c5枝化的1-脱氧野尻霉素衍生物及其制备方法和应用
技术领域
1.本发明涉及糖苷酶抑制剂技术领域,具体涉及c5枝化的1-脱氧野尻霉素衍生物及其制备方法和应用。
背景技术:
2.dnj(1-脱氧野尻霉素)是1966年从桑树中分离得到的亚氨基糖,对α-葡萄糖苷酶具有强抑制作用,对其他糖苷酶如β-葡萄糖苷酶也具有抑制活性。dnj及其衍生物具有重要的药用价值,例如,可以治疗糖尿病、高雪氏病、抗病毒、抗肿瘤、抗氧化以及降低餐后血糖(compain,p.等,iminosugars:from synthesis to therapeutic application;wiley,2007),因而在新药研发领域受到广泛关注。
3.目前,大约有300个dnj衍生物被报道,其中,上市的dnj衍生物有两个,均为n-烷基化的dnj衍生物,分别是米格列醇(n-羟乙基-dnj)和美格鲁特(n-丁基-dnj)。这大大促进了dnj衍生物的研究。对dnj的修饰可以大致分为四类:1)n-取代;2)c-取代;3)o-取代4)同位素取代。取代基的种类多种多样,例如,烷基、不饱和烃基、苯基、糖基、氟代、氯代、杂环芳基、羟基以及氨基等。修饰位点涉及dnj的各个部位:对n上的修饰主要是含1-20个碳的烷基化,烷基链的某些c原子可以被杂原子或官能团取代,也可以在末端链接其他基团,比如氨基酸、糖基和杂环芳基等((wolfsgruber,a.等,molecules 2020,25,4618;ghisaidoobe,a.等,acs med.chem.lett.2011,2,119;ghisaidoobe,a.t.等,med.chem.2014,57,9096;wennekes,t.等,j.org.chem.2007,72,1088;hoogendoorn,s等,eur.j.org.chem.2015,2015,4437;wang,l.等,acta chimica slovenica 2020,67,812)对o上的修饰主要是烷基化和糖苷化。同位素取代主要是同位素取代化合物中的氢和碳。对c上的修饰主要集中在c1位,包括烷基化和糖苷化。对c2-c4的修饰主要是氟代和氯代。目前,对c5位的修饰仅报道了少量的化合物:5-c-甲基-dnj,该化合物仅测试了对人的α-葡萄糖的活性(ic
50
=1μm)活性评价;5-c-苄基-dnj;5-c-羟甲基-dnj;5-c-羟基-dnj;5-c-氰基-dnj。(maughan,m.a.t等,angew.chem.,int.ed.2003,42,3788;pawar,n.j.等,j.org.chem.2012,77,7873;wuensche,c等,j.org.mass spectrom.1984,19,176)。这些化合物在对糖苷酶的抑制活性还不够高,另外分子极性大,水溶性好,脂溶性差,不利于透膜,存在着成药性差的缺陷。
4.另外,目前c5枝化的化合物合成方法主要有以下两种:1)以四苄基-dnj为原料,n-氯代dnj衍生物为关键中间体,通过格氏加成得到化合物前体,最后催化氢化得到化合物;该路线虽然步骤相对较短,但是原料四苄基dnj价格昂贵,产率低,尤其亚胺与格氏试剂的加成反应,收率仅有17%,难以工业化。2)以d-葡萄糖为原料,以叠氮基取代的糖衍生物为关键中间体,得到化合物。该路线虽然原料相对便宜、易得,条件温和,但是存在反应步骤较多,且多次使用剧毒的叠氮试剂,普适性低。
5.方法1):
[0006][0007]
方法2):
[0008][0009]
因此,需要寻求高活性的,成药性好的糖苷酶抑制剂及其相应的原料易得、步骤简单且利于工业化的制备方法。
技术实现要素:
[0010]
本发明的目的是为了克服现有技术存在的技术问题,提供一种具有优异糖苷酶抑制活性的c5枝化的1-脱氧野尻霉素衍生物及其制备方法和应用。
[0011]
本发明的发明人经深入研究,通过分子对接模式研究发现,采用不同的取代基对dnj(1-脱氧野尻霉素)进行c5枝化时得到的衍生物与不同的酶结合时,所结合的活性中心以及作用方式存在很大的不同,这将会导致其对酶的抑制活性存在较大的差异。例如,dnj与n-末端人麦芽糖酶-葡糖淀粉酶(ntmgam,pdb id:3l4w)结合时,其仲氨基和c-2、c-3、c-6羟基与氨基酸残基asp-443,arg-526,asp-542和asp-327,之间存在氢键作用;而5-c-甲基-dnj与ntmgam结合时,分子采取的构象相似,但参与结合作用的氨基酸残基不完全相同,为his-600、asp-542、asp-327、asp-443以及trp-406因此,c5枝化采用的基团对最终枝化后的化合物的性能产生的影响难以预期。而本发明的发明人经研究发现:长链烃基枝化的一系列的dnj(1-脱氧野尻霉素),对不同的糖苷酶均具有较好的亲和力。
[0012]
因此,为了实现上述目的,本发明第一方面提供一种c5枝化的1-脱氧野尻霉素衍生物,该1-脱氧野尻霉素衍生物为具有式(i)所示结构的化合物及其药学上可接受的盐:
[0013][0014]
其中,r1为c2-c20的烷基;
[0015]
r2为氢或羟基;
[0016]r3-r6各自独立地为h、苄基或取代苄基,其中,苄基的取代基选自c1-c6的烷氧基、c1-c6的烷基、羟基、硝基和卤素中的一种;
[0017]
所述化合物的5位碳的立体构型为r或s。
[0018]
本发明第二方面提供一种制备c5枝化的1-脱氧野尻霉素衍生物的方法,该方法包括以下步骤:
[0019]
(1)将式(ii)所示的化合物与带有r1基团的有机金属试剂进行亲核加成,得到式
(i’)所示的化合物;
[0020][0021]
(2)任选地,将式(i’)所示的化合物与脱保护试剂、氢源接触进行催化氢化反应,得到式(i”)所示的化合物或其盐;
[0022][0023]
其中,式(ii)、式(i’)和式(i”)中,所涉及的r1、r2、r3、r4、r5或r6的定义均与前述第一方面所述的定义相同。
[0024]
本发明第三方面提供一种糖苷酶抑制剂,该糖苷酶抑制剂含有前述第一方面所述的c5枝化的dnj衍生物作为活性成分。
[0025]
本发明第四方面提供一种前述第三方面所述的糖苷酶抑制剂在抑制糖苷酶中的应用。
[0026]
本发明第五方面提供一种前述第三方面所述的糖苷酶抑制剂在制备药物中的应用,该药物选自下述药物中的至少一种:1)预防和/或治疗糖尿病的药物;2)预防和/或治疗高雪氏病的药物;3)预防和/或治疗肿瘤的药物;4)抗病毒药物;5)抗菌药物;6)预防和/或治疗庞贝病的药物。
[0027]
本发明提供的c5枝化的dnj衍生物具有优异的糖苷酶抑制活性,具有很高的药用价值。
具体实施方式
[0028]
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
[0029]
本发明中,碳上的“*”本领域技术人员应当理解其表示该碳为手性碳,可以为r型或s型的构象。以下化学式中哌啶环上的碳位点处得数字表示其所在位置编号。例如,式(i)所示的化合物中的哌啶上的“5”表示该处的碳为第5位的碳,而“1”表示该处的碳为第1位的碳,其他的情况也做相似的解释。
[0030]
本发明中,“c2-c20的烷基”的具体实例可以为乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、正戊基、正己基、正庚基、正辛基、正壬基、正癸基、正十一烷基、正十二烷基、正
十三烷基、正十四烷基、正十五烷基、正十六烷基、正十七烷基、正十八烷基、正十九烷基、正二十烷基等。对于更窄碳原子范围的烷基也可以根据碳原子数的限定从该具体实例中进行选择。
[0031]
本发明中,c1-c6的烷氧基的具体示例可以为甲氧基、乙氧基、丙氧基、异丙氧基、正丁氧基、异丁氧基、叔丁氧基、正戊氧基、正己氧基等。对于更窄碳原子范围的烷基也可以根据碳原子数的限定从该具体实例中进行选择。
[0032]
本发明第一方面提供一种c5枝化的1-脱氧野尻霉素衍生物,该1-脱氧野尻霉素衍生物为具有式(i)所示结构的化合物及其药学上可接受的盐:
[0033][0034]
其中,r1为c2-c20的烷基;
[0035]
r2为氢或羟基;
[0036]r3-r6各自独立地为h、苄基或取代苄基,其中,苄基的取代基选自c1-c6的烷氧基、c1-c6的烷基、羟基、硝基和卤素(可以为f、cl、br、i)中的一种;
[0037]
所述化合物的5位碳的立体构型为r或s。
[0038]
根据本发明的一些实施方式,r1为c2-c12的烷基。
[0039]
根据本发明的一些实施方式,r2为氢。
[0040]
根据本发明的一些实施方式,r
3-r6可以各自独立地为h、苄基或取代苄基,其中,苄基的取代基可以选自c1-c4的烷氧基、c1-c4的烷基、羟基、硝基和卤素中的一种。
