一种含co4o4准立方烷结构的钴基金属有机框架化合物的制备方法及应用
技术领域
1.本发明涉及一种含co4o4准立方烷结构的钴基金属有机框架催化剂的制备方法及应用,属于催化材料技术领域。
背景技术:
2.人类社会极速发展,工业化程度越来越高,随之而来的是化石燃料大量使用带来的严峻的全球气候问题和环境污染问题。因此,寻找清洁高效的可再生能源替代传统化石能源成为当今社会的研究热点和难点。人工光合成模拟自然界光合作用的水分解过程把太阳能转化为化学能,是一种非常有应用前景的可再生能源利用技术。典型的水分解过程需要耦合两个半反应:(1)水氧化产生分子氧和还原电子;(2)质子还原为分子氢。其中,水氧化半反应需要4个电子和4个质子参与并经过一系列复杂的过程才能最终形成o-o键,需要很高的活化能,被认为是自然界光合作用和人工合成全分解水反应的瓶颈步骤。因此,开发高效、稳定、廉价丰产的水氧化催化剂是人工光合成突破的关键因素。在过去的几十年里,科学家们开发了一系列基于分子化合物、多金属氧酸盐、金属氧化物、简单的金属盐等等的催化剂用于产氧反应。
3.众所周知,在自然界中,水氧化过程发生在光系统ⅱ的产氧活性中心 mn4cao5上。该催化活性中心由一个mn3cao4立方烷片段和一个悬挂的mn原子组成。作为一种高效的催化剂,mn4cao5为科学家们开发分子水氧化催化剂提供了蓝图。科学家们花费了大量精力去模拟自然界产氧中心的结构和功能,开发了一系列的mn4ca-簇结构。但是迄今为止,只有极少数的mn-基簇结构能够在光敏剂和电子受体存在时进行光催化水氧化产氧。并且,天然产氧中心及mn4ca
-ꢀ
簇在水中稳定性较差,不利于用于机理研究和实际应用。因此,开发新型的立方烷结构水氧化催化剂具有重要理论意义和现实意义。
技术实现要素:
4.为了克服已有技术存在的不足,本发明目的是提供一种含co4o4准立方烷结构的钴基金属有机框架材料的制备方法,该金属有机框架材料能够作为助催化剂进行可见光照射下的水氧化反应和水还原反应。采用本发明所提供的制备方法得到的金属有机框架目标材料含有一维z-字形coo链结构,一维coo链中的co4o4准立方烷片段与天然光系统ⅱ的活性产氧中心mn4cao5具有相似结构,可作为催化活性位点,是一个潜在的产氧助催化剂。该金属有机框架目标材料不但在结构上模拟了天然产氧活性中心,实现了均相催化剂的多相化;而且在常用有机溶剂和水相中具有优秀的稳定性。以上性质使得其可以很好的催化目标光解水反应;更重要的是本发明提供的金属有机框架目标材料还具有制备方法简单,原料廉价等优点。
5.本发明根据目标光解水反应的需求,通过偶氮基团修饰来增强三苯胺衍生羧酸桥连配体的刚性和尺寸,与二价钴离子配位在金属有机框架中得到co4o4准立方烷簇结构。不
但在结构上模拟了天然产氧活性中心,实现了均相催化剂的多相化;同时偶氮三苯胺衍生羧酸配体的疏水性及其与co4o4簇组成的半开放空间在保证催化剂水稳定的同时增强水分子的进入和与催化活性位点的接触。多相光催化分解水产氧和产氢实验表明本发明涉及的金属有机框架材料在光催化水分解领域具有很好的应用前景。
6.为了实现上述目的,解决现有技术中存在的问题,本发明采取的技术方案是:一方面,本发明以l为桥连配体,以过渡金属盐tm中的co
2
作为节点,通过溶剂热合成方法制得含有co4o4准立方烷结构的偶氮三苯胺衍生羧酸基金属有机框架化合物tm-l,其合成路线如下:
7.l tm
→
tm-l
8.所述过渡金属盐tm选自co(no3)2·
6h2o或cocl2·
6h2o中的一种;
9.所述l选自分子式为c
40
h
28
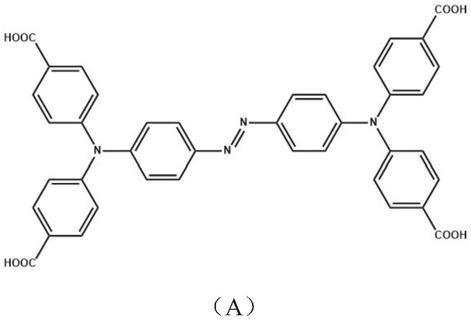
