1.本发明涉及一种吸水性树脂颗粒、吸收体、吸收性物品、吸水性树脂颗粒的通液维持率的测定方法及吸水性树脂颗粒的制造方法。
背景技术:
2.吸水性树脂用于卫生用品的领域。具体而言,用作尿布等吸收性物品中包含的吸收体的材料(例如专利文献1)。现有技术文献专利文献
3.专利文献1:日本特表2014
‑
515987号公报
技术实现要素:
本发明要解决的技术问题
4.尿布等吸收性物品具备吸收及保持尿液、经血等体液的吸收体。吸收体一般包含吸水性树脂颗粒及纸浆等纤维状物,纤维状物彼此或纤维状物与吸水性树脂颗粒相互缠绕。近年来,吸收性物品趋于薄型化,因此趋于优选吸收体中的纤维状物的比例低的吸收性物品。当吸收体中的纤维状物的比例低时,例如因穿戴吸收性物品的过程中的体重的载重等,向吸液后的吸收体施加切断力时,有时会发生吸收体在内部断裂等吸收体的变形。发生断裂的吸收体无法充分发挥吸收性能。
5.本发明的目的在于,提供一种能够抑制吸收体在吸液后发生断裂的吸水性树脂颗粒。解决技术问题的技术手段
6.对于本发明的吸水性树脂颗粒,利用通过依次包括下述(a)、(b)及(c)工序的方法测定的干粉通液速度、以及通过依次包括下述(a)、(b)、(d)及(e)工序的方法测定的溶胀凝胶通液速度,并由下述式表示的通液维持率为10%以上。通液维持率(%)=(溶胀凝胶通液速度/干粉通液速度)
×
100(a)在具备网格状底部的内径为26mm的圆筒状容器内均匀地散布吸水性树脂颗粒0.3g。(b)通过将具备网格状底部的外径为25mm、内径为19mm的圆筒状小型容器插入到上述圆筒状容器内来形成测定部。(c)将生理盐水以60ml/分钟的恒定速度投入到上述测定部的上述圆筒状小型容器内,测量从开始投入起1分钟内从上述测定部的上述底部流出的生理盐水的量,由此求出生理盐水的干粉通液速度(g/分钟)。(d)通过在容器内的40g的生理盐水中浸渍上述测定部的上述底部侧30分钟而使上述测定部内的上述吸水性树脂颗粒溶胀。
(e)向上述测定部的上述圆筒状小型容器内一次性投入20g的生理盐水,测量从开始投入起1分钟内从上述测定部的上述底部流出的生理盐水的量,由此求出生理盐水的溶胀凝胶通液速度(g/分钟)。
7.上述吸水性树脂颗粒的溶胀凝胶通液速度优选为2.5g/分钟以上。通过使吸水性树脂颗粒的溶胀凝胶通液速度在上述范围内,能够进一步抑制吸收体在吸液后发生断裂。
8.本发明还提供一种含有上述吸水性树脂颗粒的吸收体。通过使吸收体含有上述吸水性树脂颗粒,能够抑制发生断裂。
9.本发明还提供一种具备上述吸收体的吸收性物品。
10.上述吸收性物品可以是尿布。
11.本发明还提供一种吸水性树脂颗粒的通液维持率的测定方法,该吸水性树脂颗粒的通液维持率由下述式表示,该测定方法包括:通过依次包括下述(a)、(b)及(c)工序的方法测定干粉通液速度的步骤;及通过依次包括下述(a)、(b)、(d)及(e)工序的方法测定溶胀凝胶通液速度的步骤。通液维持率(%)=(溶胀凝胶通液速度/干粉通液速度)
×
100
12.(a)在具备网格状底部的内径为26mm的圆筒状容器内均匀地散布吸水性树脂颗粒0.3g。(b)通过将具备网格状底部的外径为25mm、内径为19mm的圆筒状小型容器插入到上述圆筒状容器内来形成测定部。(c)将生理盐水以60ml/分钟的恒定速度投入到上述测定部的上述圆筒状小型容器内,测量从开始投入起1分钟内从上述测定部的上述底部流出的生理盐水的量,由此求出生理盐水的干粉通液速度(g/分钟)。(d)通过在容器内的40g的生理盐水中浸渍上述测定部的上述底部侧30分钟而使上述测定部内的上述吸水性树脂颗粒溶胀。(e)向上述测定部的上述圆筒状小型容器内一次性投入20g的生理盐水,测量从开始投入起1分钟内从上述测定部的上述底部流出的生理盐水的量,由此求出生理盐水的溶胀凝胶通液速度(g/分钟)。通过上述测定方法,能够对与吸收体的耐断裂性有关的吸水性树脂颗粒的扩散性进行评价。
13.本发明还提供一种吸水性树脂颗粒的制造方法,其包括分选通过上述方法测定的通液维持率为10%以上的吸水性树脂颗粒的步骤。通过该制造方法,能够获得液体的扩散性优异,并且用于吸收体时可抑制吸液后发生断裂的吸水性树脂颗粒。
14.本发明还提供一种抑制含有上述吸水性树脂颗粒的吸收体在吸液后发生断裂的方法,该方法包括增加通过上述方法测定的吸水性树脂颗粒的通液维持率的步骤。发明效果
15.根据本发明,可获得能够抑制吸收体在吸液后发生断裂的吸水性树脂颗粒。
附图说明
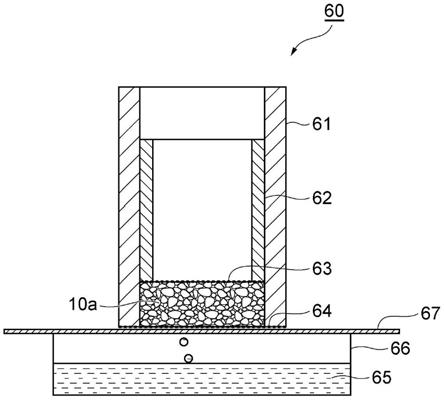
16.图1是表示吸收性物品的一个实例的剖视图。图2是表示实施例中所使用的搅拌叶片(在平板部具有狭缝的平叶片)的概况的俯
视图。图3是表示通液维持率的测定方法的示意剖视图。图4是表示无加压dw的测定方法的示意图。图5是表示吸收体断裂时间的测定方法的示意剖视图。
具体实施方式
17.以下,对本发明的适宜的实施方式进行详细说明。然而,本发明并不限定于以下的实施方式。
18.在本说明书中,将“丙烯酸”及“甲基丙烯酸”统一表述为“(甲基)丙烯酸”。“丙烯酸酯”及“甲基丙烯酸酯”也同样表述为“(甲基)丙烯酸酯”。“(聚)”是指具有前缀“聚”及不具有前缀“聚”的这两种情况。在本说明书中区间性记载的数值范围内,某区间的数值范围的上限值或下限值能够与其他区间的数值范围的上限值或下限值任意组合。在本说明书中所记载的数值范围内,可将该数值范围的上限值或下限值替换为实施例中所示的值。“水溶性”是指在25℃下在水中显示5质量%以上的溶解性。本说明书中所例示的材料可以单独使用,也可以组合使用2种以上。关于组合物中的各成分的含量,在组合物中存在多种对应于各成分的物质的情况下,只要没有特别说明,则是指存在于组合物中的该多种物质的总量。在本说明书中,只要无特别说明,则生理盐水是指浓度为0.9质量%的nacl水溶液。
19.本实施方式的吸水性树脂颗粒的由下述式表示的通液维持率为10%以上。本技术的发明人发现:相对于干燥状态下的吸水性树脂颗粒的通液速度即干粉通液速度,吸液后的溶胀状态下的吸水性树脂颗粒的通液速度即溶胀凝胶通液速度的比例越高,则越可抑制吸收体在吸液后发生断裂。通液维持率(%)=(溶胀凝胶通液速度/干粉通液速度)
×
100
20.本技术的发明人推测,若本发明的吸水性树脂颗粒的通液维持率为10%以上,则液体能够在吸收体内部更均匀地扩散。在液体未均匀地扩散于吸收体的内部的情况下,非常不易断裂的部分(几乎不吸收液体的部分)和非常容易断裂的部分(吸收了大量液体的部分)分散存在。若在穿戴吸收性物品的过程中,体重等载重施加到吸收体,则吸收体容易从该非常容易断裂的部分断裂。关于本发明的吸水性树脂颗粒,认为液体能够在吸收体内部均匀地扩散,不易产生非常容易断裂的部分,其结果,认为可抑制吸收体发生断裂。
21.关于干粉通液速度,通过依次包括下述(a)、(b)及(c)工序的方法来测定。关于溶胀凝胶通液速度,通过依次包括下述(a)、(b)、(d)及(e)工序的方法来测定。更具体的测定方法示于后述实施例中。(a)在具备网格状底部的内径为26mm的圆筒状容器内均匀地散布吸水性树脂颗粒0.3g。(b)通过将具备网格状底部的外径为25mm、内径为19mm的圆筒状小型容器插入到上述圆筒状容器内来形成测定部。(c)将生理盐水以60ml/分钟的恒定速度投入到上述测定部的上述圆筒状小型容器内,测量从开始投入起1分钟内从上述测定部的上述底部流出的生理盐水的量,由此求出生理盐水的干粉通液速度(g/分钟)。(d)通过在容器内的40g的生理盐水中浸渍上述测定部的上述底部侧30分钟而使
上述测定部内的上述吸水性树脂颗粒溶胀。(e)在上述测定部的上述圆筒状小型容器内一次性投入20g的生理盐水,测量从开始投入起1分钟内从上述测定部的上述底部流出的生理盐水的量,由此求出生理盐水的溶胀凝胶通液速度(g/分钟)。
22.本实施方式的吸水性树脂颗粒的通液维持率优选为12%以上,更优选为15%以上、20%以上、25%以上或30%以上,进一步优选为35%以上或40%以上。本实施方式的吸水性树脂颗粒的通液维持率例如可以是80%以下、70%以下、60%以下、58%以下、55%以下或50%以下。
23.本实施方式的吸水性树脂颗粒的通过上述方法测定的干粉通液速度例如可以是2g/分钟以上、5g/分钟以上、8g/分钟以上、10g/分钟以上、15g/分钟以上、18g/分钟以上或20g/分钟以上。吸水性树脂颗粒的干粉通液速度例如可以是50g/分钟以下、40g/分钟以下、35g/分钟以下、33g/分钟以下或30g/分钟以下。
