用于装载和排放氢气的基于n
‑
杂环的可逆液体有机系统、方法和过程
技术领域
1.本发明提供了一种按需储存氢气(h2)并释放氢气的系统、过程和方法,所述系统、过程和方法包括n
‑
杂环并且利用n
‑
杂环作为液体有机氢气载体(lohc)。
背景技术:
2.在过去的几个世纪期间,工业化过程为大部分人类带来了繁荣和财富。然而,与这些过程相关的一个基本障碍是化石资源的日益消耗以及废物和排放物的产生。这直接对环境造成不利影响,将来可能会严重威胁全球生活条件。因此,寻找替代性和可持续的能源系统来替代当前基于化石燃料的技术已成为社会的主要科学挑战之一。在此上下文中,长期以来,氢一直被认为是理想的替代性清洁能源载体,其具有极高的重量能量密度(较低的热值:33.3kwh/kg),并在燃烧时产生水作为唯一的副产物。氢的这些固有特性使其成为固定和移动应用两者中特别具有吸引力的候选物。
3.近来,在氢气供电的燃料电池方面已经取得了重大进展。尽管如此,作为能量载体的氢尚未得到普遍应用,这可能是由于与氢的储存和运输有关的问题。由于氢的体积能量密度低,因此氢的高效储存既至关重要又具有挑战性。传统上,氢在高压下物理地储存在气罐中,或者在低温温度下以液体形式储存。然而,储存所需的高能量输入、低体积能量密度以及潜在的安全问题在很大程度上限制了使用分子氢的应用。尽管已经进行了广泛努力来将以纳米结构化材料、金属有机框架和金属氢化物形式储存氢气,但是这些系统的氢气储存容量(hsc)低,条件严苛,能量效率低并且成本高。
4.近年来,以化学键形式储存氢气受到广泛关注,并且被视为未来“氢气经济”的有前途的途径。在此方面已经研究了几个种类金属氢化物、金属复合物和有机化合物。特别有趣的是液体有机化合物作为氢气载体,其氢气容量可能很高,接近2020年美国能源部(u.s.department of energy,美国doe,氢气储存容量为5.5wt%)和欧盟(european union,eu,氢气储存容量为5%)的目标。此外,液体有机氢气载体(lohc)具有良好的稳定性,可以长期储存且易于输送,并且可能适用于车辆中的板载使用。
5.lohc系统基于富氢有机液体的催化脱氢来形成贫h2化合物,所述贫h2化合物在催化氢化时可以重新产生富h2化合物。在早期研究中,以贫h2化合物和富h2化合物对的形式对芳香族烃和其氢化产物进行了研究。这些lohc系统的液体范围宽、热稳定性出色、价格低廉。然而,富h2烃脱氢以产生h2和对应的缺h2的芳香族烃在热力学上是不利的,其需要严苛的反应条件(温度高于250℃)。
6.基于n
‑
杂环的lohc系统中氮原子的存在可以降低氢化和脱氢的焓,因此已经对几种n
‑
杂环lohc系统进行了研究。最吸引人的实例之一是由pez和其同事开发的n
‑
乙基咔唑(nec)/十二氢
‑
n
‑
乙基咔唑(h
12
‑
nec)系统,所述系统的理论氢气储存容量(hsc)为5.80wt%(图1a)。然而,nec/h