[0041]
根据本发明的一些实施方式,r
3-r6均为氢或苄基。
[0042]
根据本发明的一些实施方式,r
3-r6均为氢。
[0043]
根据本发明的一些实施方式,所述化合物的5位碳的立体构型为r或s。
[0044]
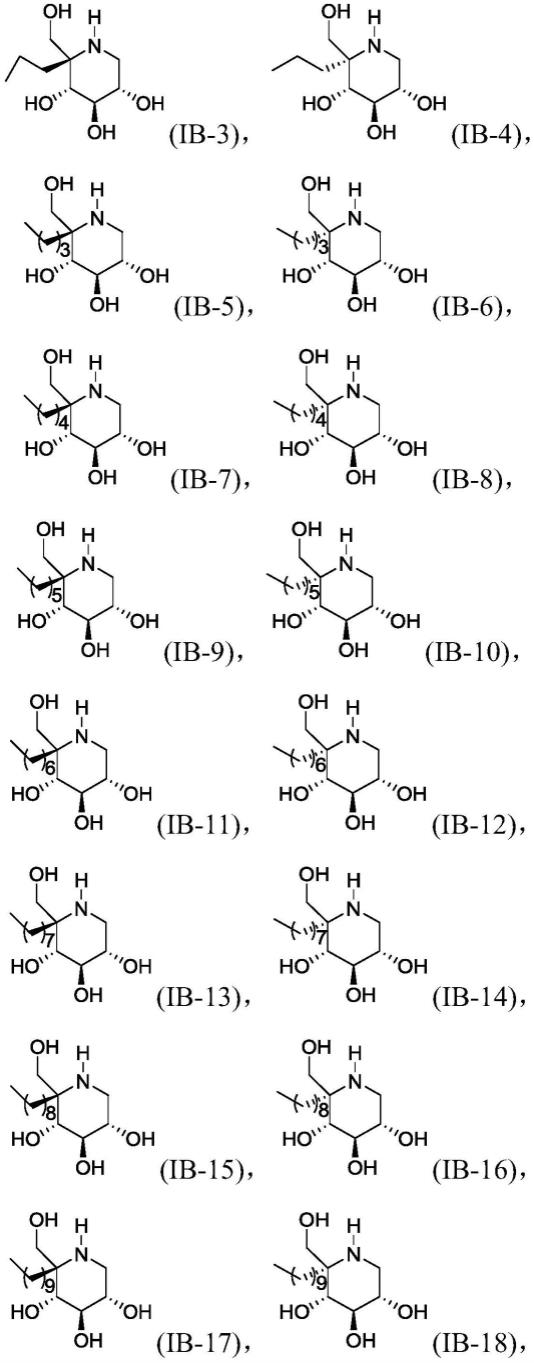
根据本发明的一些实施方式,所述化合物可以选自以下所示结构的化合物中的一种:
[0045]
[0046]
[0047]
[0048][0049]
本发明第二方面提供一种制备c5枝化的1-脱氧野尻霉素衍生物的方法,该方法包括以下步骤:
[0050]
(1)将式(ii)所示的化合物与带有r1基团的有机金属试剂进行亲核加成,得到式(i’)所示的化合物;
[0051][0052]
(2)任选地,将式(i’)所示的化合物与脱保护试剂、氢源接触进行催化氢化反应,得到式(i”)所示的化合物或其盐;
[0053]
[0054]
其中,式(ii)、式(i’)和式(i”)中,所涉及的r1、r2、r3、r4、r5或r6的定义均与前述第一方面所述的定义相同。
[0055]
本发明中,步骤(1)中通过将式(ⅱ)所示的化合物(硝酮)与带有r1基团的有机金属试剂进行亲核加成,即可使得基团r1连接至式(ⅱ)所示的化合物硝酮上,得到式(i’)所示的化合物。
[0056]
本发明中,式(ii)所示的化合物可以通过商购获得,也可以通过制备得到。其中,对式(ⅱ)所示的化合物的制备没有特别的限定,可以参照本领域常规的方式(例如可以参考boisson,j.;thomasset,a.;racine,e.;cividino,p.;banchelin sainte-luce,t.;poisson,j.f.;behr,j.b.;py,s.org.lett.2015,17,3662.)进行。
[0057]
根据本发明的一些实施方式,步骤(1)中,所述亲核加成的条件可以包括:温度为-20至50℃,优选为-10至35℃;时间为0.5-5小时,优选为0.5-2小时。
[0058]
根据本发明的一些实施方式,式(ii)所示的化合物与所述有机金属试剂的摩尔比可以为1:(1.2-50),优选为1:(1.2-10)。
[0059]
根据本发明的一些实施方式,所述有机金属试剂可以选自有机镁试剂、有机锌试剂、有机锂试剂和有机铜试剂中的至少一种,优选选自有机镁试剂和/或有机锌试剂。
[0060]
本发明中,有机镁试剂可以由式mgr1x表示,其中x为卤素(f、cl、br、i)。有机锌试剂可由式znr1x表示,其中x为卤素(f、cl、br、i)。有机锂试剂可由式lir1表示。有机铜试剂可由式cur1x表示,其中x为卤素(f、cl、br、i)。上述基团r1的选择可以根据待制备的式(i’)所示的化合物进行具体选择。
[0061]
根据本发明的一些实施方式,所述亲核加成在第一溶剂的存在下进行,所述第一溶剂为非质子溶剂,可以选自乙醚(et2o)、四氢呋喃(thf)、二氧六环和二氯甲烷(dcm)中的至少一种,优选选自四氢呋喃和/或二氧六环。
[0062]
本发明中,对所述第一溶剂的用量没有特别的限定,只要能够满足本发明的需求即可,优选地,相对于1g的式(ii)所示的化合物,所述第一溶剂的用量为2-200ml,更优选为5-50ml。
[0063]
本发明中,为了获得更好的效果,步骤(1)中,式(ⅱ)所示的化合物(硝酮)与带有r1基团的有机金属试剂的加料方式为:将所述有机金属试剂加入含有式(ii)所示化合物的溶液(优选由式(ii)所示化合物溶于第一溶剂中得到)中。
[0064]
本发明中,对亲和加成的后处理没有特别的限定,只要能够满足本发明的需求即可。例如可以按照如下方式进行后处理:采用饱和的nh4cl水溶液将反应后的体系进行淬灭,并用乙酸乙酯萃取,浓缩有机相,经柱层析分离提纯式(i’)所示的化合物。
[0065]
根据本发明的一些实施方式,步骤(2)中,所述催化氢化反应的条件可以包括:温度为0-50℃,优选为10-30℃;时间6-100小时,优选为6-48小时。
[0066]
本发明中,所述催化氢化反应所采用的催化剂为本领域常规的脱保护试剂。本发明通过所述催化氢化反应,在脱保护试剂的存在下,能够脱去羟基上的保护基。
[0067]
根据本发明的一些实施方式,所述脱保护试剂可以选自雷尼镍、pd/c(例如pd含量为10质量%的pd/c,表示为10%pd/c)、钯黑、氢氧化钯、醋酸钯、氯化钯、氧化铂、铂黑、三甲基碘硅烷、三氯化硼、三溴化硼、三氯化铝和锌/氯化铵中的至少一种,优选选自雷尼镍、pd/c、钯黑、氢氧化钯、醋酸钯、三氯化硼和三溴化硼中的至少一种。
[0068]
本发明中,当所述脱保护试剂选自雷尼镍、pd/c、钯黑、氢氧化钯、醋酸钯、氯化钯、氧化铂和铂黑中的至少一种时,式(i’)所示的化合物与脱保护试剂质量比为1:(0.1-5),优选为1:(0.1-0.2)。
[0069]
本发明中,当所述脱保护试剂选自三甲基碘硅烷、三氯化硼、三溴化硼、三氯化铝和锌/氯化铵中的至少一种时,式(i’)所示的化合物与脱保护试剂的摩尔比为1:(1-20),优选为1:(5-10)。
[0070]
根据本发明的一些实施方式,式(i’)所示的化合物与氢源的摩尔比为1:(5-100),优选为1:(10-50)。
[0071]
根据本发明的一些实施方式,所述氢源可以选自氢气、硼氢化钠和甲酸铵中的至少一种,优选为氢气。
[0072]
根据本发明的一些实施方式,所述催化氢化反应在第二溶剂的存在下进行,所述第二溶剂选自二氯甲烷、氯仿、四氢呋喃、乙醚、乙酸乙酯、乙酸、水、二氧六环、甲醇、乙醇、乙腈、甲酰胺和n,n-二甲基甲酰胺中的至少一种。
[0073]
本发明中,对所述第二溶剂的用量没有特别的限定,只要能够满足本发明的需求即可,优选地,相对于1mg的式(i’)所示的化合物,所述第二溶剂的用量为0.02-2ml,优选为0.04-1.5ml。
[0074]
本发明中,所述脱保护反应还可以在盐酸的存在下进行,其中,对盐酸的用量没有特别的限定,只要能够满足本发明的需求即可,优选地,相对于1mg式(i’)所示的化合物,盐酸的用量可以为0.01-1mmol。其中,当脱保护反应中有盐酸的存在时,步骤(2)得到式(i”)所化合物的盐酸盐。
[0075]
本发明中,对步骤(2)的后处理没有特别的限定,只要能够满足本发明的需求即可。例如可以按照如下方式进行后处理:将步骤(2)反应后的体系过滤,得到滤饼和滤液,减压(-10mpa至-0.01mpa,优选为-1mpa至-0.01mpa)下将滤液的溶剂除去,即可得到式(i”)所述的化合物或其盐。
[0076]
本发明中,脱保护后得到的产物为式(i”)所示的化合物的盐(优选为盐酸盐),通过酸性离子交换树脂处理后可以得到自由碱(式(i”)所示的化合物)。为了将式(i”)所示的化合物提取出来,该步骤还可以包括将所述催化氢化反应的产物用硅藻土进行过滤,并将滤液的溶剂除去,并加入氨水中和至碱性,将残余物用酸性离子交换树脂柱进行分离纯化,即可得到式(i”)所示的化合物。
[0077]
本发明中,步骤(1)和步骤(2)所述的方法均可以参照本领域常规的方式进行。
[0078]
本发明第三方面提供一种糖苷酶抑制剂,该糖苷酶抑制剂含有前述第一方面所述的c5枝化的dnj衍生物或其盐作为活性成分。
[0079]
本发明第四方面提供一种前述第三方面所述的糖苷酶抑制剂在抑制糖苷酶中的应用。
[0080]
根据本发明的一些实施方式,所述糖苷酶可以选自由α-葡萄糖苷酶、β-葡萄糖苷酶、α-半乳糖苷酶、β-半乳糖苷酶、α-甘露糖苷酶、β-甘露糖苷酶、α-l-岩藻糖苷酶、α-海藻糖酶、α-l-鼠李糖酶、淀粉葡萄糖苷酶以及β-葡萄糖苷酸化酶组成的至少一种糖苷酶。
[0081]
本发明第五方面提供一种前述第三方面所述的糖苷酶抑制剂在制备药物中的应用,该药物选自下述药物中的至少一种:1)预防和/或治疗糖尿病的药物;2)预防和/或治疗
高雪氏病的药物;3)预防和/或治疗肿瘤的药物;4)抗病毒药物;5)抗菌药物;6)预防和/或治疗庞贝病的药物。
[0082]
需要的时候,上述药物中还可以加入一种或多种药学上可接受的载体。所述载体包括药学领域常规的稀释剂、赋形剂、填充剂、粘合剂、湿润剂、崩解剂、吸收促进剂、表面活性剂、吸附载体、润滑剂以及任选的其他添加剂。用式(ⅰ)所示结构的化合物或其药学上可接受的水合物或药物复合物制备的药物可以制成注射液、片剂、粉剂、颗粒剂、胶囊、口服液、膏剂、霜剂等多种形式。上述各种剂型的药物均可按照药学领域的常规方法制备。
[0083]