24.本实施方式的吸水性树脂颗粒的通过上述方法测定的溶胀凝胶通液速度例如可以是0.7g/分钟以上、1.0g/分钟以上、1.5g/分钟以上、2.0g/分钟以上、2.5g/分钟以上、3.0g/分钟以上、4.0g/分钟以上、5.0g/分钟以上、6.0g/分钟以上或8.0g/分钟以上。若溶胀凝胶通液速度在上述范围内,则趋于能够进一步抑制吸收体在吸液后发生断裂。吸水性树脂颗粒的溶胀凝胶通液速度例如可以是30g/分钟以下、25g/分钟以下、20g/分钟以下、18g/分钟以下或15g/分钟以下。从这些观点考虑,溶胀凝胶通液速度优选为1~30g/分钟、3~25g/分钟或5~20g/分钟。
25.本实施方式的吸水性树脂颗粒能够具有对生理盐水的高吸水能力。吸水性树脂颗粒的生理盐水保水量例如可以是20g/g以上、23g/g以上、25g/g以上、28g/g以上、30g/g以上或32g/g以上,也可以是60g/g以下、55g/g以下、53g/g以下、50g/g以下、48g/g以下、45g/g以下、43g/g以下或40g/g以下。吸水性树脂颗粒的生理盐水保水量例如可以是20~60g/g、23~55g/g、25~50g/g、28~45g/g或30~40g/g。生理盐水保水量通过后述实施例中所记载的方法测定。
26.本实施方式的吸水性树脂颗粒的无加压dw3分钟值例如可以是14ml/g以上,也可以是16ml/g以上、18ml/g以上、20ml/g以上或25ml/g以上。无加压dw3分钟值例如可以是55ml/g以下、50ml/g以下、40ml/g以下。无加压dw3分钟值通过后述的实施例中所记载的方法来测定。
27.作为吸水性树脂颗粒的形状,可举出大致球形、破碎状、颗粒状等。吸水性树脂颗粒的中值粒径可以是250~850μm、300~700μm或300~600μm。本实施方式的吸水性树脂颗粒可以在通过后述的制造方法获得聚合物颗粒时就具有所需的粒度分布,但也可以通过进行使用了基于筛的分级的粒度调节等操作来调节粒度分布。
28.本实施方式的吸水性树脂颗粒例如能够包含交联聚合物,该交联聚合物通过包含烯属不饱和单体的单体的聚合而形成。交联聚合物具有源自烯属不饱和单体的单体单元。即,本实施方式的吸水性树脂颗粒能够具有源自烯属不饱和单体的结构单元。
29.作为使上述单体聚合的方法,可举出反相悬浮聚合法、水溶液聚合法、本体聚合法、沉淀聚合法等。其中,从容易确保所获得的吸水性树脂颗粒的良好的吸水特性及容易控制聚合反应的观点考虑,优选反相悬浮聚合法或水溶液聚合法。以下,作为使烯属不饱和单
体聚合的方法,以反相悬浮聚合法为例进行说明。
30.烯属不饱和单体优选为水溶性,例如可举出(甲基)丙烯酸及其盐、2
‑
(甲基)丙烯酰胺
‑2‑
甲基丙磺酸及其盐、(甲基)丙烯酰胺、n,n
‑
二甲基(甲基)丙烯酰胺、2
‑
羟乙基(甲基)丙烯酸酯、n
‑
羟甲基(甲基)丙烯酰胺、聚乙二醇单(甲基)丙烯酸酯、n,n
‑
二乙基氨基乙基(甲基)丙烯酸酯、n,n
‑
二乙基氨基丙基(甲基)丙烯酸酯、二乙基氨基丙基(甲基)丙烯酰胺等。当烯属不饱和单体含有氨基时,该氨基可以被季铵化。上述单体所具有的羧基及氨基等官能团能够在后述的表面交联工序中作为能够进行交联的官能团而发挥作用。这些烯属不饱和单体可以单独使用,也可以组合使用2种以上。
31.其中,从工业上容易获得的观点考虑,烯属不饱和单体优选包含选自由(甲基)丙烯酸及其盐、丙烯酰胺、甲基丙烯酰胺以及n,n
‑
二甲基丙烯酰胺组成的组中的至少一种化合物,更优选包含选自由(甲基)丙烯酸及其盐以及丙烯酰胺组成的组中的至少一种化合物。从进一步提高吸水特性的观点考虑,烯属不饱和单体进一步优选包含选自由(甲基)丙烯酸及其盐组成的组中的至少一种化合物。
32.作为单体,可以使用一部分除了上述烯属不饱和单体以外的单体。这样的单体例如能够混合于包含上述烯属不饱和单体的水溶液中使用。相对于单体总量(用于获得吸水性树脂颗粒的单体总量;例如,提供交联聚合物的结构单元的单体的总量;以下相同),烯属不饱和单体的使用量可以是70~100摩尔%,也可以是80~100摩尔%、90~100摩尔%、95~100摩尔%或100摩尔%。其中,相对于单体总量,(甲基)丙烯酸及其盐可以是70~100摩尔%,也可以是80~100摩尔%、90~100摩尔%、95~100摩尔%或100摩尔%。“(甲基)丙烯酸及其盐的比例”是指(甲基)丙烯酸及其盐的总量的比例。
33.根据本实施方式,作为吸水性树脂颗粒的一个实例,能够提供如下吸水性树脂颗粒,其包含具有源自烯属不饱和单体的结构单元的交联聚合物,该吸水性树脂颗粒中,烯属不饱和单体包含选自由(甲基)丙烯酸及其盐组成的组中的至少一种化合物,相对于用于获得吸水性树脂颗粒的单体总量,(甲基)丙烯酸及其盐的比例为70~100摩尔%。
34.通常,烯属不饱和单体适宜制成水溶液使用。通常,包含烯属不饱和单体的水溶液(以下,称为单体水溶液)中的烯属不饱和单体的浓度为20质量%以上且饱和浓度以下即可,优选为25~70质量%,更优选为30~55质量%。关于所使用的水,例如可举出自来水、蒸馏水、离子交换水等。
35.当所使用的烯属不饱和单体包含酸基时,可以用碱性中和剂中和单体水溶液的酸基而使用该单体水溶液。从提高所获得的吸水性树脂颗粒的渗透压,并进一步提高保水量等吸水特性的观点考虑,烯属不饱和单体的基于碱性中和剂的中和度为烯属不饱和单体中的酸性基团的10~100摩尔%,优选为50~90摩尔%,更优选为60~80摩尔%。作为碱性中和剂,例如可举出:氢氧化钠、碳酸钠、碳酸氢钠、氢氧化钾、碳酸钾等碱金属盐;氨等。为了简化中和操作,可以将这些碱性中和剂制成水溶液的状态来使用。上述碱性中和剂可以单独使用,也可以组合使用2种以上。例如能够通过将氢氧化钠、氢氧化钾等的水溶液滴加到上述单体水溶液中并进行混合,从而进行烯属不饱和单体的酸基的中和。
36.在反相悬浮聚合法中,在表面活性剂的存在下将单体水溶液分散于烃分散介质中,并利用自由基聚合引发剂等而进行烯属不饱和单体的聚合。
37.作为表面活性剂,例如可举出非离子表面活性剂及阴离子表面活性剂。作为非离
子表面活性剂,例如可举出山梨糖醇酐脂肪酸酯、聚甘油脂肪酸酯、蔗糖脂肪酸酯、聚氧乙烯山梨糖醇酐脂肪酸酯、聚氧乙烯甘油脂肪酸酯、山梨糖醇脂肪酸酯、聚氧乙烯山梨糖醇脂肪酸酯、聚氧乙烯烷基醚、聚氧乙烯烷基苯基醚、聚氧乙烯蓖麻油、聚氧乙烯氢化蓖麻油、烷基烯丙基甲醛缩合聚氧乙烯醚、聚氧乙烯聚氧丙烯嵌段共聚物、聚氧乙烯聚氧丙基烷基醚及聚乙二醇脂肪酸酯等。作为阴离子表面活性剂,例如可举出脂肪酸盐、烷基苯磺酸盐、烷基甲基牛磺酸盐、聚氧乙烯烷基苯基醚硫酸酯盐、聚氧乙烯烷基醚磺酸盐、聚氧乙烯烷基醚的磷酸酯及聚氧乙烯烷基烯丙基醚的磷酸酯等。其中,从w/o型反相悬浮的状态良好、容易以适宜的粒径获得吸水性树脂颗粒、工业上容易获得的观点考虑,表面活性剂优选包含选自由山梨糖醇酐脂肪酸酯、聚甘油脂肪酸酯及蔗糖脂肪酸酯组成的组中的至少一种化合物。此外,从提高所获得的吸水性树脂颗粒的吸水特性的观点考虑,表面活性剂更优选包含蔗糖脂肪酸酯。这些表面活性剂可以单独使用,也可以组合使用2种以上。
38.从充分获得相对于使用量的效果且节省费用的观点考虑,相对于烯属不饱和单体水溶液100质量份,表面活性剂的量优选为0.05~10质量份,更优选为0.08~5质量份,进一步优选为0.1~3质量份。
39.并且,可以与上述的表面活性剂一同使用高分子类分散剂。作为高分子类分散剂,例如可举出马来酸酐改性聚乙烯、马来酸酐改性聚丙烯、马来酸酐改性乙烯
‑
丙烯共聚物、马来酸酐改性epdm(乙烯
‑
丙烯
‑
二烯三元共聚物)、马来酸酐改性聚丁二烯、马来酸酐
‑
乙烯共聚物、马来酸酐
‑
丙烯共聚物、马来酸酐
‑
乙烯
‑
丙烯共聚物、马来酸酐
‑
丁二烯共聚物、聚乙烯、聚丙烯、乙烯
‑
丙烯共聚物、氧化型聚乙烯、氧化型聚丙烯、氧化型乙烯
‑
丙烯共聚物、乙烯
‑
丙烯酸共聚物、乙基纤维素、乙基羟乙基纤维素等。在这些高分子类分散剂中,尤其从单体的分散稳定性方面考虑,优选使用马来酸酐改性聚乙烯、马来酸酐改性聚丙烯、马来酸酐改性乙烯
‑
丙烯共聚物、马来酸酐
‑
乙烯共聚物、马来酸酐
‑
丙烯共聚物、马来酸酐
‑
乙烯
‑
丙烯共聚物、聚乙烯、聚丙烯、乙烯