12
‑
nec系统也有明显的缺点:反向氢化需要不同的催化剂ru/lialo2和高h2压力(68巴);贫h2化合物n
‑
乙基咔唑在室温下为固体(熔点70℃);并且n
‑
乙基的热不稳
定性导致不希望的副产物。
7.2017年,fujita和其同事报道了一种有趣的经均相ir复合物催化的lohc系统,所述系统基于含2,5
‑
二甲基吡嗪/2,5
‑
二甲基哌嗪的对二甲苯/h2o或是无溶剂的,其理论氢气储存容量为5.3wt%(图1b)。然而,在无溶剂条件下,在30巴的h2下未完全实现2,5
‑
二甲基吡嗪氢化为2,5
‑
二甲基哌嗪(78%转化率)。此外,2,5
‑
二甲基哌嗪的高熔点也是缺点。一些最近报道的基于n的杂芳香族/杂脂环lohc系统是2,6
‑
二甲基
‑
1,5
‑
萘啶/2,6
‑
二甲基十氢
‑
1,5
‑
萘啶(fujita等人,《美国化学会志(j.am.chem.soc.)》2014,136,4829
‑
4832)、吩嗪/十四氢吩嗪(forberg等人,《自然
‑
通讯(nat.commun.)》2016,7,13201)、4
‑
氨基哌啶/4
‑
氨基吡啶(cui等人,《新化学杂志(new j.chem.)》,2008,32,1027
‑
1037)、2,6
‑
二
‑
叔丁基哌啶/2,6
‑
二
‑
叔丁基吡啶(dean等人,《新化学杂志》,2011,35,417
‑
422)和2
‑
(n
‑
甲基苄基)吡啶/2
‑
[(n
‑
甲基环己基)甲基]哌啶(oh等人,《chemsuschem》2018,11,661
–
665)。然而,几乎所有这些都具有高熔点。此外,2,6
‑
二甲基
‑
1,5
‑
萘啶/2,6
‑
二甲基十氢
‑
1,5
‑
萘啶和吩嗪/十四氢吩嗪系统使用昂贵的催化剂和溶剂;4
‑
氨基哌啶/4
‑
氨基吡啶系统在平衡转化和选择性方面存在问题;6
‑
二
‑
叔丁基哌啶/2,6
‑
二
‑
叔丁基吡啶系统的氢气储存容量很低;2
‑
[(n
‑
甲基环己基)甲基]哌啶/2
‑
(n
‑
甲基苄基)吡啶的熔点低但需要高温(230℃)来进行脱氢。
[0008]
2015年,开发了基于脱氢化酰胺键形成的新lohc,这在热力学上是有利的但需要溶剂(hu等人,《自然
‑
通讯》2015,6,6859)。开发基于廉价有机液体(液体范围宽、熔点低)的氢气储存容量高、无溶剂的、在温和条件下理想地使用单一催化剂对氢化和脱氢两者进行催化的lohc系统具有重要意义并面临巨大挑战。
[0009]
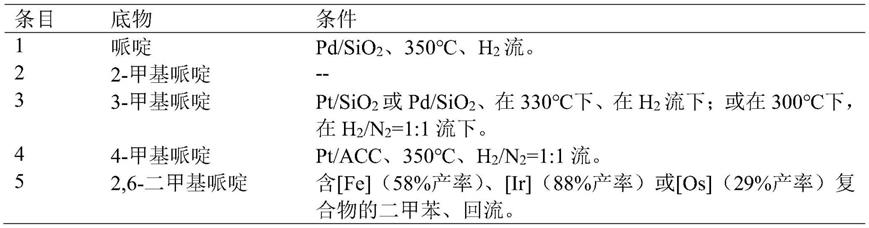
哌啶被视为lohc的潜在理想候选物,因为其充足且廉价、熔点低、液体范围宽、理论氢气储存容量高(超过us doe和欧盟的目标),如图1c所示。迄今为止,哌啶的无受体脱氢需要严苛的条件,如表1所列示的。
[0010]
表1.哌啶到吡啶的无受体脱氢.