所述药物可利用各种给药途径给药,包括但不限于口服、吸入、直肠、透皮,经粘膜肠内给药,以及皮下、肌肉或静脉注射给药。本发明所示结构的化合物或其药学上可接受的水合物或药物复合物,可以单独给药,或与其他已知治疗糖尿病、抗病毒、抗菌和抗肿瘤药物一起给药。
[0084]
以下将通过实施例对本发明进行详细描述。
[0085]
以下制备例和实施例中,在没有特别说明的情况下,用的原料均通过商购获得。
[0086]
制备例1
[0087]
本制备例用于说明式(ii)所示化合物的合成(参照boisson,j.;thomasset,a.;racine,e.;cividino,p.;banchelin sainte-luce,t.;poisson,j.f.;behr,j.b.;py,s.org.lett.2015,17,3662),其中,r
3-r6均为氢。
[0088][0089]
(1)在-10℃下,向1500ml甲醇中,滴加乙酰氯(27ml),反应半小时。转移至室温,向其中加入l-山梨糖(90g),室温反应过夜。原料消失后,加入碳酸氢钠中和至碱性,乙酸乙酯萃取,合并有机相,浓缩得粗产品iv,用200ml的n,n-二甲基甲酰胺溶解,直接用于下一步反应。
[0090]
(2)将60%的nah(120g),悬浮在200ml dmf和700ml thf组成的混合溶剂中,搅拌10分钟,至无气泡产生。并向其中滴加上一步的粗产品iv的n,n-二甲基甲酰胺(dmf)溶液,加毕,反应1小时。之后向其中加入催化量的四丁基碘化铵(1.85g),然后滴加苄基溴(261ml)。加毕,反应10分钟,原料消失。再向其中缓慢滴加饱和氯化铵水溶液淬灭,加水,乙酸乙酯萃取,合并有机相,蒸干得粗产品
ⅴ
,直接用于下一步反应。
[0091]
(3)将粗产品
ⅴ
溶于500ml的1,4-二氧六环中,加入110ml的1n盐酸,升温至90℃反应过夜。蒸除1,4-二氧六环,加水,乙酸乙酯萃取,合并有机相。加入碳酸氢钠调节至碱性,再次萃取,合并有机相,蒸干。经柱层析分离得产物ⅵ。
[0092]
(4)将20g产物ⅵ溶于200ml干燥得四氢呋喃(thf)中,加入12ml干燥的吡啶。用氩气保护。加入35.6g br2·
pph3,升温至70℃反应。原料消失后,用饱和硫代硫酸钠水溶液淬
灭,加入乙酸乙酯萃取,有机相用无水硫酸镁干燥,过滤,合并有机相。用石油醚和乙酸乙酯作为溶剂,重结晶得到大部分白色固体为三苯基氧磷,舍去固体。有机相蒸干,经柱层析分离得到产品ⅶ。
[0093]
(5)将11g产品ⅶ溶于干燥的四氢呋喃(thf)中,先加入13ml三乙胺,再加入5g盐酸羟胺,室温反应过夜。原料消失后,加水淬灭,蒸干,经柱层析分离得到硝酮ⅱ。
[0094]
实施例1
[0095]
本实施例用于说明式(ia-1)、(ia-2)、(ib-1)和(ib-2)的制备
[0096]
(1)将上述制备例1得到的式ⅱ所示的硝酮(0.97g,1.81mmol)溶于干燥的四氢呋喃(20ml)中,在冰水浴(0-5℃)下,将乙基氯化镁(5.55mmol)滴加到上述溶液中,滴加用时3分钟,继续反应35分钟,用饱和氯化铵溶液淬灭反应,乙酸乙酯(3
×
25ml)萃取,合并有机相,饱和氯化钠溶液(2
×
25ml)洗涤,无水硫酸镁干燥,减压浓缩除去溶剂,粗产物通过柱层析(硅胶200-300目)分离,得到式(ia-1)所示的化合物,收率59%,和式(ia-2)所示的化合物,收率35%。
[0097]
(2-1)将式(ia-1)所示的化合物(287mg)溶于甲醇(20ml)中,加入28.7mg的10%pd/c,加入盐酸(5ml,1n),氩气置换三次,氢气置换三次,反应12小时,过滤,弃去滤饼,得到滤液,-0.1mpa减压条件下除去溶剂得式(ib-1)所示化合物的盐酸盐,收率98%。
[0098]1h nmr(500mhz,cd3od)δ3.63(1h,d,j=12.3hz),3.56
–
3.41(2h,m),3.35(1h,t,j=8.8hz),3.26(1h,d,j=8.9hz),3.02
–
2.88(1h,m),2.74(1h,t,j=11.8hz),1.82
–
1.65(1h,m),1.64
–
1.49(1h,m),0.77(3h,t,j=7.5hz)
[0099]
经酸性离子交换树脂可以得到自由碱(式ib-1所示的化合物):
[0100]1h nmr(400mhz cd3od)δ3.74(1h,d,j=11.4hz),3.64(1h,d,j=11.4hz),3.52-3.42(1h,m),3.42-3.32(1h,m),3.29(1h,d,j=9.1hz),2.83(1h,dd,j=12.4hz,5.3hz),2.61(1h,t,j=9.1hz),1.80-1.64(1h,m),1.64-1.50(1h,m),3.29(3h,t,j=9.1hz)。
[0101]
(2-2)将式(ia-2)所示的化合物115mg溶于甲醇(25ml)中,加入17.25mg的10%pd/c,加入盐酸(3ml,1n),氩气置换三次,氢气置换三次,反应12小时,过滤,弃去滤饼,得到滤液,-0.5mpa减压条件下除去溶剂得式(ib-2)所示的化合物的盐酸盐,收率92%。
[0102]1h nmr(500mhz cd3od)δ3.93(1h,d,j=11.7hz),3.78
–
3.60(4h,m),3.32
–
3.24(1h,m),2.94(1h,t,j=11.5hz),1.96
–
1.85(1h,m),1.84
–
1.74(1h,m),1.00(3h,t,j=7.2hz)
[0103]
实施例2
[0104]
本实施例用于说明式(ia-3)、(ia-4)、(ib-3)和(ib-4)的制备
[0105]
(1)将上述制备例1得到的式ⅱ所示的硝酮(0.98g,1.82mmol)溶于干燥的乙醚(15ml)中,在冰水浴(0-5℃)下,将丙基溴化镁(4.5mmol)滴加到上述溶液中,滴加用时11分钟,继续反应40分钟,用饱和氯化铵溶液淬灭反应,乙酸乙酯(3
×
25ml)萃取,合并有机相,饱和氯化钠溶液(2
×
25ml)洗涤,无水硫酸镁干燥,减压浓缩除去溶剂,粗产物通过柱层析(硅胶200-300目)分离,得到无色糖浆,式(ia-3)所示的化合物,收率25%,和式(ia-4)所示的化合物,收率56%。
[0106]
(2-1)将式(ia-3)所示的化合物236mg溶于甲醇(25ml)中,加入70.8mg的10%pd/c,加入盐酸(7ml,1n),氩气置换三次,氢气置换三次,反应16小时,过滤,弃去滤饼,得到滤
液,-0.15mpa减压条件下除去溶剂得式(ib-3)所示的化合物的盐酸盐,收率98%。
[0107]1h nmr(500mhz cd3od)δ3.58(1h,d,j=12.3hz),3.52
–
3.43(1h,m),3.40(1h,d,j=12.3hz),3.30(1h,t,j=8.9hz),3.22(1h,d,j=9.0hz),3.00
–
2.91(1h,m),2.69(1h,t,j=11.9hz),1.59(1h,td,j=13.6hz,4.3mhz),1.43(1h,td,j=13.5hz,4.4hz),1.28
–
1.06(2h,m),0.96(3h,t,j=7.2hz)(2-2)将式(ia-4)所示的化合物36mg溶于甲醇(30ml)中,加入3.96mg的10%pd/c,加入盐酸(6ml,1n),氩气置换三次,氢气置换三次,反应18小时,过滤,弃去滤饼,得到滤液,-0.2mpa减压条件下除去溶剂得式(ib-4)所示的化合物的盐酸盐,收率92%。
[0108]1h nmr(500mhz cd3od)δ3.94(1h,d,j=11.8hz),3.76
–
3.69(1h,m),3.68
–
3.64(2h,m),3.61(1h,d,j=8.2hz),3.25(1h,dd,j=12.8hz,4.8hz),2.94(1h,dd,j=12.7hz,9.6hz),1.84
–
1.76(1h,m),1.70
–
1.64(1h,m),1.54
–
1.44(1h,m),1.42
–
1.33(1h,m),0.97(3h,t,j=7.2hz)
[0109]
实施例3
[0110]
本实施例用于说明式(ia-5)、(ia-6)、(ib-5)和(ib-6)的制备
[0111]
(1)将上述制备例1得到的式ⅱ所示的硝酮(0.2g,0.37mmol)溶于干燥的四氢呋喃(30ml)中,在冰水浴(0-5℃)下,将正丁基溴化镁(0.74mmol)滴加到上述溶液中,滴加用时10分钟,继续反应35分钟,用饱和氯化铵溶液淬灭反应,乙酸乙酯(3
×
25ml)萃取,合并有机相,饱和氯化钠溶液(2
×
25ml)洗涤,无水硫酸镁干燥,减压浓缩除去溶剂,粗产物通过柱层析(硅胶200-300目)分离,得到式(ia-5)所示的化合物,收率84%,和式(ia-6)所示的化合物,收率8%。
