1.本发明属于无机材料制备技术领域,具体涉及一种氯氧化铋晶体的制备方法,还涉及由该制备方法制得的氯氧化铋晶体。
背景技术:
2.氯氧化铋是一种银白色有珍珠光泽的粉末,常见的晶体形貌有正方形、正八边形及圆形。它避免了传统无机珠光颜料碳酸铅和砷酸氧铅的毒性,克服了云母基珠光颜料较大厚度的结构和复杂的预处理工序的缺点,具有无毒、附着性好、耐腐蚀及良好的色泽等优良性能,在化妆品、汽车内饰材料、服装饰品等领域拥有广阔的市场前景。应用在不同领域的氯氧化铋,尺寸的选择有所不同,而现有的制备方法中氯氧化铋晶体的尺寸、分散性不能够得到良好的控制,提供可以制备出尺寸可控、单分散、光泽度高、形貌规整且产率高的方法,将使氯氧化铋得到更好的实际应用。
3.公开号为cn103663378a和cn101935022a的中国专利申请分别制备了8
‑
20μm和8
‑
15μm的正方形氯氧化铋晶体,尺寸可控范围小,其在制备过程中采用氢氧化钠调控体系ph,故对氢氧化钠的滴加速度要求高,否则体系ph波动大产生副产物。
4.公开号为cn110550657a和cn104386746a的中国专利申请均采用水热法制备氯氧化铋晶体,但其耗时长,对设备要求高,固含量约为0.16%,并且难以得到大尺寸的氯氧化铋晶体。
5.公开号为cn103303975a的中国专利申请和张笛(河北科技大学.氯氧化铋珠光颜料的制备及其性能研究[d].)将铋盐滴加到去离子水中水解制得氯氧化铋晶体,工艺简单,但固含量低至0.6%,产品多为形貌不规整的氯氧化铋晶体。
[0006]
公开号为cn109749483a的中国专利申请公开了一种超声波反应制备易分散的水性氯氧化铋粉末及氯氧化铋珠光浆的方法,所制备的氯氧化铋晶体为圆片状,光泽性不优异,所需原材料较多,操作复杂。
[0007]
公开号为cn109678207a的中国专利申请公开了一种氯氧化铋珠光颜料晶体及其制备方法和应用,需要将铋单质制成铋盐,耗时长,在反应过程中可能造成其他杂质的生成,产品正方形氯氧化铋晶体有团聚现象。
[0008]
李君伟(吉林大学.珠光颜料氯氧化铋晶体的合成[d].)采用的三种不同合成氯氧化铋晶体的方法均具有一定的缺点,固相合成法制备的晶体无固定形貌,溶剂热法制备的晶体固含量低,反应结晶法得到晶体形貌差。
[0009]
上述可以看出,现有的制备方法为了获得形貌规整、无碎片、无团聚的氯氧化铋晶体,目前采用的方案只能是通过降低反应体系的固含量,减少晶体的碰撞机会和频率来缓解;制得的氯氧化铋晶体尺寸可控范围小甚至难以控制,形貌不规整,分散性差易于团聚,固含量低,光泽性差且工艺较为复杂,难以同时实现无团聚、大范围尺寸可控和高固含量等条件。
技术实现要素:
[0010]
有鉴于此,本发明有必要提供一种氯氧化铋晶体的制备方法以及氯氧化铋晶体,该制备方法可调控氯氧化铋晶体的形貌和尺寸,可制备不同尺寸的晶体,且制得的晶体分散性好、均一、无碎片、无团聚、形貌规整、光泽性优异、固含量高。且整个制备工艺操作简单,易于工业化生产。
[0011]
为了实现上述目的,本发明采用以下技术方案:
[0012]
本发明提供了一种氯氧化铋晶体的制备方法,包括以下步骤:
[0013]
将尿素、阳离子嵌段聚合物和盐酸水溶液混合搅拌,获得ph在0.8
‑
1.2的混合溶液,其中,所述阳离子嵌段聚合物由两种以上性质不同的聚合物链段组成,所述聚合物链段至少包括有一种阳离子聚合物和一种水溶性聚合物;
[0014]
向所述混合溶液中滴加铋盐溶液,反应完成后纯化、干燥,获得氯氧化铋晶体。
[0015]
进一步方案,所述阳离子聚合物选自聚甲基丙烯酸
‑2‑
(二甲氨基)乙酯、聚乙烯亚胺、聚甲基丙烯酸
‑2‑
(二异丙基氨基)乙酯、聚甲基丙烯酸
‑2‑
(二乙氨基)乙酯中的一种;
[0016]