[0011][0012]
哌啶(horrobin等人,us2765311a,1956)、3
‑
甲基哌啶(cordier等人,us4401819,1983;pianzola等人,us0092711a1,2011)和4
‑
甲基哌啶(patil等人,《国际氢能期刊(int.j.hydrogen energy.)》2017,42,16214
‑
16224)分别催化脱氢为吡啶、3
‑
甲基吡啶和4
‑
甲基吡啶需要高于300℃、使用非均相pd或pt催化剂(表1,条目1、3和4)。尚未报道2
‑
甲基哌啶无受体脱氢为2
‑
皮考啉(表1,条目2)。2,6
‑
二甲基哌啶催化脱氢为2,6
‑
二甲基吡啶仅在溶剂中通过均相fe(chakraborty等人,《美国化学会志》2014,136,8564
–
8567)、ir(fujita等人,《德国应用化学国际版(angew.chem.int.ed.)》2017,56,10886
–
10889)或os(esteruelas等人,《organometallics》2017,36,2996
‑
3004)催化剂(条目5)实现,并且尚未报道无溶剂系统。显然,使用溶剂会降低理论氢气储存容量。
[0013]
显然,开发储存和释放氢气的容量潜在地高的在相对温和的条件下理想地使用同一催化剂来进行氢气装载和排放两者的廉价且充足的有机化合物是一项重大挑战,目前尚无可接受的解决方案。因此,开发用于对哌啶/吡啶进行温和、无溶剂的、可逆的脱氢/氢化的合适催化系统不仅具有重要的理论意义还具有实际意义,并且高度期望基于哌啶的在温和条件下使用单一非均相催化剂以进行脱氢和氢化两者的、氢气储存容量高的lohc系统。
技术实现要素:
[0014]
因此,在本发明的第一方面,本发明提供了一种可逆的氢气装载和排放系统,所述系统包括:至少一种n
‑
杂环;以及一种过渡金属催化剂或过渡金属催化剂前体,其中所述系统不包括任何溶剂并且在温和温度和压力下起作用。在一些实施例中,温度介于50℃与180℃之间,压力介于1巴与80巴之间或其组合。在一些实施例中,温度介于130℃与180℃之间,压力介于1.5巴与8巴之间或其组合。在一些实施例中,n
‑
杂环是富h2化合物、贫h2化合物或其组合,并且其中所述富h2化合物是经取代的或未经取代的哌啶,并且所述贫h2化合物是经取代的或未经取代的吡啶。在一些实施例中,富h2化合物是经取代的哌啶,而贫h2化合物是经取代的吡啶。在一些实施例中,n
‑
杂环至少在介于15℃与100℃之间的温度下为液体。在一些实施例中,所述经取代的或未经取代的哌啶和所述经取代的或未经取代的吡啶两者在室温下均为液体。在一些实施例中,所述经取代的哌啶是2,6
‑
二甲基哌啶或2
‑
甲基哌啶。在一些实施例中,所述过渡金属催化剂是钯/活性炭(pd/c)。
[0015]
在一些实施例中,所述催化剂可商购获得或由催化剂前体原位产生。在一些实施例中,所述催化剂前体是pd(oac)2。在一些实施例中,同一催化剂用于氢气装载(氢化)和氢气排放(脱氢)过程两者。在一些实施例中,所述系统进一步包括催化量的至少一种酸。在一些实施例中,所述酸选自:乙酸、苯甲酸、羧基聚苯乙烯和聚丙烯酸。在一些实施例中,所述催化剂或所述催化剂前体以介于0.05w/w%到5w/w%之间的量存在。
[0016]
在本发明的第二方面,本发明提供了一种用于按需储存并释放氢气(h2)的可逆方法,所述方法包括以下步骤:
[0017]