n4o8的偶氮连接的三苯胺羧酸衍生物中的 (e)-4,4',4”,4”'-(n,n,n',n'-四苯基-对二氨基偶氮苯)甲酸;英文名称:【(e)-4,4',4”,4”'-((diazene-1,2-diylbis(4,1-phenylene))bis(azanetriyl))tetrabenzoic acid】;其分子结构式如(a)表达式:
[0010][0011]
所述制备方法具体包括以下步骤:
[0012]
步骤一、将上述桥连配体l、过渡金属盐tm按照1:3.0~5.0的摩尔比加入到体积比为1:2~2.5的水和n,n-二甲基甲酰胺的混合溶剂中;
[0013]
步骤二、将步骤一制得的反应液置于程序控温烘箱中,温度控制在120~140 ℃,时间控制在60~90h,关闭烘箱,自然冷却至室温,有晶体析出,过滤,干燥,制得目标材料tm-l;
[0014]
或将步骤一制得的反应液置于茄型烧瓶中,磁力搅拌,于油浴加热下,温度控制在120~140℃,时间控制在18~48h,关闭加热,冷却至室温,有粉体析出,离心,干燥,制得微米尺寸目标材料tm-l。
[0015]
上述方式能够获得微米尺寸的主要原因是:第一,敞口体系,反应液压力为常压;第二,在搅拌的情况下,金属有机框架化合物偏向于形成小尺寸。
[0016]
另一方面,本发明将上述方法制备的钴基金属有机框架化合物用于在可见光照射下催化水氧化产氧或水还原制氢反应。
[0017]
优选地,所述水氧化产氧或水还原制氢反应中,所述金属有机框架化合物为助催化剂。
[0018]
优选地,所述水氧化产氧或水还原制氢反应中加有光敏剂;所述光敏剂为三联吡
啶二氯化钌[ru(bpy)3]cl2。
[0019]
优选地,所述可见光照射下的水氧化实验在顶照式光反应釜中进行,加入磷酸钠缓冲溶液、所述金属有机框架化合物、[ru(bpy)3]cl2和na2s2o8,所述磷酸钠缓冲溶液ph=7,将反应釜密封,并将反应釜内空气除尽后,进行反应,反应中在反应釜上方使用带有420nm滤光片的氙灯(300w)照射,在线采样检测氧气含量。
[0020]
优选地,所述可见光照射下的水还原实验在顶照式光反应釜中进行,加入 n,n
’-
二甲基甲酰胺和去离子水的混合溶剂、所述金属有机框架材料、 [ru(bpy)3]cl2和三乙醇胺,随后将反应釜密封,并抽真空确保反应釜内空气除尽。之后,在反应釜上方使用带有420nm滤光片的氙灯(300w)照射,在线采样检测氢气。
[0021]
再一方面,本发明提供了一种上述配体l的制备方法,所述制备方法如下:
[0022]
一、将氢化钠溶于n,n
’-
二甲基甲酰胺,得到混合液i,搅拌并通氩气以除去混合液中的氧气,搅拌条件下向所述混合液中加入4,4
’-
二氨基偶氮苯,并继续室温搅拌1-2小时,得到混合液ii;
[0023]
二、向混合液ii中加入4-氟苯腈,升温至150℃,在惰性气体氛围下进行反应5小时,得到混合液iii;
[0024]
三、将混合液iii倾倒入去离子水中,有橙色沉淀析出,过滤,乙醇重结晶,得到固体粉末;
[0025]
四、取所述固体粉末加入乙醇和氢氧化钾水溶液的混合溶液中,100℃回流 5天,旋蒸除去乙醇,用稀盐酸调ph至3,有棕色沉淀析出,过滤干燥,冰乙酸重结晶得深黄色粉末,所述深黄色粉末为配体l。
[0026]
有益效果
[0027]
1、以本技术所制备的金属有机框架材料为助催化剂,在光敏剂和电子牺牲剂(即电子受体或电子供体,电子受体如na2s2o8,电子供体如三乙醇胺)的存在下进行可见光催化水氧化产氧或水还原产氢反应,其中,偶氮基团修饰的三苯胺衍生羧酸配体的疏水性及结构中的大量钴-氧配位键增强了所得金属有机框架材料的水稳定性;金属有机框架中一维coo链结构提高了载流子的迁移和co4o4准立方烷催化中心的均匀分散协同提高了助催化剂的产氧性能。