[0112]
式(ia-5),1h nmr(500mhz,cdcl3)δ7.35
–
7.23(20h,m),6.41(1h,s,br),4.92(1h,d,j=10.9hz),4.89(1h,d,j=11.2hz),4.72(1h,d,j=10.8hz),4.67(2h,s),4.60(1h,d,j=11.1hz),4.51(2h,s),4.19(1h,d,j=9.4hz),3.81(1h,t,j=8.9hz),3.76
–
3.68(2h,m),3.62(1h,dd,j=10.4hz,5.4hz),3.53(1h,d,j=9.1hz),3.12(1h,t,j=10.5hz),2.21
–
2.08(1h,m),1.66
–
1.52(2h,m),1.34
–
1.18(3h,m),0.88(3h,t,j=7.0hz)式(ia-6),1h nmr(500mhz,cdcl3)δ7.32
–
7.17(20h,m),5.41(1h,s,br),4.91(1h,d,j=10.9hz),4.87(1h,d,j=11.3hz),4.84
–
4.76(1h,m),4.71(1h,d,j=11.7hz),4.68
–
4.62(1h,m),4.51(1h,d,j=11.4hz),4.50(1h,d,j=12.1hz),4.44(1h,d,j=12.0hz),4.00
–
3.89(2h,m),3.84
–
3.72(2h,m),3.63(1h,d,j=9.5hz),3.41(1h,dd,j=12.5hz,5.5hz),2.95(1h,t,j=11.7hz,),1.70
–
1.60(3h,m),1.46
–
1.31(1h,m),1.30
–
1.18(2h,m),0.88(3h,t,j=7.2hz)。
[0113]
(2-1)将式(ia-5)所示的化合物50mg溶于甲醇(20ml)中,加入6.5mg的10%pd/c,加入盐酸(10ml 1n),氩气置换三次,氢气置换三次,反应17小时,过滤,弃去滤饼,得到滤液,-0.3mpa减压条件下除去溶剂得式(ib-5)所示的化合物的盐酸盐,收率92%。
[0114]1h nmr(500mhz cd3od)δ3.95(1h,d,j=12.1hz),3.82
–
3.71(2h,m),3.65(1h,t,j=8.5hz),3.55(1h,d,j=8.6hz),3.25(1h,dd,j=12.4hz,4.2hz),3.03(1h,t,j=11.5hz),2.07
–
1.97(1h,m),1.88
–
1.76(1h,m),1.59
–
1.33(4h,m),0.99(3h,t,j=6.9hz)。
[0115]
(2-2)将式(ia-6)所示的化合物18mg溶于甲醇(20ml)中,加入2.16mg的10%pd/c,加入盐酸(13ml,1n),氩气置换三次,氢气置换三次,反应12小时,过滤,弃去滤饼,得到滤液,-0.05mpa减压条件下除去溶剂得式(ib-6)所示的化合物的盐酸盐,收率95%。
[0116]1h nmr(500mhz cd3od)δ3.96(1h,d,j=11.6hz),3.78
–
3.59(4h,m),3.28
–
3.20(1h,m),2.94(1h,t,j=10.9hz),1.91
–
1.78(1h,m),1.78
–
1.66(1h,m),1.50
–
1.28(4h,m),0.96(3h,t,j=6.6hz)。
[0117]
实施例4
[0118]
本实施例用于说明式(ia-7)、(ia-8)、(ib-7)和(ib-8)的制备
[0119]
(1)将上述制备例1得到的式ⅱ所示的硝酮(1.03g,1.92mmol)溶于干燥的四氢呋喃(20ml)中,在冰水浴(0-5℃)下,将正戊基溴化镁(5.55mmol)滴加到上述溶液中,滴加用时15分钟,继续反应25分钟,用饱和氯化铵溶液淬灭反应,乙酸乙酯(3
×
25ml)萃取,合并有机相,饱和氯化钠溶液(2
×
25ml)洗涤,无水硫酸镁干燥,减压浓缩除去溶剂,粗产物通过柱层析(硅胶200-300目)分离,得到式(ia-7)所示的化合物,收率67%,和式(ia-8)所示的化合物,收率9%。
[0120]
式(ia-7),1h nmr(500mhz,cdcl3)δ7.32
–
7.23(20h,m,phch2o),6.38(1h,s,br,noh),4.91(2h,t,j=11.0hz),4.73(1h,d,j=10.9hz),4.67(2h,abq,j=11.7hz),4.60(1h,d,j=11.1hz),4.49(2h,s),4.18(1h,d,j=9.6hz),3.83(1h,t,j=8.9hz),3.72(2h,m),3.62(1h,dd,j=10.5hz,5.4hz),3.54(1h,d,j=9.1hz),3.14(1h,t,j=10.5hz),2.15
–
2.12(1h,m),1.62
–
1.58(2h,m),1.32
–
1.28(4h,m),1.17
–
1.15(1h,m),0.86(3h,t,j=7.0hz);
[0121]
式(ia-8),1h nmr(500mhz,cdcl3)δ7.32
–
7.17(20h,m,phch2o),5.42(1h,s,br,noh),4.91(1h,d,j=10.9hz),4.87(1h,d,j=11.3hz),4.79(1h,d,j=10.9hz),4.71(1h,d,j=11.4hz),4.65(1h,d,j=11.4hz),4.51
–
4.48(2h,m),4.44(1h,d,j=11.9hz),3.95-3.90(2h,m),3.82
–
3.74(2h,m),3.63(1h,d,j=9.5hz),3.41(1h,dd,j=12.5hz,5.5hz),2.95(1h,t,j=11.0hz),1.67
–
1.59(2h,m),1.42
–
1.36(2h,m),1.31
–
1.25(2h,m),1.22
–
1.17(2h,m),0.87(3h,t,j=7.2hz)。
[0122]
(2-1)将(ia-7)所示的化合物346mg溶于重蒸的二氯甲烷(dcm,25ml)中,用氩气保护。在-78℃下,搅拌15分钟,滴加bcl3(1m in dcm,10ml),加毕,升温至-40℃反应,4小时后,原料消失。降温至-78℃,加入甲醇,升至室温,蒸干,加入去离子水和二氯甲烷萃取,合并水相,蒸干得式(ib-7)所示的化合物的盐酸盐,收率80%。
[0123]1h nmr(500mhz cd3od)δ3.98(1h,d,j=12.3hz),3.88
–
3.82(1h,m),3.79(1h,d,j=12.3hz),3.71(1h,t,j=8.9hz),3.61(1h,d,j=8.9hz),3.35
–
3.28(1h,m),3.08(1h,t,j=11.8hz),2.02(1h,td,j=13.5hz,4.1hz),1.84(1h,td,j=13.5hz,4.2hz),1.66
–
1.46(2h,m),1.44
–
1.31(4h,m),0.95(3h,t,j=7.1hz)。
[0124]
(2-2)将(ia-8)74mg溶于重蒸的二氯甲烷(dcm,30ml)中,用氩气保护。在-78℃下,搅拌15分钟,滴加bcl3(1m in dcm,15ml),加毕,升温至-40℃反应,4小时后,原料消失。降温至-78℃,加入甲醇,升至室温,蒸干,加入去离子水和二氯甲烷萃取,合并水相,蒸干得式(ib-8)所示的化合物的盐酸盐,收率78%。
[0125]1h nmr(500mhz cd3od)δ3.95(1h,d,j=11.7hz),3.76
–
3.72(1h,m),3.70
–
3.62(3h,m),3.32
–
3.26(1h,m),2.93(1h,dd,j=12.5hz,9.9hz),1.88
–
1.79(1h,m),1.74
–
1.65(1h,m),1.52
–
1.43(1h,m),1.40
–
1.28(5h,m),0.92(3h,t,j=6.9hz)。
[0126]
实施例5
[0127]
本实施例用于说明式(ia-9)、(ia-10)、(ib-9)和(ib-10)的制备
[0128]
(1)将上述制备例1得到的式ⅱ所示的硝酮(1g,1.85mmol)溶于干燥的四氢呋喃(25ml)中,在冰水浴(0-5℃)下,将正己基溴化镁(7.4mmol)滴加到上述溶液中,滴加用时6分钟,继续反应40分钟,用饱和氯化铵溶液淬灭反应,乙酸乙酯(3
×
25ml)萃取,合并有机相,饱和氯化钠溶液(2
×