a.当期望储存氢气时,在存在第一催化剂的情况下,使经取代的或未经取代的吡啶与分子氢气(h2)在足以产生经取代的或未经取代的哌啶衍生物的条件下反应;以及
[0018]
b.当期望释放氢气时,使经取代的或未经取代的哌啶与第二催化剂在足以释放氢气的条件下反应,从而产生对应的经取代的或未经取代的吡啶衍生物和分子氢气(h2);
[0019]
其中所述第一催化剂和所述第二催化剂相同,所述方法不包括任何溶剂,并且所述方法在温和温度和氢气压力下执行。在一些实施例中,所述经取代的/未经取代的哌啶和所述经取代的/未经取代的吡啶两者在室温下仅为液体。在一些实施例中,经取代的哌啶选自:哌啶、2
‑
甲基哌啶、3
‑
甲基哌啶、4
‑
甲基哌啶、3,4
‑
二甲基哌啶、2,4
‑
二甲基哌啶、2,5
‑
二甲基哌啶和2,6
‑
二甲基哌啶。在一些实施例中,所述催化剂是钯/活性炭(pd/c)。在一些实施例中,所述催化剂可商购获得或由催化剂前体原位产生。在一些实施例中,所述催化剂前体是pd(oac)2。在一些实施例中,所述方法进一步包括至少一种酸。在一些实施例中,所述酸选自:乙酸、苯甲酸、羧基聚苯乙烯和聚丙烯酸。在一些实施例中,所述催化剂前体以介于0.05w/w%到5w/w%之间的量存在。
附图说明
[0020]
本说明书的结论部分中特别指出并明确主张了被视为本发明的主题。然而,当与附图一起阅读时,可以通过参考以下具体实施方式最佳地理解本发明的组织和操作方法以及其目的、特征和优点,在附图中:
[0021]
图1a
‑
1f描绘了基于含氮有机化合物的氢气储存系统。图1a:基于n
‑
乙基咔唑/十二氢
‑
n
‑
乙基咔唑的良好确立的液体有机氢气载体(pez等人)。图1b:基于含2,5
‑
二甲基吡嗪/2,5
‑
二甲基哌嗪的对二甲苯/h2o的或无溶剂的经均相ir复合物催化的lohc系统(fujita等人)。图1c
‑
1d:液体有机氢气载体的实例。图1e:本发明的适用于氢气储存的吡啶/哌啶化合物的实例。图1f:根据本发明的无溶剂lohc系统。
[0022]
图2a
‑
2b描绘了通过pd(oac)2/活性炭催化的2
‑
甲基哌啶的脱氢过程。图2a:2
‑
甲基哌啶脱氢的条件和结果。图2b:不同条件下的时间依赖性h2释放曲线。
[0023]
图3描绘了实验中利用的气体收集系统的示意图。
[0024]
图4描绘了本发明的通过pd催化剂催化的2
‑
甲基哌啶和2
‑
皮考啉的可逆相互转化方法。
[0025]
图5a
‑
5b描绘了本发明的系统/过程的副产物分析和选择性控制策略。图5a:建议副反应和副产物。图5b:用于通过增加亚胺或烯胺周围的位阻来控制过程的选择性的建议策略。
[0026]
图6a
‑
6b描绘了经钯基催化剂催化的2,6
‑
二甲基哌啶和2,6
‑
二甲基吡啶的相互转化。图6a:170℃(浴温,内部温度为128℃)下的时间依赖性h2释放曲线。图6b:脱氢(浅灰色)和氢化(深灰色)的转化。
[0027]
图7a
‑
7b描绘针对机械研究执行的对照实验。图7a:用pd(oac)2和活性炭(非均相)进行的脱氢。图7b:对固体进行过滤以检查均相催化。图7c:无活性炭载体的pd(oac)2催化剂。
[0028]
图8描绘了添加酸对脱氢过程的影响。右图描绘了每个实验的90%产率内的平均周转频率(atof
90
)(mol h2每mol pd每小时)。左图描绘了通过pd/c催化的2,6
‑
二甲基哌啶脱氢的时间依赖性h2释放曲线(具有和不具有其它添加剂。实验条件:[pd](0.3mol%)、酸(0.6mol%)、2,6
