1.本发明属于化学合成技术领域,具体涉及羰基取代苯并二氢呋喃及羰基取代苯并二氢吡喃化合物的合成方法。
背景技术:
2.苯并二氢呋喃、苯并二氢吡喃体系存在于大量的生物活性化合物中,具有显著的生物活性,如抗遗传毒性、抗增殖、抗癌和抗炎等,已报道的制备苯并二氢呋喃、苯并二氢吡喃类化合物的最常见的方法是邻烯丙基/异戊烯基酚的分子内环化法。羰基取代苯并二氢呋喃、苯并二氢吡喃类化合物也同样具有广泛的生物活性,例如抗癌、抗增殖以及对d
‑
氨基酸氧化酶的抑制活性。羰基取代苯并二氢呋喃、苯并二氢吡喃类化合物合成方法主要是使用i2(heterocycles.1995,41:219
‑
223)、h2so4(lett org chem.2009,6:29
‑
36;synthesis.2010,21:3745
‑
3754;eur j org chem.2015,10:2297
‑
2302)、hcl(chem pharm bull.1981,29:3033
‑
3036)、p
‑
tsoh(can j chem.1970,48:680
‑
684;rsc adv.2018,8:41377
‑
41388)、tfa(molecules.2018,23:776
‑
792)、alcl3(molecules.2018,23:776
‑
792)、zrcl4(synth commun.2004,34:3091
‑
3097)及bf3/et2o(tetrahedron lett.2007,48:7628
‑
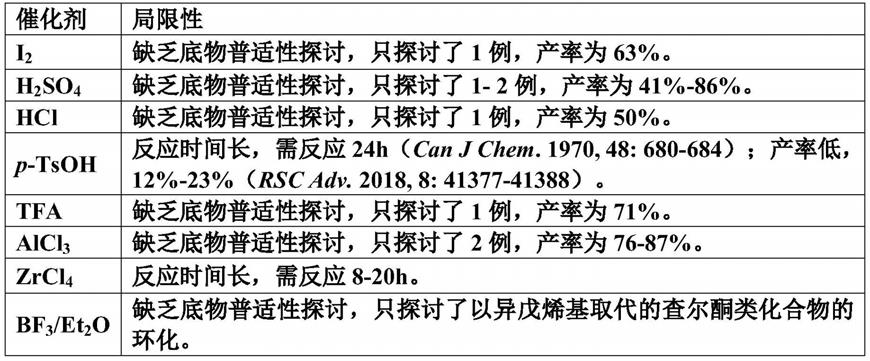
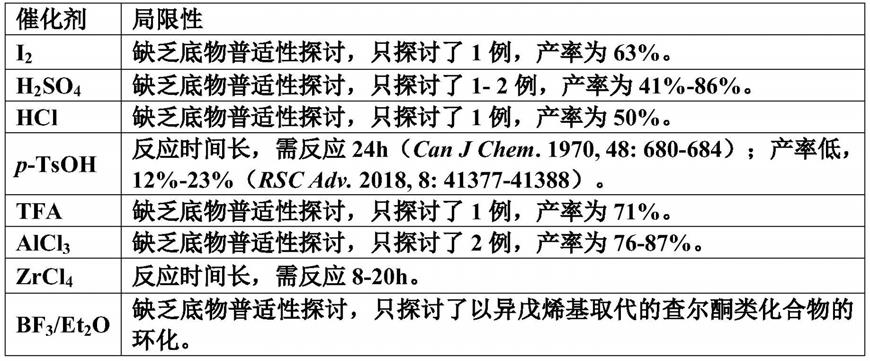
7632)进行催化环化。但是这些合成方法均存在局限性如产率低、反应时间长、未探讨底物普适性,具体局限性见表1。
3.表1已报道的羰基取代苯并二氢呋喃、苯并二氢吡喃类化合物合成方法的局限性
4.
