tioxazafen和阿塔鲁伦中间体的电化学制备方法
技术领域
1.本发明涉及杀线虫剂tioxazafen和治疗杜氏肌营养不良症(dmd)药物ataluren(阿塔鲁伦) 中间体的电氧化制备方法,具体是(z)
‑
n'
‑
羟基
‑
n
‑
(噻吩
‑2‑
基甲基)苯甲脒经电氧化制备 tioxazafen和(z)
‑
n
‑
(2
‑
氟苄基)
‑
n'
‑
羟基
‑3‑
甲基苯甲脒经电氧化制备5
‑
(2
‑
氟苯基)
‑3‑
(3
‑
甲苯 基)
‑
1,2,4
‑
噁二唑。
背景技术:
2.tioxazafen[ⅰa,化学名称为:3
‑
苯基
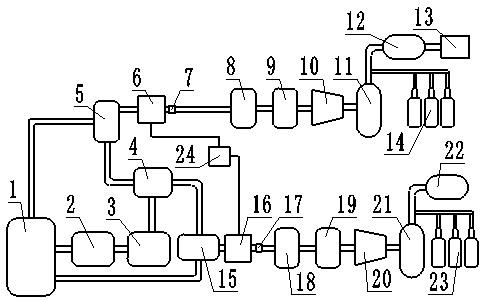
‑5‑
(2
‑
噻吩基)
‑
1,2,4
‑
噁二唑]是由孟山都公司合成的二 唑类的杀线虫剂。2013年,孟山都开发了tioxazafen。2015年,孟山都申请了tioxazafen在 北美自由贸易区(nafta)的联合评审。2017年5月1日,美国环保署(epa)批准登记了82.5% tioxazafen原药和45.9%(或541g/l)tioxazafen悬浮剂。作为新有效成分,其10年期的登记 资料保护权将终止于2027年4月30日。epa批准登记的tioxazafen悬浮剂(nemastrike),用 于玉米、大豆和棉花等三大作物,基于epa登记,nemastrike现已获得美国45个州登记, 于2018年种植季上市。
[0003]
噻唑膦(fosthiazate)和fluensufone是具有噻唑环结构的杀线虫剂,分子中含有n、s、p、 f、cl等元素,生产过程中废气和废水中含有有机硫和有机磷等化合物,对环境污染较大。 imicyafos是硫代磷酸酯类杀线虫剂,分子结构中也具有n、s、p元素,在合成过程中需要用 到二丙基硫、三氯化磷等化合物,对环境影响也较大。tioxazafen的分子结构中不含p元素, 合成过程中无需用到气味大、污染严重的原料,能达到清洁生产的效果,在不久的将来,杀 线虫剂tioxazafen必定能够代替噻唑膦、imicyafos、fluensufone成为杀线虫的主要品种。
[0004][0005]
作为一种新型、广谱、内吸性种子处理非熏蒸性杀线虫剂,tioxazafen具有全新的作用 机制,它能够通过影响线虫核糖体的活性,导致靶标线虫体内基因发生突变,从而发挥药效。 tioxazafen具有很好的选择性,只对为害寄生线虫有影响,对非靶标线虫无害。由于tioxazafen 悬浮剂在植物根部滞留时间较长,能够提供75天的持效期,因而能够有效防控两代线虫。 tioxazafen还能够增强作物根系的活力,因而能够显著提高作物产量[张明明.新型杀虫、杀螨 剂与植物生长调节剂的设计、合成.及生物活性研究.青岛科技大学博士论文,2019]。
[0006]
tioxazafen(ⅰa)的合成方法:刘安昌等[新型杀线虫剂tioxazafen的合成.农药,2014, 53(8):561
‑
563]报导了以2
‑
噻吩甲酰氯和n
‑
羟基苯甲脒为原料,乙酸丁酯为溶剂,氢氧化钠, 在室温下搅拌2h,然后升温回流继续反应4h得tioxazafen,收率86.3%。其中,合成n
‑
羟 基苯甲脒的收率82.9%,合成2
‑
噻吩甲酰氯的收率88.5%。该工艺使用了较大毒
性的苯甲腈 和具有强刺激性气味的二氯亚砜,污染环境且危害实验人员安全。
[0007][0008]
dahlb等[wo2006114400,2006年]描述了以2
‑
噻吩甲酰氯和n
‑
羟基苯甲脒为原料,以吡 啶为溶剂,回流下合成tioxazafenⅰa。该方法使用气味较强的吡啶为溶剂,严重污染环境。
[0009][0010]
deprez等[wo2013060744,2013年]描述了1,2,4
‑
噁二唑化合物的合成方法。该方法采 用高沸点的dmf为溶剂,2
‑
噻吩乙酰氯和n
‑
羟基苯甲脒回流反应得到tioxazafen。该工艺存 在dmf溶剂难于回收的问题。
[0011][0012]
miller等[wo 2014008257al,2014年]提供以苯甲腈,盐酸羟胺为原料,氢氧化钠,甲醇 为溶剂,60℃下合成了n'
‑
羟基苯甲脒,收率96.5%。向n'
‑
羟基苯甲脒的2
‑
甲基四氢呋喃溶 液中加入四丁基铵水溶液、氢氧化钠以及2
‑
噻吩羰基氯化物,70℃下反应,得tioxazafen, 收率95.0%。
[0013][0014]
kuram等[copper
‑
catalyzed direct synthesis of 1,2,4
‑
oxadiazoles from amides and organicnitriles by oxidative n
–
o bond formation.european journal of organic chemistry,2016,2016(3): 438
‑
442]描述了一种以氧气为氧化剂、铜为催化剂,噻吩甲酰胺与苯甲腈构建n—o键合成 tioxazafen的方法,收率53%。该工作使用了金属催化剂,危害环境。
[0015][0016]
市野川直辉等[cn107406437a,2017年]描述了n'
‑
羟基苯甲脒与2
‑
噻吩甲醛在氢氧化钾 作碱、叔戊醇为溶剂,102℃下反应3h得tioxazafen,收率96%。
[0017][0018]
wang等[base
‑
mediated one
‑
pot synthesis of 1,2,4
‑
oxadiazoles from nitriles,aldehydes andhydroxylamine hydrochloride without addition of extra oxidant.organic&biomolecularchemistry,2016,14(41):9814
‑
9822]以苯甲腈、醛类和盐酸羟胺为原料,一锅法合成了3,5
‑
二 取代的1,2,4
‑
噁二唑类化合物,收率73%。
[0019][0020]
张广平等[杀线虫剂tioxazafen关键中间体2
‑
噻吩甲酸的制备研究.精细化工中间体, 2020,50(4):18
‑
20]以噻吩和乙酰氯为原料制备2
‑
噻吩乙酮,随后用次氯酸钠氧化得到了 tioxazafen的关键中间体2