‑
二甲基哌啶(10mmol)、油浴(170℃)。(1)在无另外的酸的情况下反应(空心方块
□
)。(2)与乙酸反应(实心灰色三角
▲
)。(3)与ba反应:苯甲酸(实心黑色方块
■
)。(4)与对
‑
tsoh反应:4
‑
甲基苯磺酸实心灰色方块
■
)。5)与cps:羧基聚苯乙烯反应(实心点
●
)。(6)与paa:聚丙烯酸反应(空心三角
△
)。(7)与pd(oac)2和活性炭反应(空心点
○
)。
[0029]
图9a
‑
9b描绘了可信的脱氢机制(图9a);以及2
‑
皮考啉和2,6
‑
二甲基吡啶形成的建议途径(图9b)。虚线表示存在单键或双键。
[0030]
图10描绘了2
‑
甲基哌啶脱氢的时间依赖性h2释放曲线。
[0031]
图11描绘了pd(oac)2和活性炭浓度对脱氢活性的影响。条件:2,6
‑
二甲基哌啶(10mmol)、170℃(油浴温度)。
[0032]
图12描绘了当如实例7中例示的重复使用催化剂时2,6
‑
二甲基哌啶作为lohc系统提供2,6
‑
二甲基哌啶和2,6
‑
二甲基吡啶的可逆相互转化的催化剂转化率百分比。
[0033]
应当理解,为了说明的简单和清楚起见,图中所示的元件不一定按比例绘制。例如,为了清楚起见,一些元件的尺寸可能相对于其它元件被夸大。进一步地,在认为适当的
情况下,可以在附图中重复附图标记以指示相应或相似的元件。
具体实施方式
[0034]
在以下具体实施方式中,陈述了许多具体细节以便提供对本发明的透彻理解。然而,本领域的技术人员将理解的是,可以在不存在这些具体细节的情况下实践本发明。在其它情况下,并未详细描述众所周知的方法、程序和组分,以免模糊本发明。
[0035]
如本文所设想的,已经开发了一种可以装载和卸载氢气储存容量潜在地高的h2的基本上新的、可逆的系统。
[0036]
因此,本发明提供了一种用于基于富氢n
‑
杂环与至少一种过渡金属催化剂反应以形成相应贫氢n
‑
杂环和至少一个、至少两个或至少三个氢分子(针对富氢n
‑
杂环的每个分子)来按需储存氢气(h2)并释放氢气的方法、过程和系统;每一个表示根据本发明的单独实施例。在一些实施例中,催化剂是钯/活性炭(pd/c)。在其它实施例中,催化剂(用于氢化和脱氢)是pd/c、pd(oac)2活性炭。在一些实施例中,用于氢化的催化剂是ru/al2o3。在一些实施例中,系统不包括任何溶剂。在一些实施例中,氢气储存/装载在温和温度(例如,介于50
‑
200℃之间)执行。在一些实施例中,氢气储存/装载在温和氢气压力(例如,介于1.5巴与8巴之间)下执行。在一些实施例中,氢气释放/卸载在温和压力(例如,大气压)下执行。在一些实施例中,氢气释放/卸载在温和温度(例如介于100
‑
200℃之间)下执行。在其它实施例中,在用于储存并释放氢气(h2)的可逆方法中重复使用同一催化剂(照现状)。在其它实施例中,在氢气(h2)的储存和/或释放过程中再循环并使用同一催化剂。
[0037]
在装载氢分子时,贫氢n
‑
杂环与至少一个氢分子反应从而形成富氢n
‑
杂环。优选地,贫氢n
‑
杂环是经取代的或未经取代的吡啶,并且富氢n
‑
杂环是经取代的或未经取代的哌啶。在另一实施例中,贫氢n
‑
杂环是经取代的吡啶,并且富氢n
‑
杂环是经取代的哌啶。
[0038]
相应地,本发明进一步提供了一种用于基于经取代的/未经取代的吡啶的氢化和对应的经取代的/未经取代的哌啶液体
‑