技术实现要素:
5.本发明目的是提供羰基取代苯并二氢呋喃及羰基取代苯并二氢吡喃化合物的合成方法,该合成方法反应时间短,操作简单,不需要氮气保护,所用催化剂ppa廉价易得,底物普适性好。
6.为达到上述目的,本发明采用以下技术方案:
7.羰基取代苯并二氢呋喃及羰基取代苯并二氢吡喃化合物的合成方法,所述羰基取代苯并二氢呋喃、羰基取代苯并二氢吡喃化合物的结构如下式ⅱ1
‑
11所示:
[0008][0009]
合成方法如下:
[0010]
向耐压反应瓶中加入多聚磷酸ppa、溶剂n,n
‑
二甲基甲酰胺dmf和羰基取代邻烯丙基/羰基取代异戊烯基苯酚,加热搅拌反应2~10h;反应结束后分离纯化,即得羰基取代苯并二氢呋喃/羰基取代苯并二氢吡喃化合物;耐压反应瓶可有效避免溶剂挥出;
[0011]
所述羰基取代邻烯丙基、羰基取代异戊烯基苯酚的结构如下式ⅰ1
‑
11所示:
[0012][0013][0014]
前述羰基取代苯并二氢呋喃及羰基取代苯并二氢吡喃化合物的合成方法,所述羰基取代邻烯丙基/羰基取代异戊烯基苯酚与多聚磷酸ppa的摩尔比为1:5~30。
[0015]
前述羰基取代苯并二氢呋喃及羰基取代苯并二氢吡喃化合物的合成方法,所述溶剂dmf用量为1.5ml/mmol羰基取代邻烯丙基/羰基取代异戊烯基苯酚。
[0016]
前述羰基取代苯并二氢呋喃及羰基取代苯并二氢吡喃化合物的合成方法,所述加热为90~160℃油浴加热。
[0017]
前述羰基取代苯并二氢呋喃及羰基取代苯并二氢吡喃化合物的合成方法,所述反应结束后的分离纯化过程为:使用乙酸乙酯萃取反应结束后得到的物质2~3次,取有机层,依次使用水、饱和氯化钠溶液洗涤,再用无水硫酸钠干燥洗涤后的有机层,过滤;所得滤液进行减压浓缩,浓缩后的残余物再使用硅胶柱层析分离纯化,即得羰基取代苯并二氢呋喃/羰基取代苯并二氢吡喃化合物。
[0018]
前述羰基取代苯并二氢呋喃及羰基取代苯并二氢吡喃化合物的合成方法,所述硅胶柱层析洗脱剂为石油醚和乙酸乙酯,v
石油醚
/v
乙酸乙酯
=2~5:1。
[0019]
与现有技术相比,本发明的有益效果是:
[0020]
本发明提供了羰基取代苯并二氢呋喃及羰基取代苯并二氢吡喃化合物合成方法,使用多聚磷酸ppa作为催化剂催化羰基取代邻烯丙基/羰基取代异戊烯基苯酚分子内环化合成羰基取代苯并二氢呋喃/羰基取代苯并二氢吡喃化合物的方法为首次公开。本发明公开的合成方法反应时间短(2
‑
10h)、催化剂ppa廉价易得,极大降低了生产周期和成本;反应底物普适性好,产物的平均产率较高,最高可达到91%,不但为羰基取代苯并二氢呋喃及羰基取代苯并二氢吡喃化合物的合成提供了一种新方法,也为产品规模化生产及提高生产效率奠定了基础。
[0021]
为了保证本发明羰基取代的苯并二氢呋喃、苯并二氢吡喃化合物的合成方法科学、合理,发明人通过以下试验进行相应研究和筛选,最终确定了本发明的技术方案。
[0022]
以3
‑
烯丙基
‑2‑
羟基苯甲醛(ⅰ1)(0.2mmol)为反应底物,使用溶剂量为0.3ml,油浴130℃搅拌反应10h。选取了ppa摩尔当量及溶剂类别作为反应因素进行考察,考察各反应因素对产率的影响,结果如表1。
[0023]
表1各反应因素对产率的影响
[0024][0025]
由表1可知,最佳反应条件为ppa摩尔当量为5,反应溶剂为dmf。采用反应底物ⅰ2
‑ⅰ
11按前述最佳条件进行合成验证时发现,少数几个底物在ppa摩尔当量增至20、30时产率较高,因此ppa摩尔当量最终选定为5~30。
[0026]
采用优化后的ppa摩尔当量和反应溶剂dmf,合成了不同取代基的羰基取代苯并二氢呋喃/羰基取代苯并二氢吡喃化合物,详见实施例1
‑
11。
具体实施方式
[0027]
为了进一步确认该制备方法的合理性以及合成的化合物结构的准确性,采用核磁共振对实施例1
