1.本发明涉及杀菌剂氯氟醚菌唑的制备技术,具体涉及一种制备氯氟醚菌唑中间体的方法。
背景技术:
2.氯氟醚菌唑(通用名:mefentrifluconazole)是巴斯夫公司新开发的具有划时代意义的三唑类杀菌剂,于2019年正式上市,未来的市场有望超过10亿美元每年。氯氟醚菌唑具有广谱、高效、内吸、铲除和保护等作用,尤其对多种较难防治的真菌病害具有杰出的生物活性,能显著加强60多种作物病害的防治,如玉米、谷物、大豆等大田作物,以及青椒、葡萄等经济作物,也可用于草坪和种子处理等。其不但生物活性更高,且环境特性好,对哺乳动物、蜜蜂等毒性较低,安全性高。
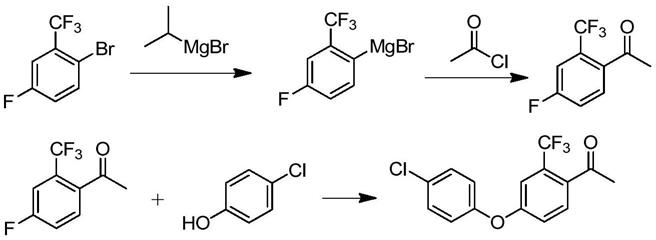
3.4-(4-氯苯氧基)-2-三氟甲基苯乙酮是合成氯氟醚菌唑的关键中间体,其经环氧化和开环取代反应制得氯氟醚菌唑,工艺简单且产率较高。目前生产4-(4-氯苯氧基)-2-三氟甲基苯乙酮所使用的技术多为参照现有技术cn103649057a中公开的技术。以2-溴-4-氟-三氟甲苯为原料,在四氢呋喃溶液中制备格氏试剂,再滴加乙酰氯,反应结束后经萃取、水洗等工艺制得4-氟-2-三氟甲基苯乙酮,接着与对氯苯酚反应合成4-(4-氯苯氧基)-2-三氟甲基苯乙酮。
[0004][0005]
在上述4-氟-2-三氟甲基苯乙酮的制备工艺中,需要用到格氏反应,条件较苛刻,同时反应后产生大量的含镁废水,难以处理。
[0006]
此外,wo2019/016115a1中公开了4-氟-2-三氟甲基苯乙酮经醚化反应合成4-(4-氯苯氧基)-2-三氟甲基苯乙酮时,不仅需要应用到钯类贵金属催化剂,废酸量大,工业化成本高,而且产物的收率较低。
[0007]
因此,现有方法在制备4-对氯苯氧基苯乙酮类化合物时,存在三废量大、生产环境恶劣、成本高等缺点。
技术实现要素:
[0008]
本发明的目的是为了克服现有技术在制备4-(4-氯苯氧基)-2-三氟甲基苯乙酮时,存在的三废量大、成本高等问题。
[0009]
为了实现上述目的,本发明提供一种制备式(i)所示的化合物的方法,该方法包
括:在极性非质子溶剂和碱性物质存在下,将式(ii)所示的化合物与对氯苯酚进行接触反应,
[0010][0011]
在本发明中,式(ii)所示的化合物可以采用现有技术中公知的方法合成,也可以是商购得到。
[0012]
在本发明中,在所述接触反应中,所述式(ii)所示的化合物与所述碱性物质和所述对氯苯酚的用量摩尔比为1:0.8-2:0.8-2。在优选情况下,所述接触反应中,所述式(ii)所示的化合物与所述碱性物质和所述对氯苯酚的用量摩尔比为1:0.9-1.2:0.9-1.2。
[0013]
在优选情况下,所述碱性物质选自氢氧化钠、氢氧化钾、碳酸钠和碳酸钾中的至少一种。更优选地,所述碱性物质为氢氧化钠、氢氧化钾或者碳酸钾。发明人发现,在该优选的具体实施方式下,本发明的方案具有进一步提高所述式(i)所示的化合物的收率的优势。
[0014]
在本发明中,所述极性非质子溶剂选自乙腈、二氧六环、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜和n-甲基吡咯烷酮中的至少一种。优选地,所述极性非质子溶剂为n,n-二甲基甲酰胺。发明人发现,在该优选的具体实施方式下,本发明的方案具有进一步提高接触反应的反应速率的优势。
[0015]
本发明中,优选情况下,所述接触反应的条件包括:温度为80-210℃,例如可以为80℃、100℃、120℃、140℃、160℃、180℃、200℃、210℃以及这些点值中的任意两个所构成的范围中的任意值,优选为100-160℃;时间为3-20h,例如可以为3h、5h、7h、9h、11h、13h、15h、17h、20h以及这些点值中的任意两个所构成的范围中的任意值,优选为5-13h。发明人发现,在该优选的具体实施方式下,式(ii)所示的化合物与对氯苯酚的接触反应更易进行。