[0028]
2、与已有技术相比,本发明涉及的助催化剂合成方法简单,易操作,助催化剂以及制备助催化剂的原料价格低廉,产率高,得到的功能材料化学性质稳定,易于大面积推广应用。
[0029]
3、本发明所提供的化合物通过偶氮基团修饰来增强三苯胺衍生羧酸桥连配体的刚性和尺寸,与二价钴离子配位在金属有机框架中得到co4o4立方烷片段作为催化活性位点,实现可见光照射下的水氧化和水还原分别产生氧气和氢气;一维coo链周围的空间增加了水分子的进入及与催化活性位点的接触,实现了均相催化剂的多相化,所得助催化剂在光催化分解水领域具有很好的应用前景。
附图说明
[0030]
图1是实施例1的材料co-l的结构示意图(图中a是co-l最小不对称单元的热椭球图;图中b是co(ⅱ)离子的配位环境图;图中c是co-l中所含一维 coo链结构)。
[0031]
图2是实施例1的材料co-l中co4o4准立方烷簇的结构示意图(图中a为 co4o4准立方烷结构及co-o键键长;图中b为co4o4准立方烷周围的配位环境)。
[0032]
图3是实施例1的材料co-l的结构示意图(图中a是co-l中配体l与二价钴离子的配位图;图中b是从c轴观察co-l的三维结构,其中粗实线为相邻一维coo链的连线;图中c是从b轴观察co-l的三维堆积结构,其中含有l的三维堆积结构,其中含有的一维孔道)。
[0033]
图4是实施例1的材料co-l中钴离子与周围配体组成的碗形空腔结构。
[0034]
图5是实施例1的材料co-l的红外光谱图(图中a为桥连配体l的红外光谱图;图中b为金属有机框架材料co-l的红外光谱图;图中c为光催化水还原反应后回收测试的红外光谱图;图中d为光催化水氧化反应后回收测试的红外光谱图)。
[0035]
图6是实施例1材料co-l稳定性实验的粉末x-射线衍射(pxrd)图(图中a为模拟图;图中b为实施例1合成;图中c为浸泡于乙醇中十天后测试所得图;图中d为浸泡于甲醇中十天后测试所得图;图中e为浸泡于去离子水中十天后测试所得图)。
[0036]
图7是实施例1的材料co-l的热分析图。
[0037]
图8是实施例5的循环伏安曲线图(图中a为空白样品;图中b为[ru(bpy)3]cl2的循环伏安曲线;图中c为金属有机框架材料co-l的循环伏安曲线图)。
[0038]
图9是实施例3的微米尺寸材料co-l的扫描电镜图(图中a为比例尺 200.0μm的图;图中b为比例尺50.0μm的图;图中c为比例尺10.0μm的图)。
[0039]
图10是实施例6的水氧化反应动力学时间跟踪曲线图。
[0040]
图11是实施例6-7、对比例3-5的水氧化反应的对照实验图。
[0041]
图12是实施例8、对比例6的还原水产氢动力学时间跟踪曲线图(图中曲线a是没有加入目标材料co-l的时间跟踪曲线图;图中曲线b是加入目标材料 co-l的时间跟踪曲线图)。
[0042]
图13是实施例6和实施例8反应后的目标材料co-l回收后的粉末x-射线衍射(xrd)图(图中a为模拟;图中b为合成;图中c中为实施例6水氧化反应后的xrd;图中d中为实施例8水还原产氢反应后的xrd)。
具体实施方式
[0043]
为了进一步说明本发明,列举以下实施实例并结合附图对本发明进行详细说明,但它并不限制各附加权利要求所定义的发明范围。(e)-4,4',4”,4”'-(n,n,n',n'
-ꢀ
四苯基-对二氨基偶氮苯)甲酸通过实施例9制备,实施例中所用其它原料均为可以通过市购获得的常规产品。
[0044]
实施例1
[0045]