所述水溶性聚合物选自聚乙二醇、聚乙烯醇、聚丙烯酸、聚丙烯酰胺、聚氧乙烯中的一种。
[0017]
进一步方案,所述阳离子嵌段聚合物与铋盐的质量比为1:(10
‑
1000)。
[0018]
进一步方案,所述混合溶液中,尿素与盐酸的质量比在1:(0.025
‑
0.5)之间,所述盐酸水溶液由浓盐酸和水按照质量比1:(15
‑
20)混合制成。
[0019]
进一步方案,在获得所述混合溶液的步骤中,将所述混合溶液加热至40
‑
90℃。
[0020]
进一步方案,所述铋盐溶液为氯化铋盐酸水溶液或硝酸铋盐酸水溶液。
[0021]
进一步方案,所述氯化铋盐酸水溶液由氯化铋、浓盐酸和去离子水按质量比1:(1.3
‑
1.8):(1
‑
2)混合制成;
[0022]
所述硝酸铋盐酸水溶液由硝酸铋、浓盐酸和去离子水按质量比为1:(1
‑
1.5):(1
‑
2)的比例混合制成。
[0023]
进一步方案,所述铋盐溶液的滴加速度为0.05
‑
0.3ml/min。
[0024]
进一步方案,所述纯化的具体工艺为:将冷却后的产物抽滤后,用去离子水和无水乙醇洗涤3
‑
4次。
[0025]
本发明还提供了一种氯氧化铋晶体,采用如前述任一项所述的制备方法制备而成。
[0026]
与现有技术相比,本发明具有以下有益效果:
[0027]
本发明中氯氧化铋晶体的制备方法,在氯氧化铋的制备过程中,以铋盐溶液作为铋源,盐酸水溶液作为氯源,通过尿素调控体系ph,从而控制氯氧化铋晶体在适宜的酸度下生长。在制备过程中,同时添加有阳离子嵌段聚合物,该阳离子嵌段聚合物至少包括一种阳离子聚合物链段和一种水溶性聚合物链段,其中,阳离子聚合物链段吸附在氯氧化铋晶体上,而水溶性聚合物链段分散在水中,以阳离子嵌段聚合物作为分散剂稳定和保护氯氧化铋晶体,制备获得了高固含量、无碎片、无团聚的氯氧化铋晶体,并且实现了氯氧化铋晶体形貌和尺寸的可控制备。
[0028]
该制备方法可以在5~100μm的范围内控制合成氯氧化铋晶体尺寸,根据氯氧化铋晶体光泽的尺寸依赖性,氯氧化铋晶体分别呈现出细柔白色、珍珠光泽、丝绸光泽、闪光珍
珠光泽、闪烁以及超闪烁等光泽,制备得到的晶体尺寸范围广,可以满足各领域对氯氧化铋晶体光泽度的应用要求。并且制得的氯氧化铋晶体的固含量可以达到10%,产量高,生产效率高,工艺简单,成本低,反应条件温和适用于工业化生产。
[0029]
由于该制备方法制得的氯氧化铋晶体尺寸可控、固含量高、光泽性和分散性好、形貌规整且性能稳定,因此可以应用在化妆品、汽车内饰材料、电子设备、体育用品和服装饰品等领域。
附图说明
[0030]
图1为实施例1中制得的尺寸为10μm的氯氧化铋晶体的显微镜图片;
[0031]
图2为实施例1中制备的氯氧化铋晶体的xrd图谱;
[0032]
图3为实施例2中制得的尺寸为15μm的氯氧化铋晶体的显微镜图片;
[0033]
图4为实施例3中制得的尺寸为20μm的氯氧化铋晶体的显微镜图片;
[0034]
图5为实施例4中制得的尺寸为40μm的氯氧化铋晶体的显微镜图片。
具体实施方式
[0035]
为了便于理解本发明,下面将结合具体的实施例对本发明进行更全面的描述。但是,本发明可以以许多不同的形式来实现,并不限于本文所描述的实施方式。相反地,提供这些实施方式的目的是使对本发明的公开内容理解的更加透彻全面。
[0036]
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施方式的目的,不是旨在于限制本发明。
[0037]
本发明第一方面提供了一种氯氧化铋晶体的制备方法,包括以下步骤:
[0038]
将尿素、阳离子嵌段聚合物和盐酸水溶液混合搅拌,获得ph在0.8
‑
1.2的混合溶液,其中,所述阳离子嵌段聚合物由两种以上性质不同的聚合物链段组成,所述聚合物链段至少包括有一种阳离子聚合物和一种水溶性聚合物;