有机氢气载体(lohc)的脱氢按需储存氢气(h2)并释放氢气的方法、过程和系统。更具体地,本发明涉及经取代的/未经取代的哌啶lohc。本发明的用于储存氢气的过程的氢气储存容量潜在地高。
[0039]
图1a
‑
1f呈现了用于在n
‑
杂环中释放和装载氢分子的过程和方法中的一些的实例。图1f呈现了本文所述的过程和方法的实例。
[0040]
富h
2 n
‑
杂环(例如,经取代的/未经取代的哌啶)经历用于形成对应的贫h
2 n
‑
杂环(例如,经取代的/未经取代的吡啶)的催化脱氢与氢气释放。贫h
2 n
‑
杂环(例如,经取代的/未经取代的吡啶)可以氢化回起到氢气储存系统的作用的对应的富h
2 n
‑
杂环(例如,经取代的/未经取代的哌啶)。这些反应可以由多种催化系统在有或没有聚合物和/或不溶性基质载体的情况下催化,所述催化系统包含过渡金属、基于过渡金属的化合物和复合物、非均相和均相催化剂。合适的催化剂的实例是钯(pd)、铂(pt)、钌(ru)、铜(cu)、铁(fe)以及含有这些金属的化合物和复合物等,优选地钯(pd)、铂(pt)和钌(ru)以及含有这些金属的化合物和复合物。优选地,催化剂是非均相催化剂,如pd/c、pd(oac)2/活性炭、ru/al2o3。在一些实施例中,催化剂相反通过使用催化剂前体原位形成,所述催化剂前体可溶(均相的)或不可溶(非均相的)于所述系统、过程或方法的反应混合物中。在一些实施例中,催化剂前体在进入反应混合物中时变为活性催化剂和/或变得与n
‑
杂环底物(或者富h2底物或贫h2底物)
或h2相互作用。在一些实施例中,所述催化剂前体是pd(oac)2。
[0041]
在一个实施例中,“过渡金属催化剂前体”是指可以原位转换为可用于本发明目的的活性催化剂的过渡金属化合物。过渡金属催化剂前体的实例包含但不限于:pd(oac)2、pdcl2、pd(tfa)2、pd(acac)2和pd2(dba)3、pt(oac)2、pt(tfa)2、pt(acac)2、ptcl2、pto2、ru(oac)2、rucl3。在一些实施例中,在进入包括n
‑
杂环和固体载体(例如活性炭)的反应混合物时,pd(oac)2催化剂前体原位转换为负载在固体载体(例如,pd/c)上的钯。在其它实施例中,当反应混合物加热到约100
–
200c的温度时,pd(oac)2催化剂前体原位转换为负载在固体载体上的钯(例如pd/c)。
[0042]
在其它实施例中,在用于储存并释放氢气(h2)的可逆方法中重复使用同一催化剂(照现状)。在其它实施例中,在氢气(h2)的储存和/或释放过程中再循环并使用同一催化剂。
[0043]
在一个实施例中,催化剂“在用于氢气(h2)的储存和释放的可逆方法中被重复使用(照现状)”是指其中氢气的储存和释放按需发生,并且在储存步骤和释放步骤两者中在不进行任何另外的处理的情况下使用同一催化剂的过程或系统。
[0044]
在一个实施例中,催化剂“在氢气(h2)的储存和/或释放过程中被再循环并使用”是指其中氢气的储存和释放按需发生,并且催化剂被再循环(即催化剂通过例如在惰性气氛下离心或过滤而被分离出来)并按需再次使用的过程或系统。
[0045]
因此,在一些实施例中,本发明涉及经富h2取代的n
‑
杂环,例如2
‑
甲基哌啶和/或2,6
‑
二甲基哌啶作为用于按需储存并释放氢气(h2)的液体有机氢气载体(lohc)的用途。
[0046]
在另一实施例中,本发明涉及一种用于释放氢气(h2)的方法,所述方法包括以下步骤:使经取代的/未经取代的哌啶衍生物(例如,2
‑
甲基哌啶或2,6
‑