将偶氮连接的三苯胺羧酸衍生物中的(e)-4,4',4”,4”'-(n,n,n',n'-四苯基-对二氨基偶氮苯)甲酸c
40
h
28
n4o8(17.3mg,0.025mmol),cocl2·
6h2o(24.0mg, 0.10mmol)溶于n,n
’-
二甲基甲酰胺(4ml)和去离子水(2ml)中搅拌均匀后,取将此溶液置于烘箱中,130℃烧制72h,关闭烘箱,冷却至室温,红褐色透明钻石状晶体产生,过滤,干燥,制得目标材料co-l,产率约56%。红外光谱峰位置 (ir):3426(w),2930(w),1680(s),1647(vs),1592(vs),1500(m),1392(vs),1315 (vs),1264(m),1179(m),1147(s),1101(w),843(m),786(s),701(w),560(m),521 (w),472(w)cm-1
。得到的目标材料结构如图1、图2、图3和图4所示。
图1-4 给出了目标材料的具体结构,从图1中a中可知,在目标材料的最小不对称单元中含有一个配体分子、三个钴原子、两个配位的氢氧根、一个配位水分子和一个配位n,n
’-
二甲基甲酰胺分子;图1中b给出了三个钴原子的配位环境,可知在钴原子周围有六个配体上的苯甲酸片段和两个配位n,n
’-
二甲基甲酰胺分子;图1中c为目标材料中的coo链结构,其中以三个钴和四个氧构筑的co3o4为重复单元,首位相连构成z-字型链。图2为目标材料中的co4o4准立方烷结构及其配位环境图,从图中可知,该准立方烷结构与产氧活性中心mn4cao5结构类似。从图3可知目标材料co-l为三维结构,其中相邻coo链之间的距离在且coo链周围有较小的一维孔道从图4中可知,在co4o4准立方烷附近存在有配体片段组成的碗形空腔,这一方面有利于水分子的进入,另一方面得益于配体片段的疏水保护,可以阻止大量水分子的侵入而破坏目标材料结构,使目标材料在纯水中保持一定的稳定能力。目标材料的红外光谱分析图如图5中b所示。粉末x-射线衍射(pxrd)图如图6中b所示。热分析图如图 7所示。由图7可知,所制备的目标材料在进行热分析时,分两步失重,第一步为目标材料中的溶剂分子的脱除,第二步为配体的分解。由此可知该目标材料具有良好的热稳定性能,在300℃以下可稳定存在。
[0046]
实施例2
[0047]
将偶氮连接的三苯胺羧酸衍生物中的(e)-4,4',4”,4”'-(n,n,n',n'-四苯基-对二氨基偶氮苯)甲酸c
40
h
28
n4o8(17.3mg,0.025mmol),co(no3)2·
6h2o(30.0mg, 0.10mmol)溶于n,n
’-
二甲基甲酰胺(4ml)和去离子水(2ml)中搅拌均匀后,取将此溶液置于烘箱中,130℃烧制72h,关闭烘箱,冷却至室温,红褐色透明钻石状晶体产生,过滤,干燥,制得目标材料co-l,产率约49%。
[0048]
实施例3
[0049]
将偶氮连接的三苯胺羧酸衍生物中的(e)-4,4',4”,4”'-(n,n,n',n'-四苯基-对二氨基偶氮苯)甲酸c
40
h
28
n4o8(34.6mg,0.05mmol),cocl2·
6h2o(48.0mg, 0.20mmol)溶于n,n
’-
二甲基甲酰胺(80ml)和去离子水(40ml)中搅拌均匀后,取将此溶液置于250ml的茄型烧瓶中,130℃回流加热24h,停止加热并冷却至室温,过滤,干燥得到橙色粉末,制得微米尺寸的目标材料co-l。得到的微米尺寸目标材料的扫面电镜图(sem)如图9所示。
[0050]
实施例4
[0051]