25ml)洗涤,无水硫酸镁干燥,减压浓缩除去溶剂,粗产物通过柱层析(硅胶200-300目)分离,得到式(ia-9)所示的化合物,收率67%,和式(ia-10)所示的化合物,收率11%。
[0129]
式(ia-9),1h nmr(400mhz,cdcl3)δ7.25-7.15(20h,m,phch2o),6.31(1h,s,br noh),4.83(2h,t,j=10.4hz),4.64(1h,d,j=10.8hz),4.58(2h,s),4.51(1h,d,j=11.1hz),4.42(2h,s),4.10(1h,d,j=9.9hz),3.74(1h,t,j=8.9hz),3.63(2h,d,j=10.2hz),3.54(1h,dd,j=10.5hz,5.4hz),3.46(1h,d,j=9.1hz),3.05(1h,t,j=10.4hz),2.07
–
1.97(1h,m),1.56
–
1.46(2h,m),1.18
–
1.09(7h,m),0.77(3h,t,j=6.7hz);
[0130]
式(ia-10),1h nmr(500mhz,cdcl3)δ7.21
–
7.08(20h,m,phch2o),6.14(1h,s,br,noh),4.82(1h,d,j=10.9hz),4.78(1h,d,j=11.3hz),4.70(1h,d,j=10.9hz),4.62(1h,d,j=11.3hz),4.56(1h,d,j=11.3hz),4.42(1h,d,j=11.3hz),4.41(1h,d,j=11.9hz),4.33(1h,d,j=11.9hz),3.90
–
3.79(2h,m),3.72(1h,t,j=9.3hz),3.68(1h,d,j=9.5hz),3.53(1h,d,j=9.4hz),3.32(1h,dd,j=12.3hz,5.5hz),2.90(1h,t,j=11.5hz),1.62
–
1.57(2h,m),1.33
–
1.30(2h,m),1.18
–
1.14(6h,m),0.79(3h,t,j=6.8hz)。
[0131]
(2-1)将式(ia-9)所示的化合物320mg溶于甲醇(15ml)中,加入41.6mg的10%pd/c,加入盐酸(9ml,1n),氩气置换三次,氢气置换三次,反应18小时,过滤,弃去滤饼,得到滤液,-0.18mpa减压条件下除去溶剂得式(ib-9)所示的化合物的盐酸盐,收率100%。
[0132]1h nmr(400mhz,cd3od)δ3.99(1h,d,j=12.2hz),3.91
–
3.76(2h,m),3.71(1h,t,j=8.8hz),3.61(1h,d,j=8.8hz),3.36
–
3.27(1h,m),3.09(1h,t,j=11.7hz),2.10
–
1.96(1h,m),1.91
–
1.78(1h,m),1.68
–
1.44(2h,m),1.43
–
1.32(6h,m),0.98
–
0.88(3h,m)。
[0133]
(2-2)将式(ia-10)所示的化合物100mg溶于甲醇(30ml)中,加入18mg的10%pd/c,加入盐酸(21ml,1n),氩气置换三次,氢气置换三次,反应12小时,过滤,弃去滤饼,得到滤液,-0.4mpa减压条件下除去溶剂得式(ib-10)所示的化合物的盐酸盐,收率71%。
[0134]1h nmr(500mhz,cd3od)δ3.94(1h,d,j=11.6hz),3.77
–
3.71(1h,m),3.70
–
3.62(3h,m),3.32
–
3.26(1h,m),2.93(1h,t,j=10.5hz),1.87
–
1.78(1h,m),1.69(1h,t,j=12.4hz),1.50
–
1.44(1h,m),1.38
–
1.30(7h,m),0.93
–
0.88(3h,m)。
[0135]
实施例6
[0136]
本实施例用于说明式(ia-11)、(ia-12)、(ib-11)和(ib-12)的制备
[0137]
(1)将上述制备例1得到的式ⅱ所示的硝酮(1g,1.85mmol)溶于干燥的四氢呋喃(30ml)中,在冰水浴(0-5℃)下,将正庚基溴化镁(7.4mmol)滴加到上述溶液中,滴加用时20分钟,继续反应35分钟,用饱和氯化铵溶液淬灭反应,乙酸乙酯(3
×
25ml)萃取,合并有机相,饱和氯化钠溶液(2
×
25ml)洗涤,无水硫酸镁干燥,减压浓缩除去溶剂,粗产物通过柱层析(硅胶200-300目)分离,得到式(ia-11)所示的化合物,收率46%,和式(ia-12)所示的化合物,收率13%。
[0138]
式(ia-11),1h nmr(400mhz,cdcl3)δ7.22
–
7.13(20h,m,phch2o),6.31(1h,noh),
4.84(1h,d,j=10.8hz),4.82(1h,d,j=10.8hz),4.63(1h,d,j=10.9hz),4.56(2h,s),4.50(1h,d,j=10.8hz),4.39(2h,s),4.06(1h,d,j=9.8hz),3.78
–
3.74(1h,m),3.69
–
3.60(2h,m),3.51(1h,dd,j=10.5hz,5.4hz),3.45(1h,d,j=9.1hz),3.06(1h,t,j=10.5hz),2.03
–
1.94(1h,m),1.54
–
1.45(2h,m),1.17
–
1.09(9h,m),0.76(3h,t,j=6.6hz);
[0139]
式(ia-12),1h nmr(400mhz,cdcl3)δ7.22
–
7.10(20h,m,phch2o),5.42(1h,s,br,noh),4.83(1h,d,j=13.7hz),4.80(1h,d,j=11.5hz),4.71(1h,d,j=10.8hz),4.64(1h,d,j=11.3hz),4.58(1h,d,j=11.3hz),4.44
–
4.35(3h,m),3.93
–
3.81(2h,m),3.71(2h,m),3.56(1h,d,j=9.4hz),3.34(1h,dd,j=12.4hz,5.3hz),2.88(1h,t,j=11.5hz),1.58(1h,t,j=7.9hz),1.32
–
1.18(11h,m),0.81(3h,t,j=6.5hz)。
[0140]
(2-1)将式(ia-11)所示的化合物542mg溶于甲醇(18ml)中,加入108mg的10%pd/c,加入盐酸(12ml,1n),氩气置换三次,氢气置换三次,反应19小时,过滤,弃去滤饼,得到滤液,-0.17mpa减压条件下除去溶剂得式(ib-11)所示的化合物的盐酸盐,收率99%。
[0141]1h nmr(500mhz cd3od)δ3.18(1h,d,j=11.8hz),3.14
–
3.04(1h,m),3.00(1h,d,j=11.9hz),2.94(1h,t,j=8.2hz),2.83(1h,d,j=8.4hz),2.56
–
2.48(1h,m),2.36
–
2.24(1h,m),1.26
–
1.14(1h,m),1.10
–
0.96(1h,m),0.85
–
0.62(2h,m),0.62
–
0.42(8h,m),0.16
–
0.04(3h,s)。
[0142]
(2-2)将式(ia-12)所示的化合物60mg溶于甲醇(20ml)中,加入6mg的10%pd/c,加入盐酸(14ml,1n),氩气置换三次,氢气置换三次,反应14小时,过滤,弃去滤饼,得到滤液,-0.3mpa减压条件下除去溶剂得式(ib-12)所示的化合物的盐酸盐,收率51%。
[0143]1h nmr(500mhz cd3od)δ3.95(1h,d,j=11.6hz),3.76
–
3.70(1h,m),3.69
–
3.60(3h,m),3.30
–
3.26(1h,m),2.93(1h,t,j=11.1hz),1.87
–
1.79(1h,m),1.69(1h,t,j=12.2hz),1.51
–
1.41(1h,m),1.39
–
1.26(9h,m),0.89(3h,t,j=6.7hz)。
[0144]
实施例7
[0145]
本实施例用于说明式(ia-13)、(ia-14)、(ib-13)和(ib-14)的制备
[0146]
(1)将上述制备例1得到的式ⅱ所示的硝酮(1.04g,1.94mmol)溶于干燥的四氢呋喃(25ml)中,在冰水浴(0-5℃)下,将正辛基溴化镁(7.8mmol)滴加到上述溶液中,滴加用时7分钟,继续反应30分钟,用饱和氯化铵溶液淬灭反应,乙酸乙酯(3
×
25ml)萃取,合并有机相,饱和氯化钠溶液(2
×
25ml)洗涤,无水硫酸镁干燥,减压浓缩除去溶剂,粗产物通过柱层析(硅胶200-300目)分离,得到式(ia-13)所示的化合物,收率69%,和式(ia-14)所示的化合物,收率12%。