‑
丙烯共聚物、氧化型聚乙烯、氧化型聚丙烯、氧化型乙烯
‑
丙烯共聚物。这些高分子类分散剂可以单独使用,也可以组合2种以上。
40.从充分获得相对于使用量的效果且节省费用的观点考虑,相对于烯属不饱和单体水溶液100质量份,高分子类分散剂的量优选为0.05~10质量份,更优选为0.08~5质量份,进一步优选为0.1~3质量份。
41.自由基聚合引发剂优选为水溶性,例如可举出:过硫酸钾、过硫酸铵、过硫酸钠等过硫酸盐;过氧化甲乙酮、4
‑
甲基
‑2‑
戊酮过氧化物、过氧化二叔丁基、过氧化叔丁基异丙苯、过氧化乙酸叔丁酯、过氧化异丁酸叔丁酯、过氧化新戊酸叔丁酯及过氧化氢等过氧化物;2,2
’‑
偶氮双(2
‑
脒基丙烷)二盐酸盐、2,2
’‑
偶氮双[2
‑
(n
‑
苯基脒基)丙烷]二盐酸盐、2,2
’‑
偶氮双[2
‑
(n
‑
烯丙基脒基)丙烷]二盐酸盐、2,2
’‑
偶氮双[2
‑
(2
‑
咪唑啉
‑2‑
基)丙烷]二盐酸盐、2,2
’‑
偶氮双{2
‑
[1
‑
(2
‑
羟基乙基)
‑2‑
咪唑啉
‑2‑
基]丙烷}二盐酸盐、2,2
’‑
偶氮双{2
‑
甲基
‑
n
‑
[1,1
‑
双(羟基甲基)
‑2‑
羟基乙基]丙酰胺}、2,2
’‑
偶氮双[2
‑
甲基
‑
n
‑
(2
‑
羟基乙基)
‑
丙酰胺]、4,4
’‑
偶氮双(4
‑
氰基戊酸)等偶氮化合物等。其中,优选过硫酸钾、过硫酸铵、过硫酸钠、2,2
’‑
偶氮双(2
‑
脒基丙烷)二盐酸盐、2,2
’‑
偶氮双[2
‑
(2
‑
咪唑啉
‑2‑
基)丙烷]二盐酸盐、2,2
’‑
偶氮双{2
‑
[1
‑
(2
‑
羟基乙基)
‑2‑
咪唑啉
‑2‑
基]丙烷}二盐酸盐。这些自由基聚合引发剂可以分别单独使用,也可以组合使用2种以上。
[0042]
相对于烯属不饱和单体1摩尔,自由基聚合引发剂的使用量可以是0.00005~0.01
摩尔。若自由基聚合引发剂的使用量为0.00005摩尔以上,则无需长时间进行聚合反应也具有效率。若使用量为0.01摩尔以下,则趋于不会发生急剧的聚合反应。
[0043]
上述自由基聚合引发剂也能够与亚硫酸钠、亚硫酸氢钠、硫酸亚铁、l
‑
抗坏血酸等还原剂同时使用而用作氧化还原聚合引发剂。
[0044]
聚合反应时,可以在用于聚合的烯属不饱和单体水溶液中包含链转移剂。作为链转移剂,例如可举出次磷酸盐类、硫醇类、硫醇酸类、仲醇类、胺类等。
[0045]
并且,为了控制吸水性树脂颗粒的粒径,可以在用于聚合的烯属不饱和单体水溶液中包含增稠剂。作为增稠剂,例如能够使用羟乙基纤维素、羟丙基纤维素、甲基纤维素、羧甲基纤维素、聚丙烯酸、聚丙烯酸(部分)中和物、聚乙二醇、聚丙烯酰胺、聚乙烯亚胺、糊精、海藻酸钠、聚乙烯醇、聚乙烯吡咯烷酮、聚环氧乙烷等。另外,若聚合时的搅拌速度相同,则烯属不饱和单体水溶液的黏度越高,所获得的颗粒的中值粒径趋于越大。
[0046]
作为烃分散介质,例如可举出:正己烷、正庚烷、2
‑
甲基己烷、3
‑
甲基己烷、2,3
‑
二甲基戊烷、3
‑
乙基戊烷、正辛烷等链状脂肪族烃;环己烷、甲基环己烷、环戊烷、甲基环戊烷、反式
‑
1,2
‑
二甲基环戊烷、顺式
‑
1,3
‑
二甲基环戊烷、反式
‑
1,3
‑
二甲基环戊烷等脂环族烃;苯、甲苯、二甲苯等芳香族烃等。这些烃分散介质可以单独使用,也可以组合使用2种以上。烃分散介质可以包含选自由碳原子数为6~8的链状脂肪族烃及碳原子数为6~8的脂环族烃组成的组中的至少一种化合物。从工业上容易获得且品质稳定的观点考虑,烃分散介质可以包含正庚烷、环己烷或这两者。并且,从相同观点考虑,作为上述烃分散介质的混合物,例如可以使用市售的exxsol heptane(exxon mobil corporation制造:含有75~85%正庚烷及同分异构体的烃)。
[0047]
从适度去除聚合热、容易控制聚合温度的观点考虑,相对于单体水溶液100质量份,烃分散介质的使用量优选为30~1000质量份,更优选为40~500质量份,进一步优选为50~300质量份。通过使烃分散介质的使用量为30质量份以上,趋于容易控制聚合温度。通过使烃分散介质的使用量为1000质量份以下,聚合的生产率趋于提高且节省费用。
[0048]
通常,聚合时虽能够发生基于自交联的内部交联,但也可以通过进一步使用内部交联剂来实施内部交联,并控制吸水性树脂颗粒的吸水特性。作为所使用的内部交联剂,例如可举出:乙二醇、丙二醇、三羟甲基丙烷、甘油、聚氧乙二醇、聚氧丙二醇、聚甘油等多元醇类的二或三(甲基)丙烯酸酯类;使上述多元醇类与马来酸、富马酸等不饱和酸进行反应而获得的不饱和聚酯类;n,n
’‑
亚甲基双(甲基)丙烯酰胺等双(甲基)丙烯酰胺类;使聚环氧化物与(甲基)丙烯酸进行反应而获得的二或三(甲基)丙烯酸酯类;使甲苯二异氰酸酯、六亚甲基二异氰酸酯等多异氰酸酯与(甲基)丙烯酸羟乙酯进行反应而获得的二(甲基)丙烯酸氨基甲酰酯类;烯丙基化淀粉、烯丙基化纤维素、邻苯二甲酸二烯丙酯、n,n’,n
”‑
三烯丙基异氰脲酸酯、二乙烯基苯等具有2个以上的聚合性不饱和基团的化合物;(聚)乙二醇二缩水甘油醚、(聚)丙二醇二缩水甘油醚、(聚)甘油二缩水甘油醚、(聚)甘油三缩水甘油醚、(聚)丙二醇聚缩水甘油醚、聚甘油聚缩水甘油醚等聚缩水甘油化合物;环氧氯丙烷、环氧溴丙烷、α
‑
甲基环氧氯丙烷等卤代环氧化合物;2,4
‑
甲苯二异氰酸酯、六亚甲基二异氰酸酯等异氰酸酯化合物等的具有2个以上的反应性官能团的化合物等。在这些内部交联剂中,优选使用聚缩水甘油化合物,更优选使用二缩水甘油醚化合物,尤其优选使用(聚)乙二醇二缩水甘油醚、(聚)丙二醇二缩水甘油醚、(聚)甘油二缩水甘油醚。这些交联剂可以单独使用,也
可以组合使用2种以上。
[0049]
从通过使所获得的聚合物适度交联而抑制水溶性的性质、并显示充分的吸水量的观点考虑,相对于每1摩尔的烯属不饱和单体,内部交联剂的量优选为0~0.03摩尔,更优选为0.00001~0.01摩尔,进一步优选为0.00002~0.005摩尔。
[0050]
能够将含有烯属不饱和单体、自由基聚合引发剂、根据需要的内部交联剂等成分的水相与含有烃类分散介质、表面活性剂、根据需要的高分子类分散剂等成分的油相混合,并在搅拌下进行加热,以在油包水体系中进行反相悬浮聚合。
[0051]
进行反相悬浮聚合时,在表面活性剂、根据需要的高分子类分散剂的存在下,使包含烯属不饱和单体的单体水溶液分散于烃分散介质中。此时,只要是在开始聚合反应之前,则表面活性剂或高分子类分散剂的添加时期可以在添加单体水溶液之前或之后。
[0052]
其中,从容易降低所获得的吸水性树脂中残留的烃分散介质的量的观点考虑,优选在使单体水溶液分散于分散有高分子类分散剂的烃分散介质中之后,进一步使表面活性剂分散,然后进行聚合。
[0053]
能够以1个阶段或2个阶段以上的多个阶段进行这种反相悬浮聚合。并且,从提高生产率的观点考虑,优选以2~3个阶段进行。
[0054]
在以2个阶段以上的多个阶段进行反相悬浮聚合的情况下,在进行第一阶段的反相悬浮聚合之后,向在第一阶段的聚合反应中获得的反应混合物中添加并混合烯属不饱和单体,通过与第一阶段相同的方法进行第二阶段以后的反相悬浮聚合即可。在第二阶段以后的各个阶段的反相悬浮聚合中,优选:除了烯属不饱和单体以外,以进行第二阶段以后的各个阶段的反相悬浮聚合时所添加的烯属不饱和单体的量为基准,在上述的各成分相对于烯属不饱和单体的摩尔比的范围内添加上述的自由基聚合引发剂或内部交联剂,由此进行反相悬浮聚合。另外,在第二阶段以后的各个阶段的反相悬浮聚合中,根据需要,可以使用内部交联剂。在使用内部交联剂的情况下,优选以供于各个阶段的烯属不饱和单体的量为基准,在上述的各成分相对于烯属不饱和单体的摩尔比的范围内添加内部交联剂,由此进行反相悬浮聚合。
[0055]
聚合反应的温度根据所使用的自由基聚合引发剂而不同,但从通过快速进行聚合且缩短聚合时间来提高经济性,并且容易地去除聚合热而顺畅地进行反应的观点考虑,聚合反应的温度优选为20~150℃,更优选为40~120℃。通常,反应时间为0.5~4小时。例如,能够根据反应体系内部的温度上升的停止来确认聚合反应的结束。由此,通常以含水凝胶的状态获得烯属不饱和单体的聚合物。
[0056]
聚合后,可以通过向所获得的含水凝胶状聚合物中添加交联剂并进行加热来实施聚合后交联。通过进行聚合后交联来提高含水凝胶状聚合物的交联度,从而能够更优选地提高吸水特性。