金属有机框架材料co-l的稳定性测试:分别取20mg实施例1所制备的金属有机框架材料co-l浸泡于纯水、甲醇和乙醇中,十天之后离心干燥,并对处理后的样品进行粉末x-射线衍射(pxrd)测试。测试结果如图6中c、d和e所示。从图6中可以看出,目标材料co-l在浸泡纯水、甲醇和乙醇中十天之后的 xrd谱图与模拟所得及合成所得谱图一致,出峰位置并未发生改变,表明目标材料依然保持结构完整。
[0052]
实施例5
[0053]
金属有机框架材料co-l的循环伏安电化学测试:将实施例3制备的微米尺寸的粉体目标材料2mg分散于1.0ml的无水乙醇中,随后滴涂在除去氧化层的玻碳电极上,在室温下自然风干备用。随后在ph为7.0,浓度为0.1m的磷酸钠缓冲溶液中测试循环伏安曲线,对电极为铂丝电极,参比电极为ag/agcl电极。电压扫描范围为0-1.6v,扫面速率为100mv
·
s-1
。所得的循环伏安曲线图如图8 所示。
[0054]
对比例1-2
[0055]
在实施例5的测试条件下,分别进行了的空白对照实验和玻碳电极在2mm [ru(bpy)3]cl2溶液中的试验。所得的循环伏安曲线图如图8所示。从图8中可知,空白对照实验并未有氧化还原峰,目标材料的起始电位约在0.7v(ag/agcl参比), [ru(bpy)3]cl2的氧化还原电位在1.1v(ag/agcl参比),可知[ru(bpy)3]cl2的氧化能力较目标材料强,可以氧化目标材料中的co(ii)为co(iii)从而进行水氧化反应。
[0056]
实施例6
[0057]
金属有机框架材料co-l可见光水氧化的动力学时间跟踪实验:在 400mlpyrex的顶照式光反应釜中加入50ml磷酸钠缓冲溶液(0.1m,ph=7),实施例3制备的微米尺寸的金属有机框架材料co-l(10mg),[ru(bpy)3]cl2(77mg,2 mm)和na2s2o8(120mg,10mm)。随后将反应釜密封,并抽真空15分钟确保反应釜内空气除尽。之后,在反应釜上方使用带有420nm滤光片的氙灯(300w)照射。每隔5分钟进行采样检测一次氧气含量。共照射20分钟,采样检测四次。实验结果如图10所示。在反应结束之后,对所回收样品分别进行红外光谱和粉末x-射线衍射测试,测试结果分别如图5中c和图13中c所示。通过对比图5 和图13的谱图可知,目标材料经光催化反应后结构依然保持,并未发生破坏。
[0058]
实施例7
[0059]
参考上述实施例6的光催化反应过程。与上述实施例6不同的是,本实施例将实施例3制备的微米尺寸的金属有机框架材料co-l替换为实施例1所制备大块状金属有机框架材料co-l(10mg),用于水氧化反应;操作过程与实施例6一样。实验结果如图11所示。
[0060]
对比例3-5
[0061]
金属有机框架材料co-l可见光水氧化反应的对照实验:参考上述实施例6 的光催化反应过程。与上述实施例6不同的是,本对比例分别测试了:(1)不含有金属有机框架材料co-l的水氧化反应;(2)不含有[ru(bpy)3]cl2的水氧化反应;(3)将金属有机框架材料co-l替换为桥连配体l(8mg)的水氧化反应。除反应时间为15分钟外,操作过程与实施例6一样。实验结果如图11所示。图11 给出了微米尺寸、大块状、不含co-l、不含[ru(bpy)3]cl2和桥连配体l的可见光水氧化反应的产氧量。从图11中不同实验的产氧量对比可知,在该反应体系中,目标材料作为助催化剂不可缺少,且当目标材料的尺寸为微米尺寸时,其催化能力得以最大发挥。
[0062]
实施例8
[0063]
金属有机框架材料co-l可见光还原水产氢的动力学时间跟踪实验:在 400mlpyrex的顶照式光反应釜中加入48ml的n,n
’-