‑
噻吩甲酸,收率87.84%。
[0021][0022]
vinaya等[one pot synthesis of 3,5
‑
diaryl substituted
‑
1,2,4
‑
oxadiazoles usinggem
‑
dibromomethylarenes.canadian journal of chemistry,2019,97(9):690
‑
696]描述了2
‑
(二溴 甲基)噻吩与苯甲酰胺肟一锅法合成tioxazafen,收率89%。
[0023][0024]
patrick等[synthesis of 1,2,4
‑
oxadiazoles via ddq
‑
mediated oxidative cyclization ofamidoximes.synthesis:international journal of methods in synthetic organic chemistry,2016, 48(12):1902
‑
1909]描述了一种以氯苯甲醛肟和噻吩
‑2‑
基甲胺反应制得(z)
‑
n'
‑
羟基
‑
n
‑
(噻吩
‑2‑ꢀ
基甲基)苯甲脒,后者经2,3
‑
二氯
‑
5,6
‑
二
of chemistry,2014,38(7): 3062
‑
3070]选择3
‑
甲基苯甲酰胺盐酸盐与2
‑
氟苯甲酸反应,形成2
‑
氟
‑
n
‑
(亚氨基(3
‑
甲苯基)甲 基)苯甲酰胺,经亲核加成—脱氨基—分子内环化反应,得到5
‑
(2
‑
氟苯基)
‑3‑
(3
‑
甲苯基)
‑
1,2,4
‑ꢀ
噁二唑(ⅰb),ⅰb经高锰酸钾氧化合成阿塔鲁伦ⅱ,收率40%。
[0035][0036]
asad等[iodine
–
induced oxidative cyclisation of n
‑
acyl amidines:a rapid synthesis of 3, 5
‑
disubstituted
‑
1,2,4
‑
oxadiazoles.chemistryselect,2016,1(15):4753
‑
4757]通过碘诱导2
‑
氟
ꢀ‑
n
‑
(亚氨基(3
‑
甲苯基)甲基)苯甲酰胺的氧化环化合成5
‑
(2
‑
氟苯基)
‑3‑
(3
‑
甲苯基)
‑
1,2,4
‑
噁二唑 (ⅰb),再经氧化合成阿塔鲁伦ⅱ,收率48%。
[0037]
技术实现要素:
[0038]
本发明的目的一方面在于提供化学结构式ⅰ所示3
‑
苯基
‑5‑
芳基
‑
1,2,4
‑
噁二唑的电氧化 制备方法,其特征在于它的制备反应如下:
[0039][0040]
其中,r选自:氢、4
‑
氟、4
‑
氯、2
‑
溴、4
‑
叔丁基、3
‑
甲基或3,4
‑
二甲基;ar选自:苯基、 2
‑
噻吩基、2
‑
氟苯基、4
‑
氯苯基、2
‑
溴苯基、4
‑
氟苯基或4
‑
甲苯基。
[0041]
电氧化制备方法是在非隔膜式电解槽中,装上阳极工作电极和阴极,以(z)
‑
n'
‑
羟基
‑
n
‑
(芳 甲基)苯甲脒、有机溶剂、碱和电解质为电解液,在一定温度下,恒电压电解一定的时间,电 氧化反应得到3
‑
苯基
‑5‑
芳基
‑
1,2,4
‑
噁二唑(ⅰ)。
[0042]
电解槽的阳极工作电极选自:碳毡电极、铂网电极或石墨电极;阳极工作电极电流密度 选自:11.0ma/cm2~25.0ma/cm2;电解槽的阴极选自:铂片电极或碳毡电极。
[0043]
恒电压选自:1.50v~4.50v;电解温度选自:15℃~55℃;电解时间选自:2h~8h;
[0044]
电解液中的有机溶剂选自:乙腈、丙酮、甲醇、甲醇/水(体积比10/1)或甲醇/六氟异丙醇 (体积比5/5)。碱选自:磷酸钾、碳酸钾、醋酸钠、吡啶或三乙胺。
[0045]
电解质选自:四丁基高氯酸铵、高氯酸锂、四氟硼酸铵、四丁基六氟磷酸铵或六氟磷酸 钾;电解质浓度选自:0.02mol/l~0.1mol/l。
[0046]
电解液中(z)
‑
n'
‑
羟基
‑
n
‑
(芳甲基)苯甲脒的浓度选自:8g/l~30g/l。
[0047]
进一步优选,电解液的配制方法为:(z)
‑
n'
‑
羟基
‑
n
‑
(芳甲基)苯甲脒溶解于有机溶剂中得 到有机溶液,所述的有机溶液与磷酸钾以摩尔比1:3混合,得到混合溶液。
[0048]
本发明的目的进一步提供式ⅰ所示3
‑
苯基
‑5‑
芳基
‑
1,2,4
‑
噁二唑选自:
[0049][0050]
本发明的目的第二方面是提供了杀线虫剂tioxazafen(ⅰa)的制备新方法,其特征在于 选择苯甲醛经肟化、氯化、与噻吩
‑2‑
基甲胺取代制备中间体(z)
‑
n'
‑
羟基
‑
n
‑
(噻吩
‑2‑
基甲基)苯 甲脒,(z)
‑
n'
‑
羟基
‑
n
‑
(噻吩
‑2‑
基甲基)苯甲脒经电氧化制备得到tioxazafen,其制备反应如下:
[0051][0052]
本发明的目的第三方面是提供了一种经如结构式ⅰb所示化合物制备阿塔鲁伦新方法,其 特征在于选择3
‑
甲基苯甲醛经肟化、氯化、与2
‑
氟苄胺取代制备中间体(z)
‑
n
‑
(2
‑
氟苄基)
‑
n'
‑ꢀ
羟基
‑3‑
甲基苯甲脒,(z)
‑
n
‑
(2
‑
氟苄基)
‑
n'
‑
羟基
‑3‑
甲基苯甲脒经电氧化制备得到化合物ⅰb,后 者氧化制备阿塔鲁伦(ⅱ),其制备反应如下:
[0053][0054]
发明的有益技术效果为:
[0055]
(1)氧化反应中无需有毒或危险的氧化剂,“电子”就是清洁的反应试剂,是发展“绿色制药 工业”的重要组成部分。
[0056]
(2)相比于传统方法,电氧化方法具有反应时间较短,温度低,且未使用有毒试剂的优势。
[0057]
(3)在工业生产中既简化了工艺流程,降低了生产成本,又安全环保,适于大规模推广应 用。
具体实施方式
[0058]
以下实施例旨在说明本发明而不是对本发明的进一步限定。
[0059]
实施例1
[0060]
tioxazafen(ⅰa)的制备
[0061][0062]
(1)(z)
‑
n'
‑
羟基
‑
n
‑
(噻吩
‑2‑
基甲基)苯甲脒的制备
[0063]
0.53g(5.0mmol)苯甲醛,0.38g(5.5mmol)盐酸羟胺,0.59g(0.75mmol)吡啶溶于20ml乙 醇,室温搅拌2h,酸洗得到苯甲醛肟粗品,脱溶后直接用于下一步反应;将苯甲醛肟溶于 10ml etoh,并分五次添加1.0g(7.5mmol)n