[0147]
式(ia-13),1h nmr(500mhz,cdcl3)δ7.36
–
7.23(20h,m,phch2o),6.40(1h,s,br,noh),4.91(2h,t,j=12.0hz),4.72(1h,d,j=10.8hz),4.67(2h,abq,j=11.6hz),4.59(1h,d,j=11.1hz),4.51(2h,s),4.19(1h,d,j=9.5hz),3.83
–
3.79(1h,m),3.77
–
3.68(2h,m),3.62(1h,dd,j=10.5hz,5.4hz),3.53(1h,d,j=9.1hz),3.12(1h,t,j=10.5hz),2.16
–
2.12(1h,m),1.62
–
1.54(2h,m),1.28
–
1.24(11h,m),0.86(3h,t,j=7.0hz);
[0148]
式(ia-14),1h nmr(500mhz,cdcl3)δ7.20
–
7.07(20h,m,phch2o),6.18(1h,noh),4.81(1h,d,j=10.9hz),4.77(1h,d,j=11.3hz),4.70(1h,d,j=10.9hz),4.61(1h,d,j=11.3hz),4.55(1h,d,j=11.3hz),4.40(2h,d,j=12.1hz),4.32(1h,d,j=11.9hz),3.88
–
3.78(2h,m),3.71(1h,d,j=9.3hz),3.68(1h,d,j=9.4hz),3.53(1h,d,j=9.4hz),3.33
(1h,dd,j=12.2hz,5.5hz),2.91(1h,t,j=11.5hz),1.58(2h,t,j=8.5hz),1.34
–
1.12(12h,m),0.80(3h,t,j=6.9hz)。
[0149]
(2-1)将式(ia-13)所示的化合物660mg溶于甲醇(27ml)中,加入72.6mg的10%pd/c,加入盐酸(13ml,1n),氩气置换三次,氢气置换三次,反应20小时,过滤,弃去滤饼,得到滤液,-0.19mpa减压条件下除去溶剂得式(ib-13)所示的化合物的盐酸盐,收率99%。
[0150]1h nmr(400mhz cd3od)δ3.92(1h,d,j=12.2hz),3.88
–
3.78(1h,m),3.76
–
3.63(2h,m),3.56(1h,d,j=8.7hz),3.29
–
3.20(1h,m),3.03(1h,t,j=11.4hz),1.96
–
1.91(1h,m),1.82
–
1.70(1h,m),1.58
–
1.37(2h,m),1.32
–
1.16(10h,m),0.86
–
0.78(3h,m)。
[0151]
(2-2)将式(ia-14)所示的化合物135mg溶于甲醇(35ml)中,加入17.8mg的10%pd/c,加入盐酸(15ml,1n),氩气置换三次,氢气置换三次,反应15小时,过滤,弃去滤饼,得到滤液,-0.6mpa减压条件下除去溶剂得式(ib-14)所示的化合物的盐酸盐,收率93%。
[0152]1h nmr(400mhz cd3od)δ3.93(1h,d,j=11.8hz),3.80
–
3.75(1h,m),3.72
–
3.64(3h,m),3.34
–
3.28(1h,m),2.98
–
2.88(1h,m),1.88
–
1.77(1h,m),1.75
–
1.64(1h,m),1.52
–
1.20(12h,m),0.87(3h,t,j=6.5hz)。
[0153]
实施例8
[0154]
本实施例用于说明式(ia-15)、(ia-16)、(ib-15)和(ib-16)的制备
[0155]
(1)将上述制备例1得到的式ⅱ所示的硝酮(0.5g,0.93mmol)溶于干燥的四氢呋喃(30ml)中,在冰水浴(0-5℃)下,将正壬基溴化镁(3.7mmol)滴加到上述溶液中,滴加用时20分钟,继续反应30分钟,用饱和氯化铵溶液淬灭反应,乙酸乙酯(3
×
25ml)萃取,合并有机相,饱和氯化钠溶液(2
×
25ml)洗涤,无水硫酸镁干燥,减压浓缩除去溶剂,粗产物通过柱层析(硅胶200-300目)分离,得到式(ia-15)所示的化合物,收率84%,和式(ia-16)所示的化合物,收率8%。
[0156]
式(ia-15)1h nmr(500mhz,cdcl3)δ7.36
–
7.23(20h,m),6.40(1h,s,br),4.93(1h,d,j=11.0hz),4.91(1h,d,j=12.4hz),4.72(1h,d,j=10.8hz),4.67(2h,abq,j=11.7hz),4.59(1h,d,j=11.1hz),4.51(2h,s),4.19(1h,d,j=9.5hz),3.81(1h,t,j=8.9hz),3.77
–
3.68(2h,m),3.62(1h,dd,j=10.4hz,5.4hz),3.53(1h,d,j=9.1hz),3.12(1h,j=10.5hz),2.18
–
2.08(1h,m),1.69
–
1.50(3h,m),1.30
–
1.16(12h,m),0.86(3h,t,j=6.9hz);
[0157]
式(ia-16)1h nmr(500mhz,cdcl3)δ7.38
–
7.14(20h,m),5.89(1h,s,br),4.90(1h,d,j=10.9hz),4.86(1h,d,j=11.3hz),4.78(1h,d,j=10.9hz),4.70(1h,d,j=11.3hz),4.64(1h,d,j=11.2hz),4.51(1h,d,j=11.3hz),4.50(1h,d,j=11.9hz),4.42(1h,d,j=11.9hz),3.99
–
3.85(2h,m),3.82
–
3.72(2h,m),3.62(1h,d,j=9.4hz),3.40(1h,dd,j=12.4hz,5.5hz),2.97(1h,t,j=11.6hz),1.72
–
1.60(2h,m),1.46
–
1.15(14h,m),0.88(3h,t,j=6.9hz)。
[0158]
(2-1)将式(ia-15)所示的化合物40mg溶于甲醇(32ml)中,加入8mg的10%pd/c,加入盐酸(14ml,1n),氩气置换三次,氢气置换三次,反应18小时,过滤,弃去滤饼,得到滤液,-0.52mpa减压条件下除去溶剂得(ib-15)所示的化合物的盐酸盐,收率94%。
[0159]1h nmr(500mhz cd3od)δ3.94(1h,d,j=11.9hz),3.75(2h,d,j=11.6hz),3.64(1h,t,j=8.0hz),3.54(1h,d,j=8.3hz),3.24(1h,d,j=8.9hz),3.02(1h,t,j=11.0hz),
1.99(1h,t,j=11.4hz),1.80(1h,t,j=11.6hz),1.64
–
1.42(2h,m),1.42
–
1.10(12h,m),0.89(3h,d,j=6.2hz)。
[0160]
(2-2)将式(ia-16)所示的化合物31mg溶于甲醇(18ml)中,加入4.7mg(的10%pd/c,加入盐酸(11ml,1n),氩气置换三次,氢气置换三次,反应27小时,过滤,弃去滤饼,得到滤液,-0.14mpa减压条件下除去溶剂得式(ib-16)所示的化合物的盐酸盐,收率93%。
[0161]1h nmr(500mhz cd3od)δ3.95(1h,d,j=11.5hz),3.78
–
3.60(4h,m),3.27(1h,d,j=9.2hz),2.94(1h,t,j=10.6hz),1.88
–
1.78(1h,m),1.70(1h,t,j=12.8hz),1.52
–
1.22(14h,m),0.90(3h,t,j=6.2hz)。
[0162]
实施例9
[0163]
本实施例用于说明式(ia-17)、(ia-18)、(ib-17)和(ib-18)的制备
[0164]
(1)将上述制备例1得到的式ⅱ所示的硝酮(0.82g,1.52mmol)溶于干燥的四氢呋喃(30ml)中,在冰水浴(0-5℃)下,将正癸基溴化镁(6.08mmol)滴加到上述溶液中,滴加用时13分钟,继续反应35分钟,用饱和氯化铵溶液淬灭反应,乙酸乙酯(3
×
25ml)萃取,合并有机相,饱和氯化钠溶液(2
×
25ml)洗涤,无水硫酸镁干燥,减压浓缩除去溶剂,粗产物通过柱层析(硅胶200-300目)分离,得到式(ia-17)所示的化合物,收率73%,和式(ia-18)所示的化合物,收率19%。
[0165]
式(ia-17)1h nmr(400mhz,cdcl3)δ7.33