二甲基甲酰胺(32ml)和去离子水(16ml)的混合溶剂,金属有机框架材料co-l(24mg),[ru(bpy)3]cl2(24mg) 和三乙醇胺(5ml)。随后将反应釜密封,并抽真空15分钟确保反应釜内空气除尽。之后,在反应釜上方使用带有420nm滤光片的氙灯(300w)照射。之后分别在光照后第1、2、4、6、8、10小时时采样检测氢气。测试结果如图12中b 所示。在反应结束之后,对所回收样品分别进行红外光谱和粉末x-射线衍射测试,测试结果分别如图5中d和图13中d所示。通过对比图5和图13的谱图可知,目标材料经光催化反应后结构依然保持,并未发生破坏。
[0064]
对比例6
[0065]
不加入金属有机框架材料co-l的可见光还原水产氢的动力学时间跟踪实验:在
400mlpyrex的顶照式光反应釜中加入48ml的n,n
’-
二甲基甲酰胺(32ml) 和去离子水(16ml)的混合溶剂,[ru(bpy)3]cl2(24mg)和三乙醇胺(5ml)。随后将反应釜密封,并抽真空15分钟确保反应釜内空气除尽。之后,在反应釜上方使用带有420nm滤光片的氙灯(300w)照射。之后分别在光照后第1、2、4、6、 8、10小时时采样检测氢气。测试结果如图12中a所示。图12给出了含有目标材料和不含目标材料的可见光水还原产氢动力学时间跟踪曲线,从图中知,当不含有目标材料co-l时,催化体系10个小时的产氢总量为13.8μmol,并且在光照8小时后产氢量几乎不再增长;而当加入目标材料co-l时,10个小时的产氢总量为88.3μmol,产氢量提高了6倍多,且产氢量还有一定增长。这说明目标材料的加入提高了体系的催化产氢能力,是一个良好的产氢助催化剂。
[0066]
实施例9
[0067]
具体实施方式的实施例中所用到的配体l即偶氮连接的三苯胺羧酸衍生物中的(e)-4,4',4”,4”'-(n,n,n',n'-四苯基-对二氨基偶氮苯)甲酸c
40
h
28
n4o8均通过以下方式提供:
[0068]
向250ml三口烧瓶中加入氢化钠1.18g和n,n
’-
二甲基甲酰胺80ml,磁力搅拌并通氩气30分钟以除去混合液中的氧气。之后在搅拌下向上述混合液中加入4,4
’-
二氨基偶氮苯2.13g,并继续室温搅拌1-2小时。然后向其中加入4-氟苯腈5.08g,升温至150℃,并保持该温度下反应5小时。该反应过程在惰性气体 (氩气)氛围下进行。待反应结束,将上述反应液倾倒入400ml去离子水中,有橙色沉淀析出,过滤,乙醇重结晶。取上述重结晶所得固体粉末3.5g加入到 250ml圆底烧瓶中,之后加入100ml乙醇和90ml氢氧化钾水溶液(含有9.0g 氢氧化钾)。100℃回流5天,旋蒸除去乙醇,用1mol/l稀盐酸调ph约到3,有棕色沉淀析出,过滤干燥,冰乙酸重结晶得深黄色粉末2.63g,即为配体l。
[0069]
配体l的核磁数据如下:1h nmr(400mhz,dmso-d6)δ=12.81(br s,acid), 7.94-7.92(d,8h),7.89-7.86(d,4h),7.26-7.24(d,4h),7.19-7.17(d,8h);
13
c nmr (126mhz,dmso-d6)δ=167.2,150.3,148.9,148.8,131.6,126.3,125.4,124.7, 124.0。
[0070]
实施例10-15
[0071]
除下面表格中的条件外,其他制备条件与实施例1相同。
[0072][0073]
实施例16-21
[0074]
微米尺寸的目标材料co-l的制备,除下面表格中条件外,其他制备条件与实施例3
相同。
[0075]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。