‑
氯丁二酰亚胺,氮气保护下搅拌4h,得n
‑
羟 基苯甲酰氯粗品;0℃下将n
‑
羟基苯甲酰氯粗品加入0.56g(5.0mmol)噻吩
‑2‑
基甲胺和0.76 g(7.5mmol)三乙胺的四氢呋喃溶液中在氮气保护下搅拌2h,室温搅拌4h,用h2o稀释, 用乙酸乙酯萃取(3
×
20ml),饱和食盐水洗涤,无水硫酸钠干燥,过滤,脱溶,经柱层析(v
乙酸乙酯/石油醚
为1/2)分离得到0.967g白色固体(z)
‑
n'
‑
羟基
‑
n
‑
(噻吩
‑2‑
基甲基)苯甲脒,收率83.3%(以 苯甲醛计),熔点130~132℃。1h nmr(400mhz,dmso
‑
d6)δ:9.89(s,1h,oh),7.40(s, 5h,苯环),7.33(d,j=4.8hz,1h,噻吩环),6.91~6.88(m,1h,噻吩环),6.75(s,1h, 噻吩环),6.31(t,j=6.8hz,1h,nh),4.31(d,j=6.8hz,2h,ch2);
13
c nmr(101mhz, dmso
‑
d6)δ:154.70,144.81,132.75,129.50,128.63,127.20,125.05,124.75,42.28。
[0064]
(2)tioxazafenⅰa的电氧化制备
[0065]
在电解槽中,以0.12g(0.5mmol)(z)
‑
n'
‑
羟基
‑
n
‑
(噻吩
‑2‑
基甲基)苯甲脒为原料,铂网为阳 极,碳毡为阴极,0.32g(1.5mmol)k3po4,0.34g(0.1m)四丁基高氯酸铵为电解质,10mlmeoh为溶剂,室温恒电压3v电解2h,用乙酸乙酯萃取(3
×
20ml),饱和食盐水洗涤,无 水硫酸钠干燥,过滤,脱溶,得到粗产品;粗产品经柱层析(v
乙酸乙酯/石油醚
为1/16)分离得到0.092g 白色固体tioxazafenⅰa,收率80.7%,熔点108~109℃。1h nmr(400mhz,dmso
‑
d6)δ: 8.06(d,j=4.8hz,1h,苯环),8.02(d,j=4.8hz,3h,苯环 噻吩环),7.58~7.51(m,3h, 苯环 噻吩环),7.32(t,j=4.4hz,1h,噻吩环);
13
c nmr(101mhz,dmso
‑
d6)δ:171.53, 168.56,134.38,133.15,132.07,129.62,127.55,126.38,124.99。
[0066]
实施例2(对照实验1)
[0067]
tioxazafen(ⅰa)的制备
[0068][0069]3‑
苯基
‑5‑
(2
‑
噻吩基)
‑
1,2,4
‑
噁二唑按文献[synthesis of 1,2,4
‑
oxadiazoles via ddq
‑
mediatedoxidative cyclization of amidoximes.synthesis:international journal of methods in syntheticorganic chemistry,2016,48(12):1902
‑
1909]方法制备:在室温下,向圆底烧瓶中添加0.05g (0.22mmol)(z)
‑
n'
‑
羟基
‑
n
‑
(噻吩
‑2‑
基甲基)苯甲脒、2.2ml dmf和0.10g(0.44mmol)ddq, 并用氮气脱气5min,然后升温至150℃并搅拌30min。将反应混合物冷却至室温,用h2o淬 灭,并用乙酸乙酯萃取(3
×
20ml),饱和食盐水洗涤,无水硫酸钠干燥,过滤,脱溶,得到粗 产品。粗产品经快速色谱法纯化(硅胶,己烷
‑
etoac,10~50%)分离得到0.082g白色固体3
‑ꢀ
苯基
‑5‑
(2
‑
噻吩基)
‑
1,2,4
‑
噁二唑ⅰa,收率53.0%,熔点108~109℃。
[0070]
实施例3
[0071]
阿塔鲁伦ⅱ的制备
[0072][0073]
(1)(z)
‑
n
‑
(2
‑
氟苄基)
‑
n'
‑
羟基
‑3‑
甲基苯甲脒的制备
[0074]
0.60g(5.0mmol)3
‑
甲基苯甲醛,0.38g(5.5mmol)盐酸羟胺,0.59g(0.75mmol)吡啶溶于20 ml乙醇,室温搅拌2h,酸洗得到苯甲醛肟粗品,脱溶后直接用于下一步反应;将苯甲醛肟 溶于10ml etoh,并分五次添加1.0g(7.5mmol)n
‑
氯丁二酰亚胺,氮气搅拌4h,得n
‑
羟基 苯甲酰氯粗品;0℃下将n
‑
羟基苯甲酰氯粗品加入0.62g(5.0mmol)(2
‑
氟苯基)甲胺和0.76 g(7.5mmol)三乙胺的四氢呋喃溶液中在氮气保护下搅拌2h,室温搅拌4h,用h2o稀释, 用乙酸乙酯萃取(3
×
20ml),饱和食盐水洗涤,无水硫酸钠干燥,过滤,脱溶,得到粗产品; 粗产品经柱层析(v
乙酸乙酯/石油醚
为1/2)分离得到0.942g白色固体(z)
‑
n
‑
(2
‑
氟苄基)
‑
n'
‑
羟基
‑3‑
甲基 苯甲脒,收率73.0%(以3
‑
甲基苯甲醛计),熔点102~103℃。1h nmr(400mhz,dmso
‑
d6) δ:9.88(d,j=4.4hz,1h,oh),7.32(t,j=7.2hz,1h,苯环),7.21~7.27(m,2h,苯 环),7.15~7.21(m,2h,苯环),7.12(d,j=7.2hz,2h,苯环),7.02~7.09(m,1h,苯环), 6.25(t,j=6.0hz,1h,nh),4.20(d,j=6.4hz,2h,ch2),2.27(s,3h,ch3);
13
c nmr (101mhz,dmso
‑
d6)δ:161.23,158.81,155.20,137.83,132.58,130.10,129.18,128.53, 128.16,125.58,124.76,115.38,115.17,21.36。
[0075]
(2)5
‑
(2
‑
氟苯基)
‑3‑
(3
‑
甲苯基)
‑
1,2,4
‑
噁二唑(ⅰb)的制备
[0076]
在电解槽中,以0.13g(0.5mmol)(z)
‑
n
‑
(2
‑
氟苄基)
‑
n'
‑
羟基
‑3‑
甲基苯甲脒为原料,铂网为 阳极,铂片为阴极,0.32g(1.5mmol)k3po4,0.34g(0.1m)四丁基高氯酸铵为电解