[0016]
本发明中,所述接触反应可以是将所述极性非质子溶剂、所述碱性物质、所述式(ii)所示的化合物与所述对氯苯酚混合,以使得所述式(ii)所示的化合物与所述对氯苯酚能够接触进行反应。
[0017]
在优选情况下,所述接触反应的过程包括:(a)将所述极性非质子溶剂、所述碱性物质和所述对氯苯酚混合并加热以得到温度为80℃以上的混合物料i;(b)将所述混合物料i与所述式(ii)所示的化合物混合并加热至100℃以上。发明人发现,在该优选的具体实施方式下,具有进一步提高接触反应的反应速率,提高接触反应的转化率和收率的优势。
[0018]
本发明中,制备式(i)所示的化合物的方法还包括:将所述接触反应后得到的物料进行除溶剂处理以得到混合物料ii,并将所述混合物料ii进行纯化处理以得到精制的所述式(i)所示的化合物。示例性地,除溶剂处理采用蒸馏的方式去除所述接触反应后得到的物料中的所述极性非质子溶剂,得到所述混合物料ii。
[0019]
优选地,所述纯化处理的过程包括:将所述混合物料ii依次进行萃取、洗涤和分离;所述萃取过程中的萃取剂优选为醚类溶剂,所述醚类溶剂具体可以为异丙醚、甲基叔丁基醚、正丁醚,进一步地,所述醚类溶剂优选为甲基叔丁基醚;所述洗涤过程中的洗涤剂优选为水或碱性水溶液。
[0020]
示例性地,所述纯化处理的过程包括将所述混合物料ii与萃取剂、洗涤剂混合后,
经分层得到含有式(i)所示的化合物的有机层,从该有机层中脱出萃取剂得到精制的所述式(i)所示的化合物。发明人发现,在该优选的具体实施方式下,本发明的方案具有进一步提高式(i)所示的化合物的纯度的优势。
[0021]
通过上述技术方案,本发明制备式(i)所示的化合物的工艺中避免使用格氏试剂和重金属催化剂,有效降低原材料的成本,减少工艺过程中产生的三废,同时产物收率高。
具体实施方式
[0022]
以下对本发明的具体实施方式进行详细说明。应当理解的是,此处所描述的具体实施方式仅用于说明和理解本发明,并不用于限制本发明。
[0023]
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
[0024]
以下将通过实施例对本发明进行详细描述。以下实施例中:
[0025]
反应物和产物的量通过液相色谱(agilent hplc 1260)测得。
[0026]
反应的转化率和选择性通过以下公式计算:
[0027]
转化率=(原料投入摩尔量-产物中残留的原料摩尔量)/原料投入摩尔量
×
100%。
[0028]
选择性=目标产物的实际摩尔量/目标产物的理论摩尔量
×
100%
[0029]
在没有特别说明的情况下,所用原料均采用市售产品,式(i)所示的化合物的含量指的是式(i)所示的化合物占最终产物的质量百分比。
[0030]
实施例1
[0031]
(1)在装有机械搅拌、温度计、冷凝管的四口瓶中,加入0.11mol,98wt%的对氯苯酚和150ml的n,n-二甲基甲酰胺,再加入0.12mol,99wt%的固体氢氧化钠,升温至120℃搅拌反应3h,并不断蒸出反应产生的水,得到混合物料i;
[0032]
(2)将步骤(1)得到的混合物料i略降温,加入0.1mol,98wt%的式(ii)所示的化合物,加热至115℃继续反应5h,利用hplc监控反应完成,得到反应物料;
[0033]
(3)将步骤(2)得到的反应物料蒸出n,n-二甲基甲酰胺,然后加入甲基叔丁基醚,再进行水洗分层得到含有式(i)所示的化合物的有机层,将该有机层脱出甲基叔丁基醚,得到精制的式(i)所示的化合物(即4-(4-氯苯氧基)-2-三氟甲基苯乙酮),其含量为98%,收率为96%(以式(ii)所示的化合物为基准计算)。
[0034]
实施例2
[0035]
(1)在装有机械搅拌、温度计、冷凝管的四口瓶中,加入0.12mol,98wt%的对氯苯酚和150ml的n,n-二甲基甲酰胺,再加入0.11mol,99wt%的固体氢氧化钠,升温至110℃搅拌反应4h,并不断蒸出反应产生的水,得到混合物料i;