[0057]
作为用于进行聚合后交联的交联剂,例如可举出:乙二醇、丙二醇、1,4
‑
丁二醇、三羟甲基丙烷、甘油、聚氧乙二醇、聚氧丙二醇、聚甘油等多元醇;(聚)乙二醇二缩水甘油醚、(聚)丙二醇二缩水甘油醚及(聚)甘油二缩水甘油醚等具有2个以上的环氧基的化合物;环氧氯丙烷、环氧溴丙烷及α
‑
甲基环氧氯丙烷等卤代环氧化合物;2,4
‑
甲苯二异氰酸酯及六亚甲基二异氰酸酯等具有2个以上的异氰酸酯基的化合物;1,2
‑
亚乙基双噁唑啉等噁唑啉化合物;碳酸亚乙酯等碳酸酯化合物;双[n,n
‑
二(β
‑
羟基乙基)]己二酰胺等羟基烷基酰胺
化合物等。其中,优选(聚)乙二醇二缩水甘油醚、(聚)甘油二缩水甘油醚、(聚)甘油三缩水甘油醚、(聚)丙二醇聚缩水甘油醚、聚甘油聚缩水甘油醚等聚缩水甘油化合物。这些交联剂可以单独使用,也可以组合使用2种以上。
[0058]
从通过使所获得的含水凝胶状聚合物适度交联而显示适宜的吸水特性的观点考虑,相对于每1摩尔的烯属不饱和单体,用于聚合后交联的交联剂的量优选为0~0.03摩尔,更优选为0~0.01摩尔,进一步优选为0.00001~0.005摩尔。
[0059]
作为聚合后交联的添加时期,只要在用于聚合的烯属不饱和单体的聚合后即可,在进行多阶段聚合的情况下,优选在多阶段聚合后添加。另外,考虑到聚合时及聚合后的发热、工序延迟所引起的滞留、添加交联剂时的体系的开放、及伴随交联剂添加而添加水等所导致的水分的变动,从含水率(后述)的观点考虑,聚合后交联的交联剂优选在[刚聚合之后的含水率
±
3质量%]的范围内进行添加。
[0060]
接着,为了从所获得的含水凝胶状聚合物中去除水分而进行干燥。通过干燥,获得包含烯属不饱和单体的聚合物的聚合物颗粒。作为干燥方法,例如可举出:(a)通过在上述含水凝胶状聚合物分散于烃分散介质的状态下,从外部进行加热而进行共沸蒸馏,并使烃分散介质回流而去除水分的方法;(b)通过倾析提取含水凝胶状聚合物,并进行减压干燥的方法;(c)利用过滤器滤取含水凝胶状聚合物,并进行减压干燥的方法等。其中,从制造工序的简便性的观点出发,优选使用(a)方法。
[0061]
例如,能够通过调节聚合反应时的搅拌机的转速、或者通过在聚合反应后或干燥初期向体系内部添加粉末状无机凝聚剂来控制吸水性树脂颗粒的粒径。通过添加凝聚剂,能够增大所获得的吸水性树脂颗粒的粒径。作为粉末状无机凝聚剂的例子,可举出二氧化硅、沸石、膨润土、氧化铝、滑石、二氧化钛、高岭土、黏土、水滑石等,其中,从凝聚效果的观点考虑,优选二氧化硅、氧化铝、滑石或高岭土。
[0062]
在反相悬浮聚合中,作为添加粉末状无机凝聚剂的方法,优选如下方法:在与聚合中使用的烃分散介质相同种类的烃分散介质或水中预先分散粉末状无机凝聚剂之后,混合于搅拌下的包含含水凝胶的烃分散介质中。
[0063]
在本实施方式的吸水性树脂颗粒的制造中,优选在干燥工序或之后的任一工序中,使用交联剂进行含水凝胶状聚合物的表面部分的交联(表面交联)。表面交联优选在含水凝胶状聚合物为特定含水率的时刻进行。表面交联的时期优选为含水凝胶状聚合物的含水率为5~50质量%的时刻,更优选为含水凝胶状聚合物的含水率为10~40质量%的时刻,进一步优选为含水凝胶状聚合物的含水率为15~35质量%的时刻。
[0064]
利用下式计算出含水凝胶状聚合物的含水率(质量%)。含水率=[ww/(ww ws)]
×
100ww:从整个聚合工序的聚合前的水溶液所含有的水分量中减去通过干燥工序而排出至体系外部的水分量,在该得到的量的基础上加上混合粉末状无机凝聚剂、表面交联剂等时根据需要使用的水分量,由此得到的含水凝胶状聚合物的水分量。ws:根据构成含水凝胶状聚合物的烯属不饱和单体、交联剂、引发剂等材料的掺入量计算的固体成分量。
[0065]
作为用于进行表面交联的表面交联剂,能够举出具有2个以上的反应性官能团的化合物。作为其实例,可举出:乙二醇、丙二醇、1,4
‑
丁二醇、三羟甲基丙烷、甘油、聚氧乙二
醇、聚氧丙二醇、聚甘油等多元醇类;(聚)乙二醇二缩水甘油醚、(聚)甘油二缩水甘油醚、(聚)甘油三缩水甘油醚、三羟甲基丙烷三缩水甘油醚、(聚)丙二醇聚缩水甘油醚、(聚)甘油聚缩水甘油醚等聚缩水甘油化合物;环氧氯丙烷、环氧溴丙烷、α
‑
甲基环氧氯丙烷等卤代环氧化合物;2,4
‑
甲苯二异氰酸酯、六亚甲基二异氰酸酯等异氰酸酯化合物;3
‑
甲基
‑3‑
氧杂环丁烷甲醇、3
‑
乙基
‑3‑
氧杂环丁烷甲醇、3
‑
丁基
‑3‑
氧杂环丁烷甲醇、3
‑
甲基
‑3‑
氧杂环丁烷乙醇、3
‑
乙基
‑3‑
氧杂环丁烷乙醇、3
‑
丁基
‑3‑
氧杂环丁烷乙醇等氧杂环丁烷化合物;1,2
‑
亚乙基双噁唑啉等噁唑啉化合物;碳酸亚乙酯等碳酸酯化合物;双[n,n
‑
二(β
‑
羟基乙基)]己二酰胺等羟基烷基酰胺化合物。其中,更优选(聚)乙二醇二缩水甘油醚、(聚)甘油二缩水甘油醚、(聚)甘油三缩水甘油醚、(聚)丙二醇聚缩水甘油醚、聚甘油聚缩水甘油醚等聚缩水甘油化合物。这些表面交联剂可以单独使用,也可以组合使用2种以上。
[0066]
从通过使所获得的含水凝胶状聚合物适度交联而显示适宜的吸水特性的观点考虑,相对于用于聚合的烯属不饱和单体1摩尔,表面交联剂的量通常为0.00001~0.02摩尔,优选为0.00005~0.01摩尔,更优选为0.0001~0.005摩尔的比。
[0067]
从充分提高聚合物颗粒的表面部分的交联密度,且提高吸水性树脂颗粒的凝胶强度的观点考虑,表面交联剂的使用量优选为0.00001摩尔以上。并且,从提高吸水性树脂颗粒的保水能力的观点考虑,优选为0.02摩尔以下。
[0068]
在表面交联反应后,通过公知的方法蒸馏去除水及烃分散介质,从而能够获得作为经表面交联的干燥品的聚合物颗粒。
[0069]
关于聚合反应,能够使用具有搅拌叶片的各种搅拌机来进行。作为搅拌叶片,能够使用平叶片、框式叶片、桨式叶片、螺旋桨式叶片、锚式叶片、涡轮式叶片、法武都拉式(pfaudler)叶片、螺带式叶片、泛能式叶片、最大叶片式(maxblend)叶片等。平叶片具有轴(搅拌轴)和配置于轴周围的平板部(搅拌部)。平板部可以具有狭缝等。在作为搅拌叶片而使用了平叶片的情况下,容易均匀地进行聚合物颗粒的交联反应,容易一边维持保水量等吸水特性,一边将通液维持率调节到适合于本发明的范围内。
[0070]
本实施方式的吸水性树脂颗粒可以仅由聚合物颗粒构成,但是能够进一步包含例如选自无机粉末、表面活性剂、氧化剂、还原剂、金属螯合剂、自由基链反应抑制剂、抗氧化剂、抗菌剂、除臭剂、凝胶稳定剂、流动性改进剂(润滑剂)等的各种追加成分。追加成分能够配置在聚合物颗粒的内部、聚合物颗粒的表面上或这两个部位。
[0071]
本实施方式的吸水性树脂颗粒优选包含无机颗粒。作为无机颗粒,例如可举出无定形二氧化硅等二氧化硅颗粒。无定形二氧化硅可以是亲水性无定形二氧化硅。例如,通过混合聚合物颗粒和无机颗粒,能够在聚合物颗粒的表面上配置无机颗粒。此处的无机颗粒通常具有比聚合物颗粒的大小微小的大小。例如,无机颗粒的平均粒径可以是0.1~50μm、0.5~30μm或1~20μm。此处的平均粒径为能够通过动态光散射法或激光衍射散射法测定的值。通过使无机颗粒的添加量在上述范围内,容易获得吸水特性良好的吸水性树脂颗粒。
[0072]
例如,相对于聚合物颗粒100质量份,作为无机颗粒添加0.05~5质量份的无定形二氧化硅,由此能够提高吸水性树脂颗粒的流动性。在吸水性树脂颗粒包含无机颗粒的情况下,无机颗粒相对于聚合物颗粒的质量的比例可以是0.05质量%以上、0.1质量%以上、0.2质量%以上、0.5质量%以上、1.0质量%以上或1.5质量%以上,也可以是5.0质量%以下、3.5质量%以下、1.5质量%以下、1.0质量%以下、0.8质量%以下、0.5质量%以下或0.3
质量%以下。
[0073]
本实施方式的吸水性树脂颗粒的制造方法可以包括分选通过上述方法测定的通液维持率为10%以上的吸水性树脂颗粒的工序。上述制造方法可以包括测定吸水性树脂颗粒的通液维持率的工序。作为所分选的吸水性树脂颗粒的性质,可以满足上述的吸水性树脂颗粒的性质(例如特定范围的生理盐水保水量、特定范围的无加压dw3分钟值等)。
[0074]
本实施方式的吸水性树脂颗粒对尿液、血液等体液的吸收性优异,例如,能够应用于纸尿布、卫生巾、卫生棉条等卫生用品、宠物垫、狗或猫的厕所掺合物等动物排泄物处理材料等领域。