质,10mlmeoh为溶剂,室温恒电压3v电解2h,用乙酸乙酯萃取(3
×
20ml),饱和食盐水洗涤,无 水硫酸钠干燥,过滤,脱溶,得到粗产品;粗产品经柱层析(v
乙酸乙酯/石油醚
为1/16)分离得到0.096g 白色固体5
‑
(2
‑
氟苯基)
‑3‑
(3
‑
甲苯基)
‑
1,2,4
‑
噁二唑ⅰb,收率75.6%,熔点99~102℃;1h nmr (400mhz,dmso
‑
d6)δ:8.17(dd,j=13.6,7.2hz,1h,苯环),7.85(d,j=8.8hz,2h, 苯环),7.76(dd,j=13.6,7.2hz,1h,苯环),7.49~7.53(m,1h,苯环),7.42~7.49(m, 2h,苯环),7.40(d,j=7.2hz,1h,苯环),2.39(s,3h,ch3);
13
c nmr(101mhz,dmso
‑
d6) δ:172.80,168.48,161.69,159.12,139.10,136.04,132.75,131.27,129.58,127.93,126.35, 125.87,124.73,117.81,117.60,112.25,21.32。
[0077]
(3)阿塔鲁伦ⅱ的制备
[0078]
按文献[synthesis of benzoic acids containing a 1,2,4
‑
oxadiazole ring.russian chemicalbulletin,2015,64(1):142
‑
145]方法制备:将0.33g(1.3mmol)5
‑
(2
‑
氟苯基)
‑3‑
(3
‑
甲苯基)
‑
1,2,4
‑ꢀ
噁二唑、0.23g(1.3mmol)醋酸钴和0.13g(1.3mmol)溴化钠溶解于40ml冰醋酸中。溶液加热 至95℃并通入空气,反应11h后冷却至-20℃,过滤得到0.313g阿塔鲁伦ⅱ,收率85.0%, 熔点241~242℃。
[0079]
实施例4(对照实验)
[0080]5‑
(2
‑
氟苯基)
‑3‑
(3
‑
甲苯基)
‑
1,2,4
‑
噁二唑(ⅰb)的制备
[0081][0082]5‑
(2
‑
氟苯基)
‑3‑
(3
‑
甲苯基)
‑
1,2,4
‑
噁二唑按文献[iodine
–
induced oxidative cyclisation ofn
‑
acyl amidines:a rapid synthesis of 3,5
‑
disubstituted
‑
1,2,4
‑
oxadiazoles.chemistry select, 2016,1(15):4753
‑
4757]方法制备:向已加入0.26g(1mmol)2
‑
氟
‑
n
‑
(亚氨基(3
‑
甲苯基)甲基)苯 甲酰胺及5mldmso的圆底烧瓶中加入0.38g(1.5mmol)碘和0.41g(3mmol)碳酸钾并将混合 物100℃下搅拌3h。反应完成后,将其冷却至室温,用10ml5%na2s2o3淬灭,然后加入15 ml盐水,并用乙酸乙酯(15
×
3ml)萃取。合并的有机层用无水硫酸钠干燥并减压浓缩。得 到的粗产物通过硅胶柱色谱纯化,使用乙酸乙酯和石油醚的混合物作为洗脱剂,得到0.220g 5
‑
(2
‑
氟苯基)
‑3‑
(3
‑
甲苯基)
‑
1,2,4
‑
噁二唑ⅰb,收率87%,熔点93~94℃。
[0083]
实施例5
[0084]
3,5
‑
二苯基
‑
1,2,4
‑
噁二唑的制备
[0085][0086]
(1)(z)
‑
n
‑
苄基
‑
n'
‑
羟基苯甲脒的制备
[0087]
按实施例1方法,反应12h,收率78.0%,熔点114~115℃;1h nmr(400mhz,dmso
‑
d6) δ:9.85(d,j=2.4hz,1h,oh),7.40
–
7.32(m,5h,苯环),7.26(t,j=7.2hz,2h,苯 环),7.19(d,j=6.8hz,1h,苯环),7.11(d,j=7.6hz,2h,苯环),6.30(t,j=7.2hz, 1h,nh),4.15(d,
j=6.8hz,2h,ch2);
13
c nmr(101mhz,dmso
‑
d6)δ:155.31,141.41, 132.90,129.41,128.81
–
128.45,127.03,46.89。
[0088]
(2)3,5
‑
二苯基
‑
1,2,4
‑
噁二唑的制备
[0089]
在电解槽中,以0.11g(0.5mmol)(z)
‑
n
‑
苄基
‑
n'
‑
羟基苯甲脒为原料,铂网为阳极,铂片 为阴极,0.32g(1.5mmol)k3po4,0.34g(0.1m)四丁基高氯酸铵为电解质,10ml meoh为 溶剂,室温恒电压3v电解2h,用乙酸乙酯萃取(3
×
20ml),饱和食盐水洗涤,无水硫酸钠 干燥,过滤,脱溶,得到粗产品。粗产品经柱层析(乙酸乙酯/石油醚为1/16)分离得到0.092g 白色固体3,5
‑
二苯基
‑
1,2,4
‑
噁二唑,收率82.7%,熔点109~111℃;1h nmr(400mhz,dmso
‑
d6) δ:8.20(d,j=7.2hz,2h,苯环),8.14~8.09(m,2h,苯环),7.75(t,j=7.2hz,1h,苯 环),7.68(t,j=7.6hz,2h,苯环),7.62(d,j=6.8hz,3h,苯环);
13
c nmr(101mhz,dmso
‑
d6)δ:175.89,168.73,133.83,132.13,130.03,129.74,128.38,127.57,126.61, 123.83。
[0090]
实施例6
[0091]3‑
(4
‑
氟苯基)
‑5‑
苯基
‑
1,2,4
‑
噁二唑的制备
[0092][0093]
(1)(z)
‑
n
‑
苄基
‑
n'
‑
羟基
‑4‑
氟苯甲脒的制备
[0094]
按实施例1方法,反应12h,收率75.0%,熔点90~92℃;1h nmr(400mhz,dmso
‑
d6) δ:9.90(s,1h,oh),7.41
–
7.34(m,2h,苯环),7.26(t,j=6.8hz,2h,苯环),7.19(dd, j=8.8,4.4hz,3h,苯环),7.11(d,j=7.2hz,2h,苯环),6.35(t,j=6.8hz,1h,nh), 4.15(d,j=7.2hz,2h,ch2);