‑
11中制备得到的羰基取代苯并二氢呋喃/羰基取代苯并二氢吡喃化合物进行核磁共振(1h nmr和
13
c nmr)检测。
[0028]
实施例1:一种羰基取代苯并二氢呋喃化合物的合成方法:
[0029]
5ml耐压反应瓶中加入ppa 1mmol和溶剂dmf 0.3ml,再加入0.2mmol的原料ⅰ1,130℃油浴搅拌反应10h;反应结束后乙酸乙酯萃取3次,依次用水、饱和氯化钠溶液洗涤有机层,再用无水硫酸钠干燥洗涤后的有机层,过滤,滤液减压浓缩后的残余物经硅胶柱层析(v
石油醚
/v
乙酸乙酯
=5:1)分离纯化,即得羰基取代苯并二氢呋喃化合物ⅱ1,产率81%。
[0030]
制备得到的羰基取代苯并二氢呋喃化合物(ⅱ1)的核磁共振(1h nmr和
13
c nmr)检测数据为:1h nmr(600mhz,cdcl3)δ10.23(s,1h),7.59(d,j=7.8hz,1h),7.38(dd,j=7.2,1.2hz,1h),6.92(t,j=7.5hz,1h),5.12(ddq,j=12.7,8.8,6.3hz,1h),3.36(dd,j=15.6,8.9hz,1h),2.85(dd,j=15.6,7.4hz,1h),1.55(d,j=6.3hz,3h).
13
c nmr(151mhz,cdcl3)δ189.00,161.97,130.93,129.53,126.99,120.36,119.51,81.72,36.01,21.83.
[0031]
实施例2:一种羰基取代苯并二氢呋喃化合物的合成方法:
[0032]
5ml耐压反应瓶中加入ppa 1mmol和溶剂dmf 0.3ml,再加入0.2mmol的原料ⅰ2,150℃油浴搅拌反应6h;反应结束后乙酸乙酯萃取3次,依次用水、饱和氯化钠溶液洗涤有机层,再用无水硫酸钠干燥洗涤后的有机层,过滤,滤液减压浓缩后的残余物经硅胶柱层析(v
石油醚
/v
乙酸乙酯
=5:1)分离纯化,即得羰基取代苯并二氢呋喃化合物ⅱ2,产率52%。
[0033]
制备得到的羰基取代苯并二氢呋喃化合物(ⅱ2)的核磁共振(1h nmr和
13
c nmr)检测数据为:1h nmr(600mhz,cdcl3)δ9.82(s,1h),7.36(s,1h),7.34(s,1h),5.19
–
5.12(m,1h),3.96(s,3h),3.43(dd,j=15.4,9.0hz,1h),2.93(dd,j=15.4,7.8hz,1h),1.58(d,j=6.3hz,3h).
13
c nmr(151mhz,cdcl3)δ190.63,153.85,145.00,130.96,128.47,121.76,111.11,82.25,56.03,36.75,21.79.
[0034]
实施例3:一种羰基取代苯并二氢呋喃化合物的合成方法:
[0035]
5ml耐压反应瓶中加入ppa 4mmol、溶剂dmf 0.3ml和0.2mmol的原料ⅰ3,90℃油浴搅拌反应10h;反应结束后乙酸乙酯萃取3次,依次用水、饱和氯化钠溶液洗涤有机层,再用无水硫酸钠干燥洗涤后的有机层,过滤,滤液减压浓缩后的残余物经硅胶柱层析(v
石油醚
/v
乙酸乙酯
=4:1)分离纯化,即得羰基取代苯并二氢呋喃化合物ⅱ3,产率78%。
[0036]
制备得到的羰基取代苯并二氢呋喃化合物(ⅱ3)的核磁共振(1h nmr和
13
c nmr)检测数据为:1h nmr(600mhz,cdcl3)δ7.84(s,1h),7.82(d,j=8.4,1.8hz,1h),6.79(d,j=8.4hz,1h),5.05(m,j=8.8,7.3,6.4hz,1h),3.38(dd,j=15.5,8.9hz,1h),2.87(dd,j=15.5,7.4hz,1h),2.56(s,3h),1.51(d,j=6.3hz,3h).