[0075]
本实施方式的吸水性树脂颗粒能够适宜地用于吸收体。本实施方式的吸收体包含吸水性树脂颗粒。从将吸收体使用于吸收性物品时获得充分的液体吸收性能的观点考虑,相对于每1平方米吸收体,吸收体中的吸水性树脂颗粒的含量优选为100~1000g(即100~1000g/m2),更优选为150~800g/m2,进一步优选为200~700g/m2。从发挥作为吸收性物品的充分的液体吸收性能、尤其是抑制漏液的观点考虑,上述含量优选为100g/m2以上。从抑制发生凝胶堵塞现象、作为吸收性物品发挥液体的扩散性能、并进一步改善液体的渗透速度的观点考虑,上述含量优选为1000g/m2以下。
[0076]
吸收体除了具备吸水性树脂颗粒以外,例如还可以进一步具备纤维状物。吸收体例如可以是包含吸水性树脂颗粒及纤维状物的混合物。相对于吸水性树脂颗粒及纤维状物的合计,吸收体中的吸水性树脂颗粒的质量比例可以是2质量%~100质量%,优选为10质量%~80质量%,更优选为20质量%~70质量%。作为吸收体的结构,例如可以是吸水性树脂颗粒及纤维状物均匀地混合的形态,也可以是在形成为片状或层状的纤维状物之间夹有吸水性树脂颗粒的形态,还可以是其他形态。
[0077]
作为纤维状物,例如可举出经微粉碎的木浆、棉花、棉绒、人造丝、乙酸纤维素等纤维素类纤维、聚酰胺、聚酯、聚烯烃等合成纤维。并且,纤维状物可以是上述纤维的混合物。
[0078]
为了进一步提高吸收体在使用前及使用期间的形态保持性,可以通过在纤维状物中添加粘合性粘结剂而使纤维彼此粘合。作为粘合性粘结剂,例如可举出热熔接性合成纤维、热熔粘合剂、粘合性乳液等。由于本实施方式的吸水性树脂颗粒在用于吸收体时的保形性优异,因此能够减少粘合性粘结剂的使用量。
[0079]
作为热熔接性合成纤维,例如可举出聚乙烯、聚丙烯、乙烯
‑
丙烯共聚物等全熔型粘结剂、由聚丙烯与聚乙烯的并列结构或芯鞘结构构成的非全熔型粘结剂。在上述非全熔型粘结剂中,仅使聚乙烯部分进行热熔接。作为热熔粘合剂,例如可举出乙烯
‑
乙酸乙烯酯共聚物、苯乙烯
‑
异戊二烯
‑
苯乙烯嵌段共聚物、苯乙烯
‑
丁二烯
‑
苯乙烯嵌段共聚物、苯乙烯
‑
乙烯
‑
丁烯
‑
苯乙烯嵌段共聚物、苯乙烯
‑
乙烯
‑
丙烯
‑
苯乙烯嵌段共聚物、非晶态聚丙烯等基础聚合物与增粘剂、增塑剂、抗氧化剂等的掺合物。
[0080]
作为粘合性乳液,例如可举出选自由甲基丙烯酸甲酯、苯乙烯、丙烯腈、丙烯酸2
‑
乙基己酯、丙烯酸丁酯、丁二烯、乙烯及乙酸乙烯酯组成的组中的至少一种以上的单体的聚合物。这些粘合性粘结剂可以单独使用,也可以组合使用2种以上。
[0081]
本实施方式的吸收体可以进一步包含无机粉末(例如无定形二氧化硅)、除臭剂、颜料、染料、抗菌剂、香料、粘着剂等添加剂。通过这些添加剂,能够对吸收体赋予各种功能。当吸水性树脂颗粒包含无机颗粒时,吸收体可以包含与吸水性树脂颗粒中的无机颗粒不同
的无机粉末。作为无机粉末,例如可举出二氧化硅、沸石、高岭土、黏土等。
[0082]
本实施方式的吸收体的形状并无特别限定,例如可以是片状。吸收体的厚度(例如,片状吸收体的厚度)例如可以是0.1~20mm、0.3~15mm。
[0083]
本实施方式的吸收性物品除了具备吸收体以外,例如还可以具备芯包片(core wrap)、透液性顶片(top sheet)、不透液性底片(back sheet)。芯包片保持吸收体的形状。透液性顶片配置在吸液对象的液体所渗入的一侧的最外部。不透液性底片配置在与吸液对象的液体所渗入的一侧为相反侧的最外部。
[0084]
作为吸收性物品,可举出尿布(例如纸尿布)、如厕训练裤、失禁垫、卫生用品(卫生巾、卫生棉条等)、吸汗垫、宠物垫、简易马桶用部件、动物排泄物处理材料等。
[0085]
图1是表示吸收性物品的一个实例的剖视图。图1所示的吸收性物品100具备吸收体10、芯包片20a、20b、透液性顶片30及不透液性底片40。在吸收性物品100中,依次层叠有不透液性底片40、芯包片20b、吸收体10、芯包片20a及透液性顶片30。在图1中,存在图示为在部件之间存在间隙的部分,但是部件之间也可以密合而不存在该间隙。
[0086]
吸收体10具有吸水性树脂颗粒10a和包含纤维状物的纤维层10b。吸水性树脂颗粒10a分散在纤维层10b内。
[0087]
芯包片20a以与吸收体10接触的状态配置于吸收体10的一面侧(图1中,吸收体10的上侧)。芯包片20b以与吸收体10接触的状态配置于吸收体10的另一面侧(图1中,吸收体10的下侧)。吸收体10配置于芯包片20a与芯包片20b之间。
[0088]
芯包片20a及芯包片20b例如具有与吸收体10相同大小的主表面。通过使用芯包片,维持吸收体的保形性,从而能够防止构成吸收体的吸水性树脂颗粒等的脱落及流动。作为芯包片,例如可举出无纺布、机织物、纸巾、具有透液孔的合成树脂膜、具有网眼的网状片材等,从经济性的观点考虑,优选使用将粉碎纸浆湿式成型而成的纸巾。
[0089]
透液性顶片30配置于吸收对象的液体所渗入的一侧的最外部。透液性顶片30以与芯包片20a接触的状态配置在芯包片20a上。不透液性底片40在吸收性物品100中配置在与透液性顶片30相反的一侧的最外部。不透液性底片40以与芯包片20b接触的状态配置于芯包片20b的下侧。透液性顶片30及不透液性底片40例如具有比吸收体10的主表面更宽的主表面,透液性顶片30及不透液性底片40的外缘部延伸到吸收体10及芯包片20a、20b的周围。
[0090]
作为透液性顶片30,可举出无纺布、多孔片等。作为无纺布,例如可举出热粘合无纺布、热风无纺布、树脂粘合无纺布、纺粘无纺布、熔喷无纺布、纺粘/熔喷/纺粘无纺布、气流成网无纺布、水刺无纺布、点粘合无纺布等。其中,优选使用热粘合无纺布、热风无纺布、纺粘无纺布、纺粘/熔喷/纺粘无纺布。
[0091]
作为透液性顶片30的构成材料,能够使用在该技术领域中公知的树脂或纤维,从使用于吸收性物品时的液体渗透性、柔软性及强度的观点考虑,可举出聚乙烯(pe)、聚丙烯(pp)等聚烯烃、聚对苯二甲酸乙二醇酯(pet)、聚对苯二甲酸丙二醇酯(ptt)、聚萘二甲酸乙二醇酯(pen)等聚酯、尼龙等聚酰胺、人造丝、其他合成树脂或合成纤维、棉、丝、麻、纸浆(纤维素)纤维等。作为构成材料,从提高透液性顶片30的强度等观点考虑,优选使用合成纤维,其中优选聚烯烃、聚酯。这些材料可以单独使用,也可以组合使用2种以上的材料。
[0092]
从提高吸收性物品的液体吸收性能的观点考虑,期望用于透液性顶片30的无纺布具有适度的亲水性。从该观点考虑,按照国际公开第2011/086843号中所记载的“无纺布的
亲水度”(根据纸浆试验方法no.68(2000))进行测定时的亲水度优选为5~200,更优选为10~150。这种具有亲水性的无纺布可以是在上述无纺布中使用了如人造纤维那样材料本身示出适度的亲水度的纤维的无纺布,也可以是使用了通过利用公知的方法对如聚烯烃纤维、聚酯纤维那样的疏水性化学纤维进行亲水化处理来赋予适度的亲水度的纤维的无纺布。
[0093]
作为化学纤维的亲水化处理的方法,例如可举出:在纺粘无纺布中,利用在疏水性化学纤维中混合有亲水剂的化学纤维并通过纺粘法获得无纺布的方法;用疏水性化学纤维制作纺粘无纺布时伴有亲水剂的方法;或使用疏水性化学纤维获得纺粘无纺布之后使亲水剂浸渍于该纺粘无纺布的方法等。作为亲水剂,可使用脂肪族磺酸盐、高级醇硫酸酯盐等阴离子表面活性剂、季铵盐等阳离子表面活性剂、聚乙二醇脂肪酸酯、聚甘油脂肪酸酯、山梨糖醇酐脂肪酸酯等非离子表面活性剂、聚氧亚烷基改性有机硅等有机硅类表面活性剂、及由聚酯类、聚酰胺类、丙烯酸类、氨基甲酸酯类树脂构成的去污剂等。
[0094]
从对吸收性物品赋予良好的液体渗透性、柔软性、强度及缓冲性,以及加快吸收性物品的液体渗透速度的观点考虑,用于透液性顶片30的无纺布优选为体积大小适度,且单位面积重量大的无纺布。无纺布的单位面积重量优选为5~200g/m2,更优选为8~150g/m2,进一步优选为10~100g/m2。并且,无纺布的厚度优选为20~1400μm,更优选为50~1200μm,进一步优选为80~1000μm。
[0095]