[0039]
向所述混合溶液中滴加铋盐溶液,反应完成后纯化、干燥,获得氯氧化铋晶体。
[0040]
该制备方法以盐酸水溶液作为氯源、铋盐溶液作为铋源,加入尿素调控反应体系ph,从而使得氯氧化铋在适宜的酸度下生长。利用阳离子嵌段聚合物来控制氯氧化铋晶体的形貌和尺寸,制得正方形薄片状的氯氧化铋晶体,该制备方法操作简便,成本低廉,可实现氯氧化铋晶体尺寸的可控制备。“阳离子嵌段聚合物”通常指的是由性质不同的两种以上的聚合物链段聚合连接在一起的一种聚合物,而本文中所述的“阳离子嵌段聚合物”指的是至少包括一种阳离子聚合物和一种水溶性聚合物链段的聚合物,其中阳离子聚合物链段可以吸附在氯氧化铋晶体上,而水溶性聚合物链段则分散在水中,从而避免氯氧化铋晶体之间的碰撞产生碎片和团聚,并且可以控制氯氧化铋晶体的尺寸。本文中所述的“阳离子嵌段聚合物”的制备没有特殊的限制,可以采用现有技术中嵌段聚合物的合成方法获得,在本发明的一个或多个实施例中,采用原子转移自由基聚合的方法制得,这里不再具体阐述。
[0041]
进一步方案,本文中所述的“阳离子聚合物”指的是具有阳离子官能团或亲电基团的有机聚合物,具体可提及的实例包括但不限于聚甲基丙烯酸
‑2‑
(二甲氨基)乙酯、聚乙烯亚胺、聚甲基丙烯酸
‑2‑
(二异丙基氨基)乙酯、聚甲基丙烯酸
‑2‑
(二乙氨基)乙酯中的一种;
[0042]
本文中所述的“水溶性聚合物”指的是一种强亲水性的高分子材料,其分子结构中含有大量的亲水基团,使得其能溶解或溶胀于水中形成水溶液或分散体系。具体可提及的实例包括但不限于聚乙二醇、聚乙烯醇、聚丙烯酸、聚丙烯酰胺、聚氧乙烯中的一种。
[0043]
进一步方案,本发明中阳离子嵌段聚合物的添加量没有特殊的限制,可根据需要制备的氯氧化铋晶体的尺寸进行调整,在本发明的一个或多个实施例中,所述阳离子嵌段聚合物与铋盐的质量比为1:(10
‑
1000),从而实现氯氧化铋晶体尺寸在5
‑
100μm的可控制备,可以理解的是这里铋盐指的是铋盐溶液中的溶质铋盐。
[0044]
进一步方案,本发明中为了提供适合氯氧化铋晶体生长的酸度环境,采用添加尿素的方式对反应体系中的酸度环境调整,因此,其添加量没有特殊的限制,保证反应体系ph在0.8
‑
1.2即可,在本发明的一个或多个实施例中,所述混合溶液中,尿素与盐酸的质量比在1:(0.025
‑
0.5)之间,所述盐酸水溶液由浓盐酸和水按照质量比1:(15
‑
20)混合制成,此外,本文中所述的“浓盐酸”均指的是质量浓度在36
‑
38%的盐酸。
[0045]
进一步方案,由于在常温条件下进行反应,产物的产率低,反应时间较长,故在本发明的一个或多个实施例中,优选的,在获得所述混合溶液的步骤中,所述混合溶液加热至温度40
‑
90℃进行反应。
[0046]
进一步方案,铋盐溶液的选择没有特殊的限制,只要能够提供铋源即可,在本发明的一个或多个实施例中,所述铋盐溶液为氯化铋盐酸水溶液或硝酸铋盐酸水溶液,由于氯化铋和硝酸铋可以稳定的溶解在盐酸水溶液中,其除了可以提供铋源,还可以进一步提供氯源,提供更加足量氯源和铋源的环境,更适宜合成形貌规整、尺寸均一、分散性优异的氯氧化铋晶体。
[0047]
进一步方案,所述氯化铋盐酸水溶液由氯化铋、浓盐酸和去离子水按质量比1:(1.3
‑
1.8):(1
‑
2)混合制成;
[0048]
所述硝酸铋盐酸水溶液由硝酸铋、浓盐酸和去离子水按质量比为1:(1
‑
1.5):(1
‑
2)的比例混合制成。可以理解的是,上述氯化铋盐酸水溶液和硝酸铋盐酸水溶液中各组分的配比没有特殊的限制,可以根据合成氯氧化铋的需要进行调整。
[0049]
进一步方案,本发明中铋盐溶液的滴加速度没有特殊的限制,可根据需要进行调整,在本发明的一个或多个实施例中,所述铋盐溶液的滴加速度为0.05
‑