–
7.22(20h,m,phch2o),6.38(1h,s,br,noh),4.94(1h,d,j=10.7hz),4.91(1h,d,j=10.8hz),4.72(1h,d,j=10.8hz),4.67(2h,s),4.59(1h,d,j=11.1hz),4.50(2h,s),4.19(1h,d,j=9.8hz),3.81(1h,t,j=8.8hz),3.78
–
3.68(2h,m),3.62(1h,dd,j=10.4hz,5.3hz),3.53(1h,d,j=9.0hz),3.12(1h,t,j=10.4hz),2.20
–
2.08(1h,m),1.64
–
1.52(2h,m),1.28
–
1.22(15h,m),0.87(3h,t,j=6.6hz);
[0166]
式(ia-18)1h nmr(400mhz,cdcl3)δ7.21
–
7.08(20h,m,phch2o),6.12(1h,br,noh),4.82(1h,d,j=10.9hz),4.78(1h,d,j=11.4hz),4.70(1h,d,j=10.8hz),4.62(1h,d,j=12.5),4.57
–
4.54(1h,m),4.43(1h,d,j=11.2hz),4.41(1h,d,j=11.8hz),4.33(1h,d,j=11.8hz),3.90
–
3.80(2h,m),3.72(1h,d,j=9.2hz),3.68(1h,d,j=9.4hz),3.54(1h,d,j=9.4hz),3.32(1h,dd,j=12.2hz,5.4hz),2.90(1h,t,j=11.5hz),1.64
–
1.54(1h,m),1.40
–
1.27(2h,m),1.21
–
1.14(15h,m),0.80(3h,t,j=6.5hz)。
[0167]
(2-1)将式(ia-17)所示的化合物494mg溶于甲醇(23ml)中,加入89mg的10%pd/c,加入盐酸(17ml,1n),氩气置换三次,氢气置换三次,反应30小时,过滤,弃去滤饼,得到滤液,-0.17mpa减压条件下除去溶剂得式(ib-17)所示的化合物的盐酸盐,收率61%。
[0168]1h nmr(500mhz cd3od)δ3.99(1h,d,j=12.1hz),3.88
–
3.78(2h,m),3.71(1h,t,j=8.5hz),3.61(1h,d,j=8.6hz),3.40
–
3.28(1h,m),3.09(1h,t,j=11.4hz),2.04(1h,t,j=11.9hz),1.85(1h,t,j=11.9hz),1.65
–
1.50(2h,m),1.40
–
1.31(14h,m),0.93(3h,t,j=6.4hz)。
[0169]
(2-2)将式(ia-18)所示的化合物110mg溶于甲醇(26ml)中,加入11mg的10%pd/c,加入盐酸(2ml,1n),氩气置换三次,氢气置换三次,反应36小时,过滤,弃去滤饼,得到滤液,-0.8mpa减压条件下除去溶剂得式(ib-18)所示的化合物的盐酸盐,收率9%。
[0170]1h nmr(500mhz cd3od)δ3.95(1h,d,j=11.7hz),3.75
–
3.69(1h,m),3.68
–
3.59(3h,m),3.24(1h,dd,j=12.7hz,4.5hz),2.96
–
2.88(1h,m),1.88
–
1.78(1h,m),1.72
–
1.64
(1h,m),1.51
–
1.40(2h,m),1.32
–
1.24(14h,m),0.89(3h,t,j=6.8hz)。
[0171]
实施例10
[0172]
本实施例用于说明式(ia-19)、(ia-20)、(ib-19)和(ib-20)的制备
[0173]
(1)将上述制备例1得到的式ⅱ所示的硝酮(1.98g,3.67mmol)溶于干燥的四氢呋喃(30ml)中,在冰水浴(0-5℃)下,将正癸基溴化镁(14.68mmol)滴加到上述溶液中,滴加用时7分钟,继续反应40分钟,用饱和氯化铵溶液淬灭反应,乙酸乙酯(3
×
25ml)萃取,合并有机相,饱和氯化钠溶液(2
×
25ml)洗涤,无水硫酸镁干燥,减压浓缩除去溶剂,粗产物通过柱层析(硅胶200-300目)分离,得到式(ia-19)所示的化合物,收率65%,和式(ia-20)所示的化合物,收率25%。
[0174]
式(ia-19)1h nmr(400mhz,cdcl3)δ7.32
–
7.23(20h,m,phch2o),6.36(1h,s,br,noh),4.94(1h,d,j=10.6hz),4.91(1h,d,j=10.2hz),4.72(1h,d,j=10.8hz),4.66(2h,s),4.60(1h,d,j=11.1hz),4.50(2h,s),4.18(1h,d,j=9.9hz),3.82(1h,t,j=8.8hz),3.75
–
3.68(2h,m),3.62(1h,dd,j=10.4hz,5.3hz),3.54(1h,d,j=9.0hz),3.13(1h,t,j=10.4hz),2.18
–
2.06(1h,m),1.65
–
1.49(2h,m),1.34
–
1.12(17h,m),0.87(3h,t,j=6.5hz);
[0175]
式(ia-20)1h nmr(500mhz,cdcl3)δ7.33
–
7.17(20h,m,phch2o),5.56(1h,s,br,noh),4.90(1h,d,j=10.9hz),4.87(1h,d,j=11.3hz),4.78(1h,d,j=10.9hz),4.71(1h,d,j=11.4hz),4.65(1h,d,j=11.4hz),4.51(1h,d,j=11.4hz),4.50(1h,d,j=12.0hz),4.44(1h,d,j=11.9hz),3.99
–
3.89(2h,m),3.80(1h,d,j=9.3hz),3.75(1h,d,j=9.4hz),3.63(1h,d,j=9.4hz),3.41(1h,dd,j=12.5hz,5.6hz),2.95(1h,t,j=11.7hz),1.68
–
1.62(2h,m),1.45
–
1.16(18h,m),0.88(3h,t,j=7.0hz)。
[0176]
(2-1)将式(ia-19)所示的化合物220mg溶于甲醇(20ml)中,加入26mg的10%pd/c,加入盐酸(5.5ml,1n),氩气置换三次,氢气置换三次,反应36h,过滤,弃去滤饼,得到滤液,-0.44mpa减压条件下除去溶剂得式(ib-19)所示的化合物的盐酸盐,收率100%。
[0177]1h nmr(500mhz cd3od)δ3.99(1h,d,j=12.2hz),3.88
–
3.78(2h,m),3.71(1h,t,j=8.7hz),3.61(1h,d,j=8.8hz),3.31(1h,dd,j=12.5hz,4.6hz),3.09(1h,t,j=11.7hz),2.08
–
2.00(1h,m),1.90
–
1.80(1h,m),1.67
–
1.46(2h,m),1.43
–
1.26(16h,m),0.93(3h,t,j=6.8hz)。
[0178]
(2-2)将式(ia-20)所示的化合物227mg溶于甲醇(24ml)中,加入36mg的10%pd/c,加入盐酸(5.5ml,1n),氩气置换三次,氢气置换三次,反应40小时,过滤,-0.37mpa减压条件下除去溶剂得式(ib-20)所示的化合物的盐酸盐,收率99%。
[0179]1h nmr(500mhz cd3od)δ3.95(1h,d,j=11.7hz),3.75
–
3.68(1h,m),3.68
–
3.66(1h,m),3.64(1h,d,j=2.6hz),3.61(1h,d,j=8.1hz),3.28
–
3.22(1h,m),2.93(1h,dd,j=12.8hz,9.4hz),1.87
–
1.78(1h,m),1.72
–
1.64(1h,m),1.50
–
1.40(1h,m),1.38
–
1.22(17h,m),0.89(3h,t,j=6.9hz)。
[0180]
实施例11
[0181]
本实施例用于说明式(ia-21)、(ia-22)、(ib-21)和(ib-22)的制备
[0182]
(1)将上述制备例1得到的式ⅱ所示的硝酮(0.96g,1.78mmol)溶于干燥的四氢呋喃(35ml)中,在冰水浴(0-5℃)下,将正癸基溴化镁(7.12mmol)滴加到上述溶液中,滴加用时6分钟,继续反应35分钟,用饱和氯化铵溶液淬灭反应,乙酸乙酯(3
×
25ml)萃取,合并有
机相,饱和氯化钠溶液(2
×