13
c nmr(101mhz,dmso
‑
d6)δ:164.06,161.61,154.44, 141.31,130.71,130.63,129.36,129.33,128.67,127.08,127.01,115.71,115.49,46.87。
[0095]
(2)3
‑
(4
‑
氟苯基)
‑5‑
苯基
‑
1,2,4
‑
噁二唑的制备
[0096]
在电解槽中,以0.12g(0.5mmol)(z)
‑
n
‑
苄基
‑
n'
‑
羟基
‑4‑
氟苯甲脒为原料,铂网为阳极, 铂片为阴极,0.32g(1.5mmol)k3po4,0.34g(0.1m)四丁基高氯酸铵为电解质,10ml meoh 为溶剂,室温恒电压3v电解2h,用乙酸乙酯萃取(3
×
20ml),饱和食盐水洗涤,无水硫酸 钠干燥,过滤,脱溶,得到粗产品。粗产品经柱层析(乙酸乙酯/石油醚为1/16)分离得到0.085 g白色固体3
‑
(4
‑
氟苯基)
‑5‑
苯基
‑
1,2,4
‑
噁二唑,收率70.8%,熔点111~114℃;1h nmr(400 mhz,dmso
‑
d6)δ:8.22
–
8.12(m,4h,苯环),7.75(t,j=7.2hz,1h,苯环),7.67(t,j= 7.6hz,2h,苯环),7.44(t,j=8.8hz,2h,苯环);
13
c nmr(101mhz,dmso
‑
d6)δ:175.94, 167.91,165.71,163.23,133.86,130.15,130.06,130.02,128.37,123.74,123.18,117.01, 116.79。
[0097]
实施例7
[0098]3‑
(4
‑
氯苯基)
‑5‑
苯基
‑
1,2,4
‑
噁二唑的制备
[0099]
[0100]
(1)(z)
‑
n
‑
苄基
‑
n'
‑
羟基
‑4‑
氯苯甲脒的制备
[0101]
按实施例1方法,反应12h,收率77.0%,熔点117~119℃;1h nmr(400mhz,dmso
‑
d6) δ:9.98(s,1h,oh),7.42(d,j=8.4hz,2h,苯环),7.35(d,j=8.4hz,2h,苯环),7.26 (t,j=7.2hz,2h,苯环),7.19(d,j=7.2hz,1h,苯环),7.11(d,j=7.2hz,2h,苯环), 6.37(t,j=7.2hz,1h,nh),4.15(d,j=7.2hz,2h,ch2);
13
c nmr(101mhz,dmso
‑
d6) δ:154.33,141.26,134.06,131.82,130.26,128.75,128.69,127.10,127.01,46.88。
[0102]
(2)3
‑
(4
‑
氯苯基)
‑5‑
苯基
‑
1,2,4
‑
噁二唑的制备
[0103]
在电解槽中,以0.13g(0.5mmol)(z)
‑
n
‑
苄基
‑
n'
‑
羟基
‑4‑
氯苯甲脒为原料,铂网为阳极, 铂片为阴极,0.32g(1.5mmol)k3po4,0.34g(0.1m)四丁基高氯酸铵为电解质,10ml meoh 为溶剂,室温恒电压3v电解2h,用乙酸乙酯萃取(3
×
20ml),饱和食盐水洗涤,无水硫酸 钠干燥,过滤,脱溶,得到粗产品。粗产品经柱层析(乙酸乙酯/石油醚为1/16)分离得到0.095 g白色固体3
‑
(4
‑
氯苯基)
‑5‑
苯基
‑
1,2,4
‑
噁二唑,收率74.2%,熔点104~106℃;1h nmr(400 mhz,dmso
‑
d6)δ:8.19(d,j=7.2hz,2h,苯环),8.11(d,j=8.4hz,2h,苯环),7.75(d, j=7.2hz,1h,苯环),7.68(dd,j=7.7,6.8hz,4h,苯环);
13
c nmr(101mhz,dmso
‑
d6) δ:176.09,167.95,136.87,133.95,130.07,129.94,129.38,128.42,125.47,123.71。
[0104]
实施例8
[0105]3‑
(2
‑
溴苯基)
‑5‑
苯基
‑
1,2,4
‑
噁二唑的制备
[0106][0107]
(1)(z)
‑
n
‑
苄基
‑
n'
‑
羟基
‑2‑
溴苯甲脒的制备
[0108]
按实施例1方法,反应12h,收率82.0%,熔点124~125℃;1h nmr(400mhz,dmso
‑
d6) δ:9.66(s,1h,oh),7.68~7.62(m,1h,苯环),7.36
–
7.26(m,3h,苯环),7.24(t,j=7.2 hz,2h,苯环),7.20~7.10(m,2h,苯环),7.07(d,j=7.2hz,2h,苯环),6.53(t,j=6.8 hz,1h,nh),3.95(d,j=6.0hz,2h,ch2);
13
c nmr(101mhz,dmso
‑
d6)δ:153.52, 141.04,133.81,132.90,132.35,131.28,128.56,127.78,127.15,123.48,46.31。
[0109]
(2)3
‑
(2
‑
溴苯基)
‑5‑
苯基
‑
1,2,4
‑
噁二唑的制备
[0110]
在电解槽中,以0.15g(0.5mmol)(z)
‑
n
‑
苄基
‑
n'
‑
羟基
‑2‑
溴苯甲脒为原料,铂网为阳极,铂 片为阴极,0.32g(1.5mmol)k3po4,0.34g(0.1m)四丁基高氯酸铵为电解质,10ml meoh 为溶剂,室温恒电压3v电解2h,用乙酸乙酯萃取(3
×
20ml),饱和食盐水洗涤,无水硫酸 钠干燥,过滤,脱溶,得到粗产品。粗产品经柱层析(乙酸乙酯/石油醚为1/16)分离得到0.090 g白色固体3
‑
(2
‑
溴苯基)
‑5‑
苯基
‑
1,2,4
‑
噁二唑,收率60.0%,熔点77~79℃;1h nmr(400mhz, dmso
‑
d6)δ:8.19(d,j=8.0hz,2h,苯环),8.03(d,j=8.0hz,2h,苯环),7.82(d,j= 8.0hz,2h,苯环),7.75(t,j=8.0hz,1h,苯环),7.68(t,j=8.0hz,2h,苯环);
13
c nmr (101mhz,dmso
‑