不透液性底片40防止吸收到吸收体10中的液体从底片40侧漏到外部。不透液性底片40能够使用以聚乙烯(pe)、聚丙烯(pp)等聚烯烃树脂为主体的不透液性膜、透气性树脂膜、在纺粘或水刺等无纺布上接合有透气性树脂膜的复合膜、用高强度纺粘无纺布夹住耐水性熔喷无纺布而得到的纺粘/熔喷/纺粘(sms)无纺布等。从确保柔软性,以不损害吸收性物品的穿着感的观点考虑,底片40能够使用以低密度聚乙烯(ldpe)树脂为主体的单位面积重量为10~50g/m2的树脂膜。并且,使用透气性材料时,能够降低穿戴时的闷热,从而也能够降低对穿着者带来的不适感。
[0096]
吸收体10、芯包片20a、20b、透液性顶片30及不透液性底片40的大小关系并无特别限定,可根据吸收性物品的用途等适当调节。并且,使用芯包片20a、20b保持吸收体10的形状的方法并无特别限定,如图1所示,可以利用多个芯包片夹持吸收体,也可以利用一片芯包片覆盖吸收体。
[0097]
吸收体10可以粘合于透液性顶片30。通过粘合吸收体10与透液性顶片30,液体更顺畅地导入到吸收体中,因此容易获得漏液的防止效果更加优异的吸收性物品。当吸收体10被芯包片夹持或覆盖时,优选至少粘合芯包片与透液性顶片30,更优选进一步粘合芯包片与吸收体10。作为粘合方法,例如可举出:相对于透液性顶片30,沿其宽度方向以规定间隔将热熔粘合剂涂布成纵向条纹状、螺旋状等形状来进行粘合的方法;使用选自淀粉、羧甲基纤维素、聚乙烯醇、聚乙烯吡咯烷酮及其他水溶性高分子的水溶性粘结剂来进行粘合的方法等。并且,当吸收体10包含时,可采用通过该热熔接性合成纤维的热熔接来进行粘合的方法。
[0098]
本发明还提供一种由下述式表示的吸水性树脂颗粒的通液维持率的测定方法。通液维持率(%)=(溶胀凝胶通液速度/干粉通液速度)
×
100
[0099]
关于干粉通液速度,通过依次包括下述(a)、(b)及(c)工序的方法来测定。
seika chemica1s company,limited,hec aw
‑
15f)、作为自由基聚合引发剂的2,2
’‑
偶氮双(2
‑
脒基丙烷)二盐酸盐0.092g(0.339毫摩尔)及过硫酸钾0.018g(0.067毫摩尔)、作为内部交联剂的乙二醇二缩水甘油醚0.0046g(0.026毫摩尔)并进行溶解,从而制备了第一阶段的水溶液。
[0105]
将所制备的水溶液添加到上述可拆式烧瓶内的反应液中并搅拌了10分钟。接着,在正庚烷6.62g中加热溶解作为表面活性剂的蔗糖硬脂酸酯(mitsubishi
‑
chemical foods corporation,ryoto sugar ester s
‑
370,hlb:3)0.736g来制备了表面活性剂溶液。将表面活性剂溶液进一步添加到反应液中,一边将搅拌机的转速设为425rpm来进行搅拌一边用氮气充分地置换了体系内部。然后,通过将烧瓶浸渍于70℃的水浴中进行升温,并进行60分钟的聚合,由此获得了第一阶段的聚合浆液。
[0106]
另一方面,向另一个内容积为500ml的烧杯中加入作为烯属不饱和单体的80.5质量%的丙烯酸水溶液128.8g(1.44摩尔),从外部冷却的同时,滴加27质量%的氢氧化钠水溶液159.0g来进行了75摩尔%的中和。然后,加入作为自由基聚合引发剂的2,2
’‑
偶氮双(2
‑
脒基丙烷)二盐酸盐0.129g(0.476毫摩尔)及过硫酸钾0.026g(0.096毫摩尔)、以及作为内部交联剂的乙二醇二缩水甘油醚0.0116g(0.067毫摩尔)并进行溶解,从而制备了第二阶段的水溶液。
[0107]
一边将搅拌机的转速设为650rpm来进行搅拌,一边将上述可拆式烧瓶体系内部冷却至25℃。接着,将上述第二阶段的水溶液全部添加到第一阶段的聚合浆液中,并将体系内部用氮气置换了30分钟。然后,再次将烧瓶浸渍于70℃的水浴中进行升温,并进行60分钟的聚合反应,从而获得了含水凝胶状聚合物。
[0108]
在搅拌下,将45质量%的二乙烯三胺五乙酸五钠水溶液0.589g添加到第二阶段的聚合后的含水凝胶状聚合物中。然后,将烧瓶浸渍于设定为125℃的油浴中,通过正庚烷与水的共沸蒸馏,一边使正庚烷回流,一边将207.9g的水提取到体系外。然后,向烧瓶中添加作为表面交联剂的2质量%的乙二醇二缩水甘油醚水溶液4.42g(0.507毫摩尔),并在83℃下保持了2小时。
[0109]
然后,通过在125℃下使正庚烷蒸发并干燥而获得了聚合物颗粒(干燥品)。使该聚合物颗粒通过孔径为850μm的筛,且将相对于聚合物颗粒的质量为0.2质量%的无定形二氧化硅(oriental silicas corporation,tokusil np
‑
s)与聚合物颗粒混合,从而获得了包含无定形二氧化硅的吸水性树脂颗粒232.3g。吸水性树脂颗粒的中值粒径为361μm。
[0110]
[实施例2]将提取到体系外的水量变更为224.3g,除此以外,以与实施例1相同的方式,获得了231.0g的吸水性树脂颗粒。吸水性树脂颗粒的中值粒径为342μm。
[0111]
[实施例3]将提取到体系外的水量变更为234.6g,除此以外,以与实施例1相同的方式,获得了232.1g的吸水性树脂颗粒。吸水性树脂颗粒的中值粒径为355μm。
[0112]
[实施例4]作为第一阶段的水溶液的制备中的自由基聚合引发剂未使用2,2
’‑
偶氮双(2
‑
脒基丙烷)二盐酸盐,将过硫酸钾的使用量变更为0.0736g(0.272)毫摩尔;将作为内部交联剂的乙二醇二缩水甘油醚的使用量变更为0.010g(0.057毫摩尔);作为第二阶段的水溶液的
制备中的自由基聚合引发剂未使用2,2
’‑
偶氮双(2
‑
脒基丙烷)二盐酸盐,将过硫酸钾的使用量变更为0.090g(0.333毫摩尔);并将提取到体系外的水量变更为247.3g,除此以外,以与实施例1相同的方式,获得了231.5g的吸水性树脂颗粒。吸水性树脂颗粒的中值粒径为359μm。
[0113]
[实施例5]将第一阶段的水溶液的制备中的作为内部交联剂的乙二醇二缩水甘油醚的使用量变更为0.010g(0.057毫摩尔);并将提取到体系外的水量变更为238.5g,除此以外,以与实施例1相同的方式,获得了222.4g的吸水性树脂颗粒。吸水性树脂颗粒的中值粒径为354μm。
[0114]
[实施例6]作为第一阶段的水溶液的制备中的自由基聚合引发剂未使用2,2
’‑
偶氮双(2
‑
脒基丙烷)二盐酸盐,将过硫酸钾的使用量变更为0.0736g(0.272毫摩尔);将作为内部交联剂的乙二醇二缩水甘油醚的使用量变更为0.010g(0.057毫摩尔);作为第二阶段的水溶液的制备中的自由基聚合引发剂未使用2,2
’‑
偶氮双(2
‑
脒基丙烷)二盐酸盐,将过硫酸钾的使用量变更为0.090g(0.334毫摩尔);并将提取到体系外的水量变更为257.6g,除此以外,以与实施例1相同的方式,获得了229.8g的吸水性树脂颗粒。吸水性树脂颗粒的中值粒径为362μm。
[0115]
[实施例7]将提取到体系外的水量变更为201.3g;将作为表面交联剂的2质量%乙二醇二缩水甘油醚水溶液的使用量变更为6.62g(0.761毫摩尔),除此以外,以与实施例1相同的方式,获得了224.0g的吸水性树脂颗粒。吸水性树脂颗粒的中值粒径为356μm。
[0116]
[比较例1]准备了具备回流冷凝器、滴液漏斗、氮气导入管及搅拌机的内径为11cm、容积为2l的圆底圆筒型可拆式烧瓶,所述搅拌机为具有两层叶片直径为5cm的4片斜桨式叶片的搅拌叶片。向该烧瓶中加入作为烃分散介质的正庚烷293g,并添加作为高分子类分散剂的马来酸酐改性乙烯
‑
丙烯共聚物(mitsui chemicals,inc.,hi
‑
wax 1105a)0.736g,由此获得了混合物。通过对该混合物进行搅拌,且同时升温至80℃来溶解分散剂之后,将混合物冷却至50℃。
[0117]
另一方面,向内容积为300ml的烧杯中加入作为烯属不饱和单体的80.5质量%的丙烯酸水溶液92.0g(1.03摩尔),从外部冷却的同时,滴加20.9质量%的氢氧化钠水溶液147.7g来进行了75摩尔%的中和。然后,加入作为增稠剂的羟乙基纤维素0.092g(sumitomo seika chemicals company,limited,hec aw
‑
15f)、作为自由基聚合剂的过硫酸钾0.0736g(0.272毫摩尔)及作为内部交联剂的乙二醇二缩水甘油醚0.010g(0.057毫摩尔)并进行溶解,从而制备了第一阶段的水溶液。
[0118]