13
c nmr(151mhz,cdcl3)δ196.72,163.85,130.56,130.46,127.76,125.63,108.92,81.04,36.37,26.40,21.79.
[0037]
实施例4:一种羰基取代苯并二氢呋喃化合物的合成方法:
[0038]
5ml耐压反应瓶中加入ppa 4mmol和溶剂dmf 0.3ml,再加入0.2mmol的原料ⅰ4,90℃油浴搅拌反应10h;反应结束后乙酸乙酯萃取3次,依次用水、饱和氯化钠溶液洗涤有机层,再用无水硫酸钠干燥洗涤后的有机层,过滤,滤液减压浓缩后的残余物经硅胶柱层析(v
石油醚
/v
乙酸乙酯
=3:1)分离纯化,即得羰基取代苯并二氢呋喃化合物ⅱ4,产率71%。
[0039]
制备得到的羰基取代苯并二氢呋喃化合物(ⅱ4)的核磁共振(1h nmr和
13
c nmr)检测数据为:1h nmr(600mhz,cdcl3)δ7.69(d,j=8.7hz,1h),6.40(d,j=8.7hz,1h),5.16
–
5.10(m,1h),3.32(dd,j=15.2,9.0hz,1h),2.79(dd,j=15.2,7.2hz,1h),2.60(s,3h),1.54(d,j=6.3hz,3h).
13
c nmr(151mhz,cdcl3)δ196.54,162.52,157.21,130.52,114.76,113.15,108.68,81.44,33.31,30.71,22.07.
[0040]
实施例5:一种羰基取代苯并二氢呋喃化合物的合成方法:
[0041]
5ml耐压反应瓶中加入ppa 6mmol、溶剂dmf 0.3ml和0.2mmol的原料ⅰ5,160℃油浴搅拌反应10h;反应结束后乙酸乙酯萃取2次,依次用水、饱和氯化钠溶液洗涤有机层,再用无水硫酸钠干燥洗涤后的有机层,过滤,滤液减压浓缩后的残余物经硅胶柱层析(v
石油醚
/v
乙酸乙酯
=5:1)分离纯化,即得羰基取代苯并二氢呋喃化合物ⅱ5,产率90%。
[0042]
制备得到的羰基取代苯并二氢呋喃化合物(ⅱ5)的核磁共振(1h nmr和
13
c nmr)检测数据为:1h nmr(600mhz,cdcl3)δ12.82(s,1h),7.90(s,1h),7.79
–
7.77(m,2h),7.68
–
7.65(m,2h),7.56(td,j=7.5,1.2hz,2h),7.49
–
7.43(m,4h),5.18(dp,j=9.2,6.4hz,1h),3.42(dd,j=15.6,9.3hz,1h),2.89(dd,j=15.6,6.7hz,1h),1.49(d,j=6.3hz,3h).
13
c nmr(151mhz,cdcl3)δ200.35,192.81,165.50,163.27,139.19,138.08,137.81,132.53,131.95,129.68,128.97,128.43,128.03,114.59,114.34,113.82,83.41,32.89,21.96.
[0043]
实施例6:一种羰基取代苯并二氢吡喃化合物的合成方法:
[0044]
5ml耐压反应瓶中加入ppa 1mmol和溶剂dmf 0.3ml,再加入0.2mmol的原料ⅰ6,110℃油浴搅拌反应2h;反应结束后乙酸乙酯萃取3次,依次用水、饱和氯化钠溶液洗涤有机层,再用无水硫酸钠干燥洗涤后的有机层,过滤,滤液减压浓缩后的残余物经硅胶柱层析(v
石油醚
/v
乙酸乙酯
=3:1)分离纯化,即得羰基取代苯并二氢吡喃化合物ⅱ6,产率80%。
[0045]
制备得到的羰基取代苯并二氢吡喃化合物(ⅱ6)的核磁共振(1h nmr和
13
c nmr)检测数据为:1h nmr(600mhz,cdcl3)δ13.09(s,1h),7.85(d,j=15.4hz,1h),7.64(s,1h),7.61(d,j=8.5hz,2h),7.48(d,j=15.4hz,1h),6.91(d,j=8.6hz,2h),6.39(s,1h),5.38(s,1h),2.80(t,j=6.7hz,2h),1.87(t,j=6.7hz,2h),1.39(s,6h).