0.3ml/min,具体的说,当滴加速度低于0.05ml/min时,起始形成的晶核数目较少,有利于晶体生长,但反应时间长,效率低;而当滴加速度高于0.3ml/min时,虽然也能够产出氯氧化铋晶体,但晶体尺寸不够均一。
[0050]
进一步方案,本发明中所述的纯化工艺没有特殊的限制,其均可以采用本领域中的常规的方式,包括冷却后分离、洗涤、干燥,在本发明的一个或多个实施例中,所述纯化的具体工艺为:将冷却后的产物抽滤后,用去离子水和无水乙醇洗涤3
‑
4次。
[0051]
本发明第二方面提供了一种氯氧化铋晶体,采用如本发明第一方面任一项所述的制备方法制备而成。通过本发明第一方面所述的制备方法制得的氯氧化铋晶体为正方形薄片状,固含量可高达10%,形貌规整、尺寸可控(5~100μm),分散性和均一性好,具有优异的光泽性,具有良好的工业化应用前景。
[0052]
下面结合具体的实施例对本发明的技术方案进行更加清楚完整的说明。
[0053]
实施例1
[0054]
将15g尿素、0.32g聚甲基丙烯酸
‑2‑
(二甲氨基)乙酯
‑
b
‑
聚乙烯醇和质量比为1:15的盐酸水溶液加入到三口瓶中,100rpm转速下搅拌,升温至90℃后,获得ph在0.8
‑
1.2的混合溶液;
[0055]
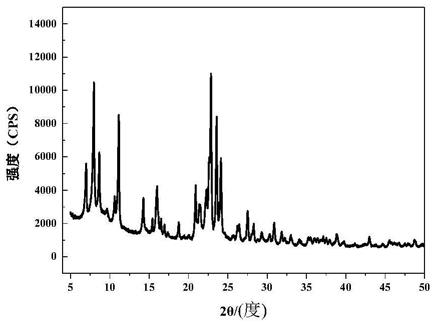
将硝酸铋、浓盐酸及去离子水按质量比为1:1:2的比例混合制成的铋盐溶液以0.08ml/min的滴加速度加入到三口瓶中反应。将得到的产物冷却至室温,抽滤,用去离子水和无水乙醇洗涤3到4次,烘干后得到10μm形貌规整、分散性好的片状氯氧化铋晶体,如图1中所示的。图2为本实施例中氯氧化铋晶体的xrd图谱,可以看出,该样品在11.98
°
、24.10
°
、25.86
°
、32.50
°
、33.45
°
处均具有biocl典型的(001)、(002)、(101)、(110)和(102)特征衍射峰与标准的jcpdf06
‑
0249卡片的衍射峰的位置都相同,说明产品中无杂质。样品的衍射峰处于细长型且衍射峰的强度很高,说明制备的氯氧化铋产品的结晶度好。
[0056]
本实施例中得到的产品为具有柔和珍珠光泽的白色粉末。
[0057]
实施例2
[0058]
将18g尿素、0.29g聚乙烯亚胺
‑
聚丙烯酰胺
‑
b
‑
聚乙二醇和1:20的盐酸水溶液加入到三口瓶中,100rpm转速下搅拌,升温至80℃后,获得ph在0.8
‑
1.2的混合溶液;
[0059]
将氯化铋、浓盐酸及去离子水按质量比为1:1.3:1的比例混合制成的铋盐水溶液以0.05ml/min的滴加速度加入到三口瓶中反应。反应结束后,冷却至室温,得到固含量为10.3%的氯氧化铋晶体的分散体。将其抽滤,用去离子水和无水乙醇洗涤3到4次,烘干后得到15μm片状氯氧化铋晶体,如图3中所示的。
[0060]
本实施例中得到的产品为具有明亮的珍珠光泽的白色粉末。
[0061]
实施例3
[0062]
将20g尿素、0.26g甲基丙烯酸
‑2‑
(二甲氨基)乙酯
‑
b
‑
聚乙二醇和1:18的盐酸水溶液加入到三口瓶中,100rpm转速下搅拌,升温至85℃后,获得ph在0.8
‑
1.2的混合溶液;
[0063]
将硝酸铋、浓盐酸及去离子水按质量比为1:1.5:1的比例混合制成的铋盐水溶液以0.3ml/min的滴加速度加入到三口瓶中反应。反应结束后,冷却至室温,得到固含量为11.2%的氯氧化铋晶体的分散体。将其抽滤,用去离子水和无水乙醇洗涤3到4次,烘干后得到20μm的片状氯氧化铋晶体,如图4中所示的。
[0064]
本实施例中得到的产品为具有闪烁光泽的白色粉末。
[0065]
实施例4
[0066]
将23g尿素、0.21g聚甲基丙烯酸
‑2‑
(二异丙基氨基)乙酯
‑
b
‑