[0036]
(2)将步骤(1)得到的混合物料i略降温,加入0.1mol,98wt%的式(ii)所示的化合物,加热至105℃继续反应7h,利用hplc监控反应完成,得到反应物料;
[0037]
(3)将步骤(2)得到的反应物料蒸出n,n-二甲基甲酰胺,然后加入甲基叔丁基醚,再进行水洗分层得到含有式(i)所示的化合物的有机层,将该有机层脱出甲基叔丁基醚,得
到精制的式(i)所示的化合物(即4-(4-氯苯氧基)-2-三氟甲基苯乙酮),其含量为97.8%,收率为95.6%(以式(ii)所示的化合物为基准计算)。
[0038]
实施例3
[0039]
(1)在装有机械搅拌、温度计、冷凝管的四口瓶中,加入0.11mol,98wt%的对氯苯酚和150ml的n,n-二甲基甲酰胺,再加入0.09mol,99wt%的固体氢氧化钠,升温至155℃搅拌反应2h,并不断蒸出反应产生的水,得到混合物料i;
[0040]
(2)将步骤(1)得到的混合物料i略降温,加入0.1mol,98wt%的式(ii)所示的化合物,加热至140℃继续反应6h,利用hplc监控反应完成,得到反应物料;
[0041]
(3)将步骤(2)得到的反应物料蒸出n,n-二甲基甲酰胺,然后加入甲基叔丁基醚,再进行水洗分层得到含有式(i)所示的化合物的有机层,将该有机层脱出甲基叔丁基醚,得到精制的式(i)所示的化合物(即4-(4-氯苯氧基)-2-三氟甲基苯乙酮),其含量为97.2%,收率为95%(以式(ii)所示的化合物为基准计算)。
[0042]
实施例4
[0043]
(1)在装有机械搅拌、温度计、冷凝管的四口瓶中,加入0.11mol,98wt%的对氯苯酚和150ml的n,n-二甲基甲酰胺,再加入0.12mol,99wt%的固体氢氧化钠,升温至90℃搅拌反应6h,并不断蒸出反应产生的水,得到混合物料i;
[0044]
(2)将步骤(1)得到的混合物料i略降温,加入0.1mol,98wt%的式(ii)所示的化合物,加热至100℃继续反应12h,利用hplc监控反应完成,得到反应物料;
[0045]
(3)将步骤(2)得到的反应物料蒸出n,n-二甲基甲酰胺,然后加入甲基叔丁基醚,再进行水洗分层得到含有式(i)所示的化合物的有机层,将该有机层脱出甲基叔丁基醚,得到精制的式(i)所示的化合物(即4-(4-氯苯氧基)-2-三氟甲基苯乙酮),其含量为97%,收率为94.7%(以式(ii)所示的化合物为基准计算)。
[0046]
实施例5
[0047]
(1)在装有机械搅拌、温度计、冷凝管的四口瓶中,加入0.11mol,98wt%的对氯苯酚和150ml的n,n-二甲基甲酰胺,再加入0.12mol,99wt%的固体氢氧化钠,升温至200℃搅拌反应2h,并不断蒸出反应产生的水,得到混合物料i;
[0048]
(2)将步骤(1)得到的混合物料i略降温,加入0.1mol,98wt%的式(ii)所示的化合物,加热至190℃继续反应2h,利用hplc监控反应完成,得到反应物料;
[0049]
(3)将步骤(2)得到的反应物料蒸出n,n-二甲基甲酰胺,然后加入异丙醚,再进行水洗分层得到含有式(i)所示的化合物的有机层,将该有机层脱出异丙醚,得到精制的式(i)所示的化合物(即4-(4-氯苯氧基)-2-三氟甲基苯乙酮),其含量为96.8%,收率为94.4%(以式(ii)所示的化合物为基准计算)。
[0050]
通过上述实施例的结果可以看出,本发明制备4-(4-氯苯氧基)-2-三氟甲基苯乙酮(即式(i)所示的化合物)的工艺中能够在避免使用格氏试剂和重金属催化剂的前提下实现产物收率高的优势,同时,还能有效降低原材料的成本,减少工艺过程中产生的三废。
[0051]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。