25ml)洗涤,无水硫酸镁干燥,减压浓缩除去溶剂,粗产物通过柱层析(硅胶200-300目)分离,得到(ia-21)所示的化合物,收率73%和(ia-22)所示的化合物,收率11%。
[0183]
式(ia-21),1h nmr(500mhz,cdcl3)δ7.32
–
7.23(20h,m,phch2o),6.37(1h,s,br,noh),4.93(1h,d,j=11.2hz),4.91(1h,d,j=11.8hz),4.73(1h,d,j=10.8hz),4.66(2h,s),4.60(1h,d,j=11.1hz),4.49(2h,s),4.18(1h,d,j=9.2hz),3.86
–
3.80(1h,m),3.76
–
3.68(2h,m),3.64
–
3.59(1h,m),3.54(1h,d,j=8.9hz),3.14(1h,t,j=10.5hz),2.18
–
2.08(1h,m),1.65
–
1.50(2h,m),1.34
–
1.12(19h,m),0.91
–
0.83(3h,m);
[0184]
式(ia-22),1h nmr(500mhz,cdcl3)δ7.24
–
7.09(20h,m,phch2o),5.83(1h,s,br,noh),4.82(1h,d,j=10.9hz),4.78(1h,d,j=11.3hz),4.71(1h,d,j=10.9hz),4.62(1h,d,j=11.3hz),4.56(1h,d,j=11.4hz),4.41(2h,d,j=11.5hz),4.34(1h,d,j=11.9hz),3.90
–
3.80(2h,m),3.72(1h,d,j=9.3hz),3.68(1h,d,j=9.5hz),3.54(1h,d,j=9.4hz),3.33(1h,dd,j=12.4hz,5.5hz),2.89(1h,t,j=11.6hz),1.58(2h,t,j=8.5hz),1.36
–
1.14(20h,m),0.80(3h,t,j=6.9hz)。
[0185]
(2-1)将式(ia-21)所示的化合物264mg溶于甲醇(25ml)中,加入34mg的10%pd/c,加入盐酸(6.5ml,1n),氩气置换三次,氢气置换三次,反应36小时,过滤,弃去滤饼,得到滤液,-0.78mpa减压条件下除去溶剂得式(ib-21)所示的化合物的盐酸盐,收率89%。
[0186]1h nmr(500mhz cd3od)δ3.98(1h,d,j=12.2hz),3.86
–
3.77(2h,m),3.69(1h,t,j=8.8hz),3.59(1h,d,j=8.9hz),3.35
–
3.28(1h,m),3.07(1h,dd,j=12.3hz,11.3hz),2.06
–
1.98(1h,m),1.88
–
1.80(1h,m),1.64
–
1.47(2h,m),1.41
–
1.26(18h,m),0.92(3h,t,j=6.9hz)。
[0187]
(2-2)将式(ⅰa-22)所示的化合物35mg溶于甲醇(35ml)中,加入6mg的10%pd/c,加入盐酸(4.5ml 1n),氩气置换三次,氢气置换三次,反应40小时,过滤,弃去滤饼,得到滤液,-0.85mpa减压条件下除去溶剂得式(ib-22)所示的化合物的盐酸盐,收率99%。
[0188]1h nmr(500mhz cd3od)δ 3.95(1h,d,j=11.8hz),3.76
–
3.69(1h,m),3.69
–
3.60(3h,m),3.28
–
3.24(1h,m),2.94(1h,dd,j=12.6hz,9.7hz),1.88
–
1.78(1h,m),1.73
–
1.64(1h,m),1.52
–
1.40(1h,m),1.40
–
1.22(19h,m),0.88(3h,t,j=6.8hz)。
[0189]
对比例1
[0190]
本对比例用于说明式(dia-1)、(dia-2)、(dib-1)和(dib-2)的制备
[0191]
(1)将上述制备例1得到的式ⅱ所示的硝酮(1.01g,1.88mmol)溶于干燥的四氢呋喃(15ml)中,在冰水浴(0-5℃)下,将甲基氯化镁(3.0m in thf,1.85ml,5.55mmol)滴加到上述溶液中,滴加用时14分钟,继续反应0.5小时,饱和氯化铵溶液淬灭反应,乙酸乙酯(3
×
25ml)萃取,合并有机相,饱和氯化钠溶液(2
×
25ml)洗涤,无水硫酸镁干燥,减压浓缩除去溶剂,粗产物通过柱层析(硅胶200-300目)分离,得到白色固体得到(dia-1)所示的化合物,收率31%,和(dia-2)所示的化合物,收率35%。
[0192]
式(dia-1),1h nmr(400mhz cdcl3)7.20-7.05(20h,m,phch2o),5.86(1h,s,noh),4.84-4.31(8h,m,phch2o),3.64-3.58(3h,m),3.45(1h,d,j=9.2hz),3.35-3.32(2h,m),2.87(1h,t,j=10.8hz),0.93(3h,s)。
[0193]
式(dia-2),1h nmr(500mhz cdcl3)7.33-7.23(20h,m,phch2o),6.48(1h,s,noh),
4.92-4.51(8h,m,phch2o),3.84-3.80(3h,m),3.71(1h,s),3.48(2h,d,j=7.2hz),3.13(1h,t,j=11.1hz),1.41(3h,s)。
[0194]
(2-1)将式(dia-1)所示的化合物(116mg)溶于甲醇(15ml)中,加入19mg的10%pd/c,加入盐酸(10ml 1n),氩气置换三次,氢气置换三次,反应48h,过滤,弃去滤饼,得到滤液,-0.8mpa减压条件下除去溶剂得式(dib-1)所示的化合物的盐酸盐(82mg,99%)。
[0195]1h nmr(500mhz cd3od)δ 3.85(1h,d,j=11.7hz),3.78-3.76(1h,m),3.63-3.58(3h,m),3.33-3.29(1h,m),3.02(1h,t,j=12.0hz),1.31(3h,s)。
[0196]
(2-2)将式(ⅰa-2)所示的化合物(320mg)溶于甲醇(25ml)中,加入60mg的10%pd/c,加入盐酸(4.5ml 1n),氩气置换三次,氢气置换三次,反应48h,过滤,-9mpa减压条件下除去溶剂得式(dib-2)所示的化合物的盐酸盐。(37.9mg,85%)。
[0197]1h nmr(500mhz cd3od)δ 3.96(1h,d,j=12.1hz),3.86-3.83(1h,m),3.72-3.66(2h,m),3.56(1h,d,j=9.0hz),3.35-3.29(1h,m),3.06(1h,t,j=11.6hz),1.54(3h,s)。
[0198]
测试例
[0199]
以下测试例用于说明本发明的c5枝化的dnj衍生物的抑制活性
[0200]
(1)试验材料及来源
[0201]
供试化合物:本发明所提供的式(ib-1)-(ib-22)所示的c5枝化的dnj衍生物以及作为对照化合物的dnj、式(dib-1)和式(dib-2)所示的化合物。
[0202]
试验材料:以下测试所用到的4-硝基酚吡喃糖苷基质、二糖和糖苷酶(α-葡萄糖苷酶、β-葡萄糖苷酶、α-半乳糖苷酶、β-半乳糖苷酶、α-甘露糖苷酶、β-甘露糖苷酶、α-l-岩藻糖苷酶、α-海藻糖酶、α-l-鼠李糖酶、淀粉葡萄糖苷酶以及β-葡萄糖苷酸化酶等)均购自sigma-aldrich。
[0203]
(2)测试方法
[0204]
活性测试以4-硝基酚吡喃糖苷为基质,在每种酶的最佳活性ph下进行测试。将基质、酶溶液(0.1-0.5mg/ml)和抑制剂(本发明的c5枝化的dnj衍生物和对照化合物)在37℃下培养30分钟,然后再紫外可见分光光度计中启动反应,测试其对400nm波长光的吸收。最后,使用grafit程序进行数据分析(参见leatherbarrow,r.j.grafit 4.0;erithacussoftware:staines,uk,1998)。
[0205]
(3)评价结果
[0206]
本发明提供的c5枝化的dnj衍生物以及对照化合物对糖苷酶的抑制活性结果如表1所示。
[0207]
表1中,ic
50
(μm)表示抑制50%酶的活性的抑制剂浓度。百分数表示1000μm下的抑制率。nd表示未测试化合物对该酶的活性。以下用到的糖苷酶均购买自sigma-aldrich。
[0208]
表1
[0209][0210]
续表1
[0211]
[0212][0213]
续表1
[0214][0215]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。