聚丙烯酸和1:15的盐酸水溶液加入到三口瓶中,100rpm转速下搅拌,升温至70℃后,获得ph在0.8
‑
1.2的混合溶液;
[0067]
将氯化铋、浓盐酸及去离子水按质量比为1:1.8:1的比例混合制成的铋盐水溶液以0.12ml/min的滴加速度加入到三口瓶中反应。反应结束后,冷却至室温,得到固含量为10.1%的氯氧化铋晶体的分散体。将其抽滤,用去离子水和无水乙醇洗涤3到4次,烘干后得到40μm的片状氯氧化铋晶体,如图5中所示的。
[0068]
本实施例中得到的产品为具有闪烁珍珠光泽的白色粉末。
[0069]
实施例5
[0070]
将20g尿素、0.16g聚甲基丙烯酸
‑2‑
(二异丙基氨基)乙酯
‑
b
‑
聚乙烯醇和1:16的盐酸水溶液加入到三口瓶中,100rpm转速下搅拌,升温至85℃后,获得ph在0.8
‑
1.2的混合溶
液;
[0071]
将硝酸铋、浓盐酸及去离子水按质量比为1:1.3:2的比例混合制成的铋盐水溶液以0.3ml/min的滴加速度加入到三口瓶中反应。反应结束后,冷却至室温,得到固含量为11.3%的氯氧化铋晶体的分散体。将其抽滤,用去离子水和无水乙醇洗涤3到4次,烘干后得到53μm的片状氯氧化铋晶体。
[0072]
本实施例中得到的产品为具有超闪烁光泽的白色粉末。
[0073]
实施例6
[0074]
将27g尿素、0.14g聚甲基丙烯酸
‑2‑
(二乙氨基)乙酯
‑
b
‑
聚氧乙烯
‑
b
‑
聚乙烯醇和1:17的盐酸水溶液加入到三口瓶中,180rpm转速下搅拌,升温至40℃后,获得ph在0.8
‑
1.2的混合溶液;
[0075]
将氯化铋、浓盐酸及去离子水按质量比为1:1.8:2的比例混合制成的铋盐水溶液以0.05ml/min的滴加速度加入到三口瓶中反应。反应结束后,冷却至室温,得到固含量为12.0%的氯氧化铋晶体的分散体。将其抽滤,用去离子水和无水乙醇洗涤3到4次,烘干后得到62μm的片状氯氧化铋晶体。
[0076]
本实施例中得到的产品为具有闪烁珍珠光泽的白色粉末。
[0077]
实施例7
[0078]
将35g尿素、0.07g聚甲基丙烯酸
‑2‑
(二甲氨基)乙酯
‑
b
‑
聚丙烯酰胺和1:15的盐酸水溶液加入到三口瓶中,260rpm转速下搅拌,升温至60℃后,获得ph在0.8
‑
1.2的混合溶液;
[0079]
将硝酸铋、浓盐酸及去离子水按质量比为1:1:1的比例混合制成的铋盐水溶液以0.26ml/min的滴加速度加入到三口瓶中反应。反应结束后,冷却至室温,得到固含量为10.1%的氯氧化铋晶体的分散体。将其抽滤,用去离子水和无水乙醇洗涤3到4次,烘干后得到73μm的片状氯氧化铋晶体。
[0080]
本实施例中得到的产品为具有闪烁珍珠光泽的白色粉末。
[0081]
实施例8
[0082]
将26g尿素、0.02g聚甲基丙烯酸
‑2‑
(二异丙基氨基)乙酯
‑
b
‑
聚甲基丙烯酸
‑2‑
(二甲氨基)乙酯
‑
b
‑
聚丙烯酰胺和1:16的盐酸水溶液加入到三口瓶中,88rpm转速下搅拌,升温至90℃后,获得ph在0.8
‑
1.2的混合溶液;
[0083]
将硝化铋、浓盐酸及去离子水按质量比为1:1.5:2的比例混合制成的铋盐水溶液以0.22ml/min的滴加速度加入到三口瓶中反应。反应结束后,冷却至室温,得到固含量为10.9%的氯氧化铋晶体的分散体。将其抽滤,用去离子水和无水乙醇洗涤3到4次,烘干后得到100μm的片状氯氧化铋晶体。
[0084]
本实施例中得到的产品为具有超闪烁光泽的白色粉末。
[0085]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0086]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。