d6)δ:176.11,168.07,133.93,132.85,130.05,129.53,128.42,125.78, 123.71。
[0111]
实施例9
[0112]3‑
(4
‑
叔丁基苯基)
‑5‑
苯基
‑
1,2,4
‑
噁二唑的制备
[0113][0114]
(1)(z)
‑
n
‑
苄基
‑
n'
‑
羟基
‑4‑
叔丁基苯甲脒的制备
[0115]
按实施例1方法,反应12h,收率68.0%,熔点143~146℃;1h nmr(400mhz,dmso
‑
d6) δ:9.82(s,1h,oh),7.39(d,j=8.4hz,2h,苯环),7.28(dd,j=15.8,8.4hz,4h,苯 环),7.17(dd,j=21.6,7.2hz,3h,苯环),6.20(t,j=7.2hz,1h,nh),4.17(d,j=7.2 hz,2h,ch2),1.27(s,9h,c4h9);
13
c nmr(101mhz,dmso
‑
d6)δ:155.24,151.92,141.47, 130.03,128.68,128.25,127.01,125.42,46.94,34.86,31.49。
[0116]
(2)3
‑
(4
‑
叔丁基苯基)
‑5‑
苯基
‑
1,2,4
‑
噁二唑的制备
[0117]
在电解槽中,以0.14g(0.5mmol)(z)
‑
n
‑
苄基
‑
n'
‑
羟基
‑4‑
叔丁基苯甲脒为原料,铂网为阳极, 铂片为阴极,0.32g(1.5mmol)k3po4,0.34g(0.1m)四丁基高氯酸铵为电解质,10ml meoh 为溶剂,室温恒电压3v电解2h,用乙酸乙酯萃取(3
×
20ml),饱和食盐水洗涤,无水硫酸 钠干燥,过滤,脱溶,得到粗产品。粗产品经柱层析(乙酸乙酯/石油醚为1/16)分离得到0.085 g无色油状液体3
‑
(4
‑
叔丁基苯基)
‑5‑
苯基
‑
1,2,4
‑
噁二唑,收率61.1%,;1h nmr(400mhz, dmso
‑
d6)δ:8.16(d,j=7.2hz,2h,苯环),8.00(d,j=8.0hz,2h,苯环),7.71(t,j=6.8 hz,1h,苯环),7.63(t,j=7.6hz,2h,苯环),7.56(d,j=8.4hz,2h,苯环),1.29(s, 9h,ch3);
13
c nmr(101mhz,dmso
‑
d6)δ:175.65,168.60,154.80,133.64,129.90,128.28, 127.36,126.37,123.88,35.11,31.26。
[0118]
实施例10
[0119]3‑
(3
‑
甲基苯基)
‑5‑
苯基
‑
1,2,4
‑
噁二唑的制备
[0120][0121]
(1)(z)
‑
n
‑
苄基
‑
n'
‑
羟基
‑3‑
甲基苯甲脒的制备
[0122]
按实施例1方法,反应12h,收率78.0%,熔点107~109℃;1h nmr(400mhz,dmso
‑
d6) δ:9.81(s,1h,oh),7.25(dd,j=15.2,7.6hz,3h,苯环),7.16
‑
7.21(m,2h,苯环), 7.13(d,j=10.8hz,4h,苯环),6.25(t,j=6.8hz,1h,nh),4.14(d,j=7.2hz,2h, ch2),2.27(s,3h,ch3);
13
c nmr(101mhz,dmso
‑
d6)δ:155.41,141.47,137.78,132.82, 130.02,129.16,128.57,127.05,125.69,46.96,21.38。
[0123]
(2)3
‑
(3
‑
甲基苯基)
‑5‑
苯基
‑
1,2,4
‑
噁二唑的制备
[0124]
在电解槽中,以0.12g(0.5mmol)(z)
‑
n
‑
苄基
‑
n'
‑
羟基
‑3‑
甲基苯甲脒为原料,铂网为阳极, 铂片为阴极,0.32g(1.5mmol)k3po4,0.34g(0.1m)四丁基高氯酸铵为电解质,10ml meoh 为溶剂,室温恒电压3v电解2h,用乙酸乙酯萃取(3
×
20ml),饱和食盐水洗涤,无水硫酸 钠干燥,过滤,脱溶,得到粗产品。粗产品经柱层析(乙酸乙酯/石油醚为1/16)分离得到0.075 g白色固体3
‑
(3
‑
甲基苯基)
‑5‑
苯基
‑
1,2,4
‑
噁二唑,收率64.7%,熔点87~89℃;1h nmr(400 mhz,dmso
‑
d6)δ:8.19(d,j=8.0hz,2h,苯环),7.90(d,j=8.0hz,2h,苯环),7.75
(t, j=6.0hz,1h,苯环),7.67(t,j=8.0hz,2h,苯环),7.41
‑
7.52(m,2h,苯环),2.42(s, 3h,ch3);
13
c nmr(101mhz,dmso
‑
d6)δ:175.77,168.78,139.09,133.74,132.70,129.98, 129.57,128.34,127.95,126.56,124.72,123.87,21.35。
[0125]
实施例11
[0126]3‑
(3,4
‑
二甲基苯基)
‑5‑
苯基
‑
1,2,4
‑
噁二唑的制备
[0127][0128]
(1)(z)
‑
n
‑
苄基
‑
n'
‑
羟基
‑
3,4
‑
二甲基苯甲脒的制备
[0129]
按实施例1方法,反应12h,收率64.0%,熔点116~118℃;1h nmr(400mhz,dmso
‑
d6) δ:9.76(s,1h,oh),7.27(t,j=7.2hz,2h,苯环),7.19(d,j=7.2hz,1h,苯环),7.12 (t,j=8.8hz,4h,苯环),7.07(d,j=7.6hz,1h,苯环),6.20(t,j=6.8hz,1h,nh), 4.15(d,j=6.8hz,2h,ch2),2.20(s,3h,ch3),2.18(s,3h,ch3);
13
c nmr(101mhz, dmso
‑
d6)δ:155.40,141.54,137.56,136.42,130.35,129.63,128.65,127.02,125.96, 46.97,19.71。
[0130]
(2)3
‑
(3,4
‑
二甲基苯基)
‑5‑
苯基
‑
1,2,4
‑
噁二唑的制备
[0131]
在电解槽中,以0.12g(0.5mmol)(z)
‑
n
‑
苄基
‑
n'
‑
羟基
‑
3,4