13
c nmr(151mhz,cdcl3)δ191.76,164.13,161.38,157.90,143.76,131.04,130.53,127.90,118.18,115.98,114.25,112.63,104.93,75.94,32.80,26.99,21.83.
[0046]
实施例7:一种羰基取代苯并二氢吡喃化合物的合成方法:
[0047]
5ml耐压反应瓶中加入ppa 1mmol、溶剂dmf 0.3ml和0.2mmol的原料ⅰ7,110℃油浴搅拌反应2h;反应结束后乙酸乙酯萃取2次,依次用水、饱和氯化钠溶液洗涤有机层,再用无水硫酸钠干燥洗涤后的有机层,过滤,滤液减压浓缩后的残余物经硅胶柱层析(v
石油醚
/v
乙酸乙酯
=3:1)分离纯化,即得羰基取代苯并二氢吡喃化合物ⅱ7,产率77%。
[0048]
制备得到的羰基取代苯并二氢吡喃化合物(ⅱ7)的核磁共振(1h nmr和
13
c nmr)检测数据为:1h nmr(600mhz,cdcl3)δ14.84(s,1h),7.85(d,j=15.6hz,1h),7.78(d,j=
15.6hz,1h),7.53(d,j=8.6hz,2h),6.89(d,j=8.6hz,2h),5.90(s,1h),5.73(s,1h),3.90(s,3h),2.65(t,j=6.8hz,2h),1.83(t,j=6.8hz,2h),1.38(s,6h).
13
c nmr(151mhz,cdcl3)δ192.59,165.55,160.75,160.73,157.61,142.02,130.29,128.48,125.39,115.88,105.52,102.03,91.78,76.21,55.70,32.16,26.75,16.12.
[0049]
实施例8:一种羰基取代苯并二氢吡喃化合物的合成方法:
[0050]
5ml耐压反应瓶中加入ppa 1mmol和溶剂dmf 0.3ml,再加入0.2mmol的原料ⅰ8,130℃油浴搅拌反应2h;反应结束后乙酸乙酯萃取3次,依次用水、饱和氯化钠溶液洗涤有机层,再用无水硫酸钠干燥洗涤后的有机层,过滤,滤液减压浓缩后的残余物经硅胶柱层析(v
石油醚
/v
乙酸乙酯
=2:1)分离纯化,即得羰基取代苯并二氢吡喃化合物ⅱ8,产率81%。
[0051]
制备得到的羰基取代苯并二氢吡喃化合物(ⅱ8)的核磁共振(1h nmr和
13
c nmr)检测数据为:1h nmr(600mhz,cdcl3)δ11.51(s,1h),8.21(d,j=8.9hz,2h),7.06(d,j=9.0hz,2h),6.71(s,1h),6.28(s,1h),3.92(s,3h),2.93(t,j=6.7hz,2h),1.92(t,j=6.7hz,2h),1.41(s,6h).
13
c nmr(151mhz,cdcl3)δ175.18,160.99,160.37,158.38,153.91,144.97,135.78,129.16,123.65,114.14,103.54,99.83,76.12,55.43,31.71,26.65,16.25.