将所制备的水溶液添加到上述可拆式烧瓶内的反应液中并搅拌了10分钟。接着,在正庚烷6.62g中加热溶解作为表面活性剂的蔗糖硬脂酸酯(mitsubishi
‑
chemical foods corporation,ryoto sugar ester s
‑
370,hlb:3)0.736g来制备了表面活性剂溶液。将表面活性剂溶液进一步添加到反应液中,一边将搅拌机的转速设为550rpm来进行搅拌一边用氮气充分地置换了体系内部。然后,通过将烧瓶浸渍于70℃的水浴中进行升温,并进行60分钟
的聚合,由此获得了第一阶段的聚合浆液。
[0119]
另一方面,向另一个内容积为500ml的烧杯中加入作为烯属不饱和单体的80.5质量%的丙烯酸水溶液128.8g(1.44摩尔),从外部冷却的同时,滴加27质量%的氢氧化钠水溶液159.0g来进行了75摩尔%的中和。然后,加入作为自由基聚合引发剂的过硫酸钾0.090g(0.333毫摩尔)并使其溶解,制备了第二阶段的水溶液。
[0120]
一边将搅拌机的转速设为1000rpm来进行搅拌,一边将上述可拆式烧瓶体系内部冷却至25℃。接着,将上述第二阶段的水溶液全部添加到第一阶段的聚合浆液中,并将体系内部用氮气置换了30分钟。然后,再次将烧瓶浸渍于70℃的水浴中进行升温,并进行了60分钟的聚合反应。然后,作为用于聚合后交联的交联剂添加2质量%的乙二醇二缩水甘油醚水溶液0.580g(0.067毫摩尔),获得了含水凝胶状聚合物。
[0121]
在搅拌下,将45质量%的二乙烯三胺五乙酸五钠水溶液0.265g添加到第二阶段的聚合后的含水凝胶状聚合物中。然后,将烧瓶浸渍于设定为125℃的油浴中,通过正庚烷和水的共沸蒸馏,一边使正庚烷回流,一边将259.4g的水提取到体系外。然后,向烧瓶中添加作为表面交联剂的2质量%的乙二醇二缩水甘油醚水溶液6.30g(0.723毫摩尔),并在83℃下保持了2小时。
[0122]
然后,通过在125℃下使正庚烷蒸发并干燥而获得了聚合物颗粒(干燥品)。使该聚合物颗粒通过孔径为850μm的筛,且将相对于聚合物颗粒的质量为0.5质量%的无定形二氧化硅(oriental silicas corporation,tokusil np
‑
s)与聚合物颗粒混合,从而获得了包含无定形二氧化硅的吸水性树脂颗粒230.8g。吸水性树脂颗粒的中值粒径为349μm。
[0123]
[比较例2]作为第二阶段的水溶液的制备中的内部交联剂添加乙二醇二缩水甘油醚0.012g(0.067毫摩尔);未使用用于聚合后交联的2质量%的乙二醇二缩水甘油醚水溶液;将提取到体系外的水量变更为278.9g;将作为表面交联剂的2质量%的乙二醇二缩水甘油醚水溶液的使用量变更为4.42g(0.507毫摩尔);并将相对于聚合物颗粒的无定形二氧化硅的混合量变更为0.2质量%,除此以外,以与比较例1相同的方式,获得了230.8g的吸水性树脂颗粒。吸水性树脂颗粒的中值粒径为358μm。
[0124]
[比较例3]将第一阶段的水溶液的制备中的作为内部交联剂的乙二醇二缩水甘油醚的使用量变更为0.011g(0.063毫摩尔);作为第二阶段的水溶液的制备中的内部交联剂添加乙二醇二缩水甘油醚乙二醇二缩水甘油醚0.013g(0.075毫摩尔);未使用用于聚合后交联的交联剂;将提取到体系外的水量变更为262.6g;将作为表面交联剂的2质量%的乙二醇二缩水甘油醚水溶液的使用量变更为9.93g(1.14毫摩尔);且未混合无定形二氧化硅,除此以外,以与比较例1相同的方式,获得了230.3g的吸水性树脂颗粒。吸水性树脂颗粒的中值粒径为349μm。
[0125]
关于所获得的吸水性树脂颗粒,通过以下方法,对通液维持率、生理盐水保水量、中值粒径、无加压dw3分钟值及吸收体的断裂性进行了评价。另外,在本实施例中使用的生理盐水为0.9质量%的nacl水溶液。
[0126]
<通液维持率的测定>[溶胀凝胶通液速度的测定]
使用图3所示的仪器来进行溶胀凝胶通液速度的测定。测定部60具有:丙烯酸树脂制圆筒状容器61,其在底部具备250目的尼龙网片64;及丙烯酸树脂制圆筒状小型容器62,其装在圆筒状容器61的内部,并且同样地在底部具备尼龙网片63。圆筒状容器61的内径为26mm、外径为40mm、高度为140mm。圆筒状小型容器62的内径为19mm、外径为25mm、高度为120mm、质量为30g。圆筒状小型容器62能够在圆筒状容器61的内部上下无阻力地移动。
[0127]
以下述方式进行了溶胀凝胶通液速度的测定。在圆筒状容器61中均匀地散布0.3g的吸水性树脂颗粒10a,从其上部插入圆筒状小型容器62,用尼龙网片63及尼龙网片64夹住吸水性树脂颗粒10a,由此形成了具有干燥状态的吸水性树脂颗粒10a的测定部60。
[0128]
另外准备装有40
±
0.1g的生理盐水的培养皿,将测定部60的底部侧浸渍于生理盐水中30分钟,由此使吸水性树脂颗粒10a吸收生理盐水而使其饱和溶胀,获得了具有溶胀凝胶的测定部60。
[0129]
在预先测定了空质量(wa)的内径约为90mm的培养皿66上放置具有10cm见方的大小的、孔径为2mm的金属丝网67,并在金属丝网67上放置了上述包含溶胀凝胶的测定部60。从圆筒状小型容器62的上部,向测定部60内一次性投入20g的生理盐水65。测定包含从开始投入起1分钟内通过了溶胀凝胶的生理盐水65的培养皿66的质量(wb),由下述式求出通液速度(g/分钟)。将结果示于表1。通液速度(g/分钟)=wb
‑
wa
[0130]
[干粉通液速度的测定]在干粉通液速度的测定中,使用了在25℃、湿度45%(rh)下保管的干燥状态的吸水性树脂颗粒。在25℃、湿度45%(rh)下进行了通液速度的测定。以与上述溶胀凝胶通液速度的测定相同的方式,形成了具有干燥状态的吸水性树脂颗粒10a的测定部60。使用自动泵(integra dose it p910)以60ml/分钟的恒定速度从圆筒状小型容器62的上部向测定部60内投入生理盐水1分钟。将通过测定部60内的吸水性树脂颗粒10a并从测定部60下部流出的液体积存于另一个容器中,并测定所积存的液体的质量,由此求出从开始投入起1分钟内通过了干粉的液体量(通液速度(g/分钟))。将结果示于表1。
[0131]
根据所测定的溶胀凝胶通液速度及干粉通液速度来计算出通液维持率。将结果示于表1。通液维持率(%)=(溶胀凝胶通液速度/干粉通液速度)
×
100
[0132]
<生理盐水保水量的测定>将称取了2.0g吸水性树脂颗粒的棉袋(阔幅棉布60号,横100mm
×
纵200mm)设置在500ml容积的烧杯内。以不产生结块的方式,向装有吸水性树脂颗粒的棉袋中一次性注入500g生理盐水,用橡皮筋绑住棉袋的上部,静置30分钟,从而使吸水性树脂颗粒溶胀。利用将离心力设定为167g的脱水机(kokusan co.ltd.制造,产品编号:h
‑
122),对经过30分钟后的棉袋进行1分钟脱水,并测定了脱水后的包含溶胀凝胶的棉袋的质量wc(g)。不添加吸水性树脂颗粒而进行相同操作,测定棉袋在湿润时的皮重wd(g),并由下式计算出生理盐水保水量。将结果示于表1。生理盐水保水量(g/g)=[wc
‑
wd]/2.0
[0133]
<中值粒径(粒度分布)的测定>将50g吸水性树脂颗粒用于中值粒径(粒度分布)的测定。从上至下依次组合了作
为jis标准筛的孔径为850μm的筛、孔径为500μm的筛、孔径为425μm的筛、孔径为300μm的筛、孔径为250μm的筛、孔径为180μm的筛、孔径为150μm的筛及托盘。
[0134]
向所组合的最上侧的筛中加入吸水性树脂颗粒,使用ro
‑
tap型振动器(sieve factory iida co.,ltd.制造)、依据jis z 8815(1994)进行了分级。分级之后,将残留在各个筛上的吸水性树脂颗粒的质量以相对于总量的质量百分比的形式算出,求出粒度分布。关于该粒度分布,从粒径大的开始依次对筛上进行累计,将筛的孔径与残留在筛上的吸水性树脂颗粒的质量百分比的累计值的关系绘制在对数概率纸上。通过将概率纸上的标绘点以直线连接,从而将相当于累计质量百分比为50质量%的粒径作为中值粒径。
[0135]
