21,133.00,129.56,129.28,129.23,128.91,127.55,127.46,125.65,124.54,124.51,75.89,33.72.
实施例8化合物3h的合成:
将0.2mmol(0.055g)化合物1hn-(2-乙烯基苯基)萘甲酰胺,0.12mmol(0.037g)二苯基二硒醚2a和0.4mmol(0.132g)四丁基四氟硼酸铵溶于乙腈(10ml)溶剂中,得到均相溶液a,添加到注射泵a中;注射泵a的注射流速为56.25μl/min;施加电流为10ma;微反应器反应体积v=225μl,反应时间4min;在微反应器反应历经一个周期后,收集反应液体,以hplc的方法计算产物收率为59%。反应液体用乙酸乙酯稀释五倍后,经水洗、干燥、过滤后,用乙酸乙酯/石油醚(1:30)的混合溶剂淋洗下分离得到产物3h。1hnmr(400mhz,chloroform-d)δ8.55(s,1h),8.18(dd,j=8.7,1.7hz,1h),7.89–7.84(m,1h),7.81–7.75(m,2h),7.49–7.41(m,4h),7.31–7.24(m,2h),7.15–7.07(m,4h),7.01–6.96(m,1h),5.56(dd,j=8.1,5.1hz,1h),3.43–3.36(m,1h),3.24–3.18(m,1h).13cnmr(101mhz,chloroform-d)δ155.25,138.03,133.90,131.97,131.72,128.69,128.47,128.32,128.19,127.84,126.87,126.68,126.48,126.27,125.48,125.32,124.14,123.76,123.64,123.53,74.40,32.49.
实施例9化合物3i的合成:
将0.2mmol(0.047g)化合物1in-(4-甲基-2-乙烯基苯基)苯甲酰胺,0.12mmol(0.037g)二苯基二硒醚2a和0.4mmol(0.132g)四丁基四氟硼酸铵溶于乙腈(10ml)溶剂中,得到均相溶液,添加到注射泵中;注射泵的注射流速为56.25μl/min;施加电流为10ma;微反应器反应体积v=225μl,反应时间4min;在微反应器反应历经一个周期后,收集反应液体,以hplc的方法计算产物收率为77%。反应液体用乙酸乙酯稀释五倍后,经水洗、干燥、过滤后,用乙酸乙酯/石油醚(1:30)的混合溶剂淋洗下分离得到产物3i。1hnmr(400mhz,chloroform-d)δ8.16–8.12(m,2h),7.51–7.48(m,2h),7.47–7.39(m,3h),7.23–7.19(m,4h),7.12–7.08(m,1h),6.81(s,1h),5.54(dd,j=8.3,4.8hz,1h),3.43–3.36(m,1h),3.26–3.21(m,1h),2.30(s,3h).13cnmr(101mhz,chloroform-d)δ155.52,136.46,136.36,133.00,132.49,131.28,129.87,129.54,129.16,128.20,128.05,127.25,124.97,124.92,124.50,75.35,33.58,21.20.
实施例10化合物3j的合成:
将0.2mmol(0.052g)化合物1jn-(5-氯-2-乙烯基苯基)苯甲酰胺,0.12mmol(0.037g)二苯基二硒醚2a和0.4mmol(0.132g)四丁基四氟硼酸铵溶于乙腈(10ml)溶剂中,得到均相溶液a,添加到注射泵a中;注射泵a的注射流速为56.25μl/min;施加电流为10ma;微反应器反应体积v=225μl,反应时间4min;在微反应器反应历经一个周期后,收集反应液体,以hplc的方法计算产物收率为81%。反应液体用乙酸乙酯稀释五倍后,经水洗、干燥、过滤后,用乙酸乙酯/石油醚(1:30)的混合溶剂淋洗下分离得到产物3j。1hnmr(400mhz,chloroform-d)δ8.16–8.11(m,2h),7.53–7.47(m,3h),7.45–7.41(m,2h),7.31(d,j=2.1hz,1h),7.26–7.19(m,3h),7.12–7.08(m,1h),6.97–6.93(m,1h),5.55(dd,j=7.7,5.4hz,1h),3.41–3.35(m,1h),3.24–3.18(m,1h).13cnmr(101mhz,chloroform-d)δ157.14,140.31,134.72,133.09,131.93,131.85,129.26,129.13,128.30,127.45,126.21,125.64,125.05,122.91,75.05,33.27.
实施例11化合物3k的合成:
将0.2mmol(0.045g)化合物1an-(2-乙烯基苯基)苯甲酰胺,0.12mmol(0.026g)二乙基二硒醚2k和0.4mmol(0.132g)四丁基四氟硼酸铵溶于乙腈(10ml)溶剂中,得到均相溶液,添加到注射泵中;注射泵的注射流速为56.25μl/min;施加电流为10ma;微反应器反应体积v=225μl,反应时间4min;在微反应器反应历经一个周期后,收集反应液体,以hplc的方法计算产物收率为68%。反应液体用乙酸乙酯稀释五倍后,经水洗、干燥、过滤后,用乙酸乙酯/石油醚(1:30)的混合溶剂淋洗下分离得到产物3k。1hnmr(400mhz,chloroform-d)δ8.22–8.16(m,2h),7.52–7.43(m,3h),7.37–7.31(m,2h),7.23–7.18(m,1h),7.15–7.11(m,1h),5.60(dd,j=7.7,5.6hz,1h),3.13–3.07(m,1h),2.96–2.90(m,1h),2.52(q,j=7.5hz,2h),1.33(t,j=7.5hz,3h).13cnmr(101mhz,chloroform-d)δ156.33,138.98,132.52,131.49,129.23,128.27,128.15,126.39,125.05,125.03,124.57,76.19,28.77,18.51,15.68.
实施例12化合物3l的合成:
将0.2mmol(0.045g)化合物1an-(2-乙烯基苯基)苯甲酰胺,0.12mmol(0.045g)二(4-甲氧基苯基)二硒醚2l和0.4mmol(0.132g)四丁基四氟硼酸铵溶于乙腈(10ml)溶剂中,得到均相溶液,添加到注射泵中;注射泵的注射流速为56.25μl/min;施加电流为10ma;微反应器反应体积v=225μl,反应时间4min;在微反应器反应历经一个周期后,收集反应液体,以hplc的方法计算产物收率为78%。反应液体用乙酸乙酯稀释五倍后,经水洗、干燥、过滤后,用乙酸乙酯/石油醚(1:30)的混合溶剂淋洗下分离得到产物3l。1hnmr(400mhz,chloroform-d)δ8.18–8.13(m,2h),7.50–7.41(m,5h),7.33–7.29(m,2h),7.18–7.13(m,1h),7.03–6.99(m,1h),6.79–6.75(m,2h),5.53(dd,j=8.2,4.9hz,1h),3.76(s,3h),3.35–3.29(m,1h),3.17–3.11(m,1h).13cnmr(101mhz,chloroform-d)δ159.53,156.27,138.97,135.97,132,45,131.44,129.21,128.19,126.39,125.07,124.85,124.47,119.17,114.91,75.22,55.24,34.54.
本发明提供了一种通过连续电化学微反应器装置制备1,3-苯并噁嗪衍生物的方法的思路及方法,具体实现该技术方案的方法和途径很多,以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。本实施例中未明确的各组成部分均可用现有技术加以实现。
本文用于企业家、创业者技术爱好者查询,结果仅供参考。