[0052]
实施例9:一种羰基取代苯并二氢吡喃化合物的合成方法:
[0053]
5ml耐压反应瓶中加入ppa 1mmol、溶剂dmf 0.3ml和0.2mmol的原料ⅰ9,110℃油浴搅拌反应4h;反应结束后乙酸乙酯萃取3次,依次用水、饱和氯化钠溶液洗涤有机层,再用无水硫酸钠干燥洗涤后的有机层,过滤,滤液减压浓缩后的残余物经硅胶柱层析(v
石油醚
/v
乙酸乙酯
=3:1)分离纯化,即得羰基取代苯并二氢吡喃化合物ⅱ9,产率69%。
[0054]
制备得到的羰基取代苯并二氢吡喃化合物(ⅱ9)的核磁共振(1h nmr和
13
c nmr)检测数据为:1h nmr(600mhz,cdcl3)δ11.80(s,1h),7.20(dd,j=8.4,2.0hz,1h),7.16(s,1h),6.85(d,j=8.4hz,1h),5.99(s,1h),5.34(dd,j=13.3,2.8hz,1h),3.08(dd,j=17.0,13.3hz,1h),2.83(t,j=6.7hz,2h),2.79(dd,j=17.0,2.9hz,1h),2.66
–
2.55(m,2h),1.85(t,j=6.7hz,2h),1.82
–
1.73(m,2h),1.38(s,6h),1.37(s,3h),1.35(s,3h).
13
c nmr(151mhz,cdcl3)δ196.12,162.99,161.45,160.04,154.47,129.72,127.47,125.41,121.12,117.56,102.64,100.81,97.41,78.99,76.12,74.58,43.19,32.66,31.91,27.16,26.94,22.57,16.36.
[0055]
实施例10:一种羰基取代苯并二氢吡喃化合物的合成方法:
[0056]
5ml耐压反应瓶中加入ppa 1mmol、溶剂dmf 0.3ml和0.2mmol的原料ⅰ10,130℃油浴搅拌反应2h;反应结束后乙酸乙酯萃取3次,依次用水、饱和氯化钠溶液洗涤有机层,再用无水硫酸钠干燥洗涤后的有机层,过滤,滤液减压浓缩后的残余物经硅胶柱层析(v
石油醚
/v
乙酸乙酯
=2:1)分离纯化,即得羰基取代苯并二氢吡喃化合物ⅱ10,产率91%。
[0057]
制备得到的羰基取代苯并二氢吡喃化合物(ⅱ10)的核磁共振(1h nmr和
13
c nmr)检测数据为:1h nmr(600mhz,cdcl3)δ13.74(s,1h),6.84(s,1h),6.37(s,1h),6.25(s,1h),5.30(t,j=6.4hz,1h),4.12(d,j=6.3hz,2h),3.83(s,3h),2.73(t,j=6.8hz,2h),1.87
–
1.84(m,5h),1.71(s,3h),1.39(s,6h).
13
c nmr(151mhz,cdcl3)δ182.04,160.73,160.62,155.89,154.72,154.45,142.40,136.94,132.08,123.27,112.15,103.78,102.87,101.62,94.02,76.04,62.03,31.89,26.75,26.55,25.83,18.22.
[0058]
实施例11:一种羰基取代苯并二氢吡喃化合物的合成方法:
[0059]
5ml耐压反应瓶中加入ppa 1mmol和溶剂dmf 0.3ml,再加入0.2mmol的原料ⅰ11,130℃油浴搅拌反应2h;反应结束后乙酸乙酯萃取3次,依次用水、饱和氯化钠溶液洗涤有机层,再用无水硫酸钠干燥洗涤后的有机层,过滤,滤液减压浓缩后的残余物经硅胶柱层析(v
石油醚
/v
乙酸乙酯
=2:1)分离纯化,即得羰基取代苯并二氢吡喃化合物ⅱ11,产率89%。
[0060]
制备得到的羰基取代苯并二氢吡喃化合物(ⅱ11)的核磁共振(1h nmr和
13
c nmr)检测数据为:1h nmr(600mhz,cdcl3)δ13.76(s,1h),6.82(s,1h),6.41(s,1h),6.27(s,1h),3.53(t,j=6.8hz,2h),2.74(t,j=6.8hz,2h),1.91(t,j=6.8hz,2h),1.86(t,j=6.8hz,2h),1.41(s,6h),1.39(s,6h).
13
c nmr(151mhz,cdcl3)δ182.67,160.51,160.44,154.93,153.27,151.54,137.79,121.33,111.22,103.53,103.03,100.50,93.99,75.94,75.51,32.89,31.92,26.77,26.49,22.37,16.08.
[0061]
羰基取代苯并二氢呋喃/羰基取代苯并二氢吡喃化合物ⅱ1
‑
11结构式按编号对应如下:
[0062]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。