‑
二甲基苯甲脒为原料,铂网为阳 极,铂片为阴极,0.32g(1.5mmol)k3po4,0.34g(0.1m)四丁基高氯酸铵为电解质,10mlmeoh为溶剂,室温恒电压3v电解2h,用乙酸乙酯萃取(3
×
20ml),饱和食盐水洗涤,无 水硫酸钠干燥,过滤,脱溶,得到粗产品。粗产品经柱层析(乙酸乙酯/石油醚为1/16)分离得 到0.095g白色固体3
‑
(3,4
‑
二甲基苯基)
‑5‑
苯基
‑
1,2,4
‑
噁二唑,收率76.0%,熔点112~114℃; 1
h nmr(400mhz,dmso
‑
d6)δ:8.18(d,j=7.2hz,2h,苯环),7.87(s,1h,苯环),7.81 (d,j=8.0hz,1h,苯环),7.74(t,j=7.6hz,1h,苯环),7.67(t,j=7.6hz,2h,苯环), 7.35(d,j=8.0hz,1h,苯环),2.33(s,3h,ch3),2.31(s,3h,ch3);
13
c nmr(101mhz, dmso
‑
d6)δ:175.67,168.78,140.89,137.81,133.75,130.74,130.02,128.35,125.08, 124.12,123.91,19.86。
[0132]
实施例12
[0133]5‑
(2
‑
氟苯基)
‑3‑
苯基
‑
1,2,4
‑
噁二唑的制备
[0134][0135]
(1)(z)
‑
n
‑
(2
‑
氟苄基)
‑
n'
‑
羟基苯甲脒的制备
[0136]
按实施例1方法,反应12h,收率83.0%,熔点100~103℃;1h nmr(400mhz,dmso
‑
d6) δ:9.99
–
9.90(m,1h,oh),7.41
–
7.28(m,6h,苯环),7.28
–
7.21(m,1h,苯环),7.15(t, j=7.6hz,1h,苯环),7.05(t,j=9.4hz,1h,苯环),6.28(d,j=6.0hz,1h,nh),4.21 (d,j=4.4hz,2h,ch2);
13
c nmr(101mhz,dmso
‑
d6)δ:1161.23,158.80,155.12,132.66, 129.49,129.17,128.66,128.46,128.07,124.76,115.39,115.18。
[0137]
(2)5
‑
(2
‑
氟苯基)
‑3‑
苯基
‑
1,2,4
‑
噁二唑的制备
[0138]
在电解槽中,以0.12g(0.5mmol)(z)
‑
n
‑
(2
‑
氟苄基)
‑
n'
‑
羟基苯甲脒为原料,铂网
为阳极, 铂片为阴极,0.32g(1.5mmol)k3po4,0.34g(0.1m)四丁基高氯酸铵为电解质,10ml meoh 为溶剂,室温恒电压3v电解2h,用乙酸乙酯萃取(3
×
20ml),饱和食盐水洗涤,无水硫酸 钠干燥,过滤,脱溶,得到粗产品。粗产品经柱层析(乙酸乙酯/石油醚为1/16)分离得到0.090 g白色固体5
‑
(2
‑
氟苯基)
‑3‑
苯基
‑
1,2,4
‑
噁二唑,收率75.0%,熔点100~102℃;1h nmr(400 mhz,dmso
‑
d6)δ:8.21(s,1h,苯环),8.06
‑
8.12(m,2h,苯环),7.78(d,j=3.6hz, 1h,苯环),7.45
‑
7.64(m,5h,苯环);
13
c nmr(101mhz,dmso
‑
d6)δ:172.90,168.46, 161.71,159.15,136.11,132.17,131.32,129.74,127.59,126.44,125.92,117.85,117.65, 112.27。
[0139]
实施例13
[0140]5‑
(4
‑
氟苯基)
‑3‑
苯基
‑
1,2,4
‑
噁二唑的制备
[0141][0142]
(1)(z)
‑
n
‑
(4
‑
氟苄基)
‑
n'
‑
羟基苯甲脒的制备
[0143]
按实施例1方法,反应12h,收率80.0%,熔点90~93℃;1h nmr(400mhz,dmso
‑
d6) δ:9.86(d,j=2.8hz,1h,oh),7.35(dd,j=6.4,5.6hz,5h,苯环),7.10(dt,j=19.2, 8.0hz,4h,苯环),6.34(t,j=6.7hz,1h,nh),4.12(d,j=6.9hz,2h,ch2);
13
c nmr (101mhz,dmso
‑
d6)δ:162.69,160.28,155.15,137.57,132.90,129.41,128.96,128.58, 115.45,115.24,46.21。
[0144]
(2)5
‑
(4
‑
氟苯基)
‑3‑
苯基
‑
1,2,4
‑
噁二唑的制备
[0145]
在电解槽中,以0.12g(0.5mmol)(z)
‑
n
‑
(4
‑
氟苄基)
‑
n'
‑
羟基苯甲脒为原料,铂网为阳极, 铂片为阴极,0.32g(1.5mmol)k3po4,0.34g(0.1m)四丁基高氯酸铵为电解质,10ml meoh 为溶剂,室温恒电压3v电解2h,用乙酸乙酯萃取(3
×
20ml),饱和食盐水洗涤,无水硫酸 钠干燥,过滤,脱溶,得到粗产品。粗产品经柱层析(乙酸乙酯/石油醚为1/16)分离得到0.085 g白色固体5
‑
(4
‑
氟苯基)
‑3‑
苯基
‑
1,2,4
‑
噁二唑,收率70.8%,熔点108~110℃;1h nmr(400 mhz,dmso
‑
d6)δ:8.28
–
8.21(m,2h,苯环),8.08(d,j=6.4hz,2h,苯环),7.60(d,j =6.8hz,3h,苯环),7.50(t,j=8.0hz,2h,苯环);
13
c nmr(101mhz,dmso
‑
d6)δ: 175.05,168.74,166.68,164.17,132.15,131.29,129.73,127.57,126.55,120.57,117.43, 117.21。
[0146]
实施例14
[0147]5‑
(4
‑
氯苯基)
‑3‑
苯基
‑
1,2,4
‑
噁二唑的制备
[0148][0149]
(1)(z)
‑
n
‑
(4
‑
氯苄基)
‑
n'
‑
羟基苯甲脒的制备
[0150]
按实施例1方法,反应12h,收率82.0%,熔点110~112℃;1h nmr(400mhz,dmso
‑