<无加压dw的测定>使用图4所示的测定装置对吸水性树脂颗粒的无加压dw进行了测定。对1种吸水性树脂颗粒实施5次测定,求出了除了最低值和最高值以外的3个测定值的平均值。该测定装置具有滴定管部1、导管5、测定台13、尼龙网片15、台架11及夹具3。滴定管部1具有:标有刻度的滴定管21;密封滴定管21的上部开口的橡胶塞23;与滴定管21的下部的前端连接的旋塞22;与滴定管21的下部连接的空气导入管25;及旋塞24。滴定管部1用夹具3固定。平板状的测定台13具有形成于其中央部的直径为2mm的贯穿孔13a,且利用高度可变的台架11进行支撑。测定台13的贯穿孔13a与滴定管部1的旋塞22通过导管5连接。导管5的内径为6mm。
[0136]
在温度为25℃、湿度为60
±
10%的环境下进行测定。首先,关闭滴定管部1的旋塞22和旋塞24,且将已调节为25℃的生理盐水50从滴定管21上部的开口加入到滴定管21中。生理盐水的浓度(0.9质量%)为以nacl水溶液的质量为基准的浓度。用橡胶塞23密封滴定管21的开口之后,打开旋塞22及旋塞24。用0.9质量%的盐水50填满导管5内部以防止气泡进入。调节测定台13的高度,以使到达贯穿孔13a内的生理盐水的水面的高度与测定台13的上表面的高度相同。调节之后,由滴定管21的刻度读取滴定管21内的生理盐水50的水面的高度,并将该位置设为零点(0秒时的读取值)。
[0137]
在测定台13上的贯穿孔13a的附近铺上尼龙网片15(100mm
×
100mm,250目,厚度约为50μm),且在其中央部放置内径为30mm、高度为20mm的缸体(cylinder)。将1.00g的吸水性树脂颗粒10a均匀地散布于该缸体中。然后,小心取下缸体,获得了吸水性树脂颗粒10a以圆形分散在尼龙网片15的中央部的样品。接着,以吸水性树脂颗粒10a不会散逸的程度快速移动载置有吸水性树脂颗粒10a的尼龙网片15,以使其中心处在贯穿孔13a的位置,开始测定。将气泡最先从空气导入管25导入到滴定管21内的时刻作为开始吸水(0秒)。
[0138]
以0.1ml单位依次读取滴定管21内的生理盐水50的减少量(即,吸水性树脂颗粒10a所吸收的生理盐水的量),并读取了从吸水性树脂颗粒10a开始进行吸水起3分钟后的生理盐水50的减少量we(g)。根据we,并通过下述式求出了无加压dw的3分钟值。无加压dw为每1.00g吸水性树脂颗粒10a的吸水量。将结果示于表1。无加压dw的3分钟值(ml/g)=we/1.00
[0139]
<吸收体断裂性的测定>[评价用吸收性物品的制作]使用otec co.,ltd制造的pad former,通过空气抄造均匀地混合吸水性树脂颗粒10g及粉碎纸浆6.67g,制作了大小为40cm
×
12cm的吸收体。通过喷洒,向放置在大小为42cm
×
14cm、单位面积重量为16g/m2的纸巾上的上述吸收体上散布0.4g离子交换水,并在该吸收体上进一步放置40cm
×
12cm的纸巾来制作了层叠体。接着,将具有2mm孔径的大小为62cm
×
22cm的金属丝网放置在上述层叠体上,使用冲压机(小型空气式冲压机,imoto machinery co.,ltd)以0.141mpa的压力对层叠体进行了冲压。冲压后,从层叠体上取下了金属丝网。
[0140]
利用辊切机(roller cutter)从冲压后的层叠体的中心部沿长度方向分别切取15cm的距离,将长度设为30cm。利用单位面积重量为22g/m2、34cm
×
14cm的2片聚乙烯制透气型多孔透液片(以下,也称为“透液片”)夹住该层叠体,并利用热封机(fuji impulse sealer,型号:fi
‑
450
‑
5型,fuji impulse co.,ltd制造)压接夹住层叠体的2片透液片的4个边,从而获得了评价用吸收性物品100’。压接时,关于纸巾从吸收体中冒出的部分,利用透液片夹住冒出的纸巾并进行压接。
[0141]
[试验液的制备]向10l的容器中加入适量的蒸馏水,并添加氯化钠100g、氯化钙二水合物3.0g及氯化镁六水合物6.0g,进行了溶解。接着,添加聚氧乙烯壬基苯基醚0.25g,进一步追加蒸馏水,使整体质量成为10kg。此外,用少量的蓝色1号进行着色,由此制备了断裂性测定用试验液。
[0142]
[吸收体断裂时间的测定1]在温度为25
±
2℃的室内,利用图5所示的u字形仪器51(进深14cm,丙烯酸制)进行吸收体断裂时间的测定。u字形仪器51的开口宽度w为21cm、深度d为18.5cm,以开口部朝上的方式使用。通过在u字的底部放置14cm
×
6.5cm的厚纸52,形成了宽度6.5cm
×
进深14cm的平坦的底部。使评价用吸收性物品100’的中心与厚纸52的中央对齐,以使评价用吸收性物品100’的长边方向沿着u字形仪器51的曲线的方式,将评价用吸收性物品100’放置在u字形仪器51上。在评价用吸收性物品100’的中心放置内径2cm、外径3cm、高度6.5cm、质量60g的圆筒形缸体53,将已调节为25
±
1℃的80ml的上述试验液投入到缸体53内。在投入试验液的过程中,以将试验液的液面距离缸体底部的高度维持为约5cm的方式持续投入。
[0143]
投入到缸体53内的试验液均被评价用吸收性物品100’吸收后,从评价用吸收性物品100’上取下了缸体53。从开始投入试验液起3分钟后,再次将缸体53放置于放置在u字形仪器51上的评价用吸收性物品100’的中心,以与上述相同的方式投入80ml的试验液(第二次),使评价用吸收性物品100’对其进行吸收。
[0144]
从开始第一次投入试验液起6分钟后,在与评价用吸收性物品100’相同大小的厚纸(单位面积重量为3500g/m2)上放置评价用吸收性物品100’,并利用粘着胶带贴附长度方向的两端,由此将评价用吸收性物品100’固定在厚纸上。向具有3处用于抽出空气的1cm切口的带夹头的聚乙烯制透明袋(40cm
×
28cm,厚度0.04mm,uni
‑
pack,seisannipponsha ltd制造,k
‑
4)中,放入了被固定的评价用吸收性物品100’。向上述透明袋中放入评价用吸收性物品100’时,以袋的角与评价用吸收性物品100’的角对齐的方式放入,折叠袋的空余部分并用粘着胶带固定,以使整体的大小与评价用吸收性物品100’大致相同的方式进行了填充。
[0145]
从开始第一次投入试验液起11分钟后,以离心力27.5g(转速405r/分钟)对所填充的评价用吸收性物品100’进行离心。关于离心,通过如下方式进行:以使评价用吸收性物品
100’的表面与旋转轴呈直角,且旋转轴为评价用吸收性物品100’的中心的方式,用胶带将所填充的评价用吸收性物品100’固定在直径为30cm的转盘上。
[0146]
从开始离心至第4分钟为止,每1分钟中断离心一次,从开始离心起4分钟后,每2分钟中断离心一次,并肉眼确认了是否存在吸液后的吸收体的断裂。将直至观察到断裂为止的合计离心时间作为断裂时间。断裂时间越长,则能够判断为性能越良好。测定进行至20分钟,当在该过程中未确认到吸收体的断裂时,评价为“20分钟以上”。将结果示于表1。
[0147]
[吸收体断裂时间的测定2]在第二次投入试验液之后,进一步在3分钟之后以相同的方式投入80ml的试验液(第三次),将之后的操作各延迟3分钟来进行,除此以外,以与上述吸收体断裂时间的测定1相同的方式,对实施例中获得的吸收体的断裂性进行了评价。将结果示于表1。
[0148]
[表1]
[0149]
确认到:使用了通液维持率为一定以上的实施例的吸水性树脂颗粒的吸收体,其在测定1(投入2次试验液)中的断裂性测定中断裂时间为20分钟以上,抑制了吸液后的吸收体发生断裂。确认到:在条件更为严苛的测定2(投入3次试验液)中的断裂性测定中,实施例1~实施例3、实施例7、特别是实施例1及实施例7的断裂性进一步得到了抑制。附图标记说明
[0150]
1:滴定管部;3:夹具;5:导管;10:吸收体;10a:吸水性树脂颗粒;10b:纤维层;11:台架;13:测定台;13a:贯穿孔;15、63、64:尼龙网片;20a、20b:芯包片;21:滴定管;22:旋塞;23:橡胶塞;24:旋塞;25:空气导入管;30:透液性顶片;40:不透液性底片;50:生理盐水;51:u字形仪器;52:厚纸;53:圆筒形缸体;60:测定部;61:圆筒状容器;62:圆筒状小型容器;65:生理盐水;66:培养皿;67:金属丝网;100、100’:吸收性物品;200:搅拌叶片;200a:轴;200b:平板部;s:狭缝。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。