d6) δ:9.87(s,1h,oh),7.36(d,j=6.8hz,3h,苯环),7.31(d,j=8.0hz,4h,苯环),7.12 (d,j=
8.4hz,2h,苯环),6.40(t,j=6.8hz,1h,nh),4.14(d,j=7.2hz,2h,ch2);
[0151]
(2)5
‑
(4
‑
氯苯基)
‑3‑
苯基
‑
1,2,4
‑
噁二唑的制备
[0152]
在电解槽中,以0.13g(0.5mmol)(z)
‑
n
‑
(4
‑
氯苄基)
‑
n'
‑
羟基苯甲脒为原料,铂网为阳极, 铂片为阴极,0.32g(1.5mmol)k3po4,0.34g(0.1m)四丁基高氯酸铵为电解质,10ml meoh 为溶剂,室温恒电压3v电解2h,用乙酸乙酯萃取(3
×
20ml),饱和食盐水洗涤,无水硫酸 钠干燥,过滤,脱溶,得到粗产品。粗产品经柱层析(乙酸乙酯/石油醚为1/16)分离得到0.100 白色固体5
‑
(4
‑
氯苯基)
‑3‑
苯基
‑
1,2,4
‑
噁二唑,收率78.1%,熔点125~127℃;1h nmr(400mhz, dmso
‑
d6)δ:8.18(d,j=8.4hz,2h,苯环),8.08(d,j=6.0hz,2h,苯环),7.72(d,j= 8.8hz,2h,苯环),7.61(t,j=5.6hz,3h,苯环);
13
c nmr(101mhz,dmso
‑
d6)δ:175.05, 168.79,138.68,132.18,130.20,129.74,127.57,126.48,122.70。
[0153]
实施例15
[0154]5‑
(2
‑
溴苯基)
‑3‑
苯基
‑
1,2,4
‑
噁二唑的制备
[0155][0156]
(1)(z)
‑
n
‑
(2
‑
溴苄基)
‑
n'
‑
羟基苯甲脒的制备
[0157]
按实施例1方法,反应12h,收率75.0%,熔点110~112℃;1h nmr(400mhz,dmso
‑
d6) δ:9.84(d,j=2.4hz,1h,oh),7.42(d,j=8.4hz,2h,苯环),7.27
‑
7.37(m,5h,苯 环),7.04(d,j=8.0hz,2h,苯环),6.38(t,j=6.8hz,1h,nh),4.09(d,j=7.2hz, 2h,ch2);
13
c nmr(101mhz,dmso
‑
d6)δ:155.05,140.98,132.82,131.49,129.35,128.58, 120.03,46.27。
[0158]
(2)5
‑
(2
‑
溴苯基)
‑3‑
苯基
‑
1,2,4
‑
噁二唑的制备
[0159]
在电解槽中,以0.15g(0.5mmol)(z)
‑
n
‑
(2
‑
溴苄基)
‑
n'
‑
羟基苯甲脒为原料,铂网为阳极, 铂片为阴极,0.32g(1.5mmol)k3po4,0.34g(0.1m)四丁基高氯酸铵为电解质,10ml meoh 为溶剂,室温恒电压3v电解2h,用乙酸乙酯萃取(3
×
20ml),饱和食盐水洗涤,无水硫酸 钠干燥,过滤,脱溶,得到粗产品。粗产品经柱层析(乙酸乙酯/石油醚为1/16)分离得到0.105 g白色固体5
‑
(2
‑
溴苯基)
‑3‑
苯基
‑
1,2,4
‑
噁二唑,收率70.0%,熔点60~62℃;1h nmr(400mhz, dmso
‑
d6)δ:8.08(t,j=8.4hz,4h,苯环),7.86(d,j=8.4hz,2h,苯环),7.60(q,j=6.0 hz,3h,苯环);
13
c nmr(101mhz,dmso
‑
d6)δ:175.18,168.80,133.14,132.19,130.27, 129.74,127.65,126.48,123.03。
[0160]
实施例16
[0161]5‑
(4
‑
甲基苯基)
‑3‑
苯基
‑
1,2,4
‑
噁二唑的制备
[0162][0163]
(1)(z)
‑
n
‑
(4
‑
甲基苄基)
‑
n'
‑
羟基苯甲脒的制备
[0164]
按实施例1方法,反应12h,收率78.0%,熔点101~103℃;1h nmr(400mhz,dmso
‑
d6) δ:9.84(s,1h,oh),7.36(s,5h,苯环),7.06(d,j=8.0hz,2h,苯环),7.00(d,j=8.0 hz,2h,苯环),6.21(t,j=6.8hz,1h,nh),4.10(d,j=6.8hz,2h,ch2),2.24(s,3h, ch3);
13
c nmr
(101mhz,dmso
‑
d6)δ:155.34,138.31,136.08,132.95,129.31,128.58, 127.01,46.70,21.10。
[0165]
(2)5
‑
(4
‑
甲基苯基)
‑3‑
苯基
‑
1,2,4
‑
噁二唑的制备
[0166]
在电解槽中,以0.12g(0.5mmol)(z)
‑
n
‑
(4
‑
甲基苄基)
‑
n'
‑
羟基苯甲脒为原料,铂网为阳极, 铂片为阴极,0.32g(1.5mmol)k3po4,0.34g(0.1m)四丁基高氯酸铵为电解质,10ml meoh 为溶剂,室温恒电压3v电解2h,用乙酸乙酯萃取(3
×
20ml),饱和食盐水洗涤,无水硫酸 钠干燥,过滤,脱溶,得到粗产品。粗产品经柱层析(乙酸乙酯/石油醚为1/16)分离得到0.075 g白色固体5
‑
(4
‑
甲基苯基)
‑3‑
苯基
‑
1,2,4
‑
噁二唑,收率64.7%,熔点113~115℃;1h nmr(400 mhz,dmso
‑
d6)δ:8.09(dd,j=7.6,3.2hz,4h,苯环),7.61(d,j=7.2hz,3h,苯环), 7.48(d,j=8.0hz,2h,苯环),2.44(s,3h,苯环);
13
c nmr(101mhz,dmso
‑
d6)δ:175.98, 168.67,144.28,132.08,130.59,129.72,128.36,127.56,126.71,121.14,21.73。
[0167]
在本说明书中,本发明已参照其特定的实施例作了描述。但是,很显然仍可以作出各种 修改和变换而不背离本发明的精神和范围。因此,说明书应被认为是说明性的而非限制性的。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。