二甲基哌啶)与催化剂(例如,pd/c、pd(oac)2/活性炭)在足以释放氢气的条件下反应,从而产生对应的经取代的/未经取代的吡啶衍生物(例如,2
‑
甲基吡啶(2
‑
皮考啉)或2,6
‑
二甲基吡啶(2,6
‑
二甲基吡啶))和分子氢气(h2)。在一些实施例中,所述方法用催化量的酸执行。在一些实施例中,所述方法在温和温度(例如,介于100
‑
200℃之间)下执行。在一些实施例中,所述方法在温和压力(例如,大气压)下执行。在一些实施例中,所述方法不包括任何溶剂。
[0047]
在本发明的上下文中,术语“释放”、“排放”和“卸载”可互换使用并且对应于通过化合物的催化脱氢过程由富氢有机化合物产生氢分子。
[0048]
在另一实施例中,本发明涉及一种用于储存氢的方法,所述方法包括以下步骤:在存在催化剂(例如,pd/c、pd(oac)2/活性炭、ru/al2o3)的情况下,使经取代的/未经取代的吡啶衍生物(例如,2
‑
甲基吡啶(2
‑
皮考啉)或2,6
‑
二甲基吡啶(2,6
‑
二甲基吡啶))与分子氢气(h2)在足以产生经取代的/未经取代的哌啶衍生物(例如,2
‑
甲基哌啶或2,6
‑
二甲基哌啶)作为氢气储存系统的条件下反应。在一些实施例中,所述方法用催化量的酸执行。在一些实施例中,所述方法在温和温度(例如,介于100
‑
200℃之间)下执行。在一些实施例中,所述方法在温和氢气压力(例如,介于1.5巴与8巴之间)下执行。在一些实施例中,所述方法不含有任何溶剂。
[0049]
在本发明的上下文中,术语“储存”、“装料”和“装载”可互换使用并且对应于通过化合物的催化氢化过程将氢分子掺入到贫氢有机化合物中。
[0050]
在另一实施例中,本发明涉及一种按需储存氢气(h2)并释放氢气的方法,所述方
法包括以下步骤:(a)当期望储存氢气时,在存在第一催化剂(例如,pd/c、pd(oac)2/活性炭、ru/al2o3)的情况下,使经取代的/未经取代的吡啶衍生物(例如,2
‑
甲基吡啶(2
‑
皮考啉)或2,6
‑
二甲基吡啶(2,6
‑
二甲基吡啶))与分子氢气(h2在足以产生经取代的/未经取代的哌啶衍生物(例如,2
‑
甲基哌啶或2,6
‑
二甲基哌啶)的条件下反应;以及(b)当期望释放氢气时,使经取代的/未经取代的哌啶衍生物(例如,2
‑
甲基哌啶或2,6
‑
二甲基哌啶)与第二催化剂(例如,pd/c、pd(oac)2/活性炭)在足以释放氢气的条件下反应,从而产生对应的经取代的/未经取代的吡啶衍生物(例如,2
‑
甲基吡啶(2
‑
皮考啉)或2,6
‑
二甲基吡啶(2,6
‑
二甲基吡啶))和分子氢气(h2)。所述第一催化剂和所述第二催化剂可以相同或不同。在一个优选实施例中,所述第一催化剂和所述第二催化剂相同。在其它实施例中,在用于储存并释放氢气(h2)的可逆方法中重复使用同一催化剂(照现状)。在其它实施例中,在氢气(h2)的储存和/或释放过程中再循环并使用同一催化剂。
[0051]
在一些实施例中,所述方法用催化量的酸执行。在一些实施例中,所述方法在温和温度(例如,介于100
‑
200℃、130
‑
180℃、150
‑
170℃之间)下执行。在一些实施例中,用于储存氢气的方法在温和氢气压力(例如,介于1.5巴与8巴之间)下执行。在一些实施例中,用于释放氢气的方法在温和压力(例如,大气压)下执行。在一些实施例中,所述方法不包括任何溶剂。
[0052]
本发明提供了一种可逆氢气装载和排放系统,所述系统包括:至少一种n
‑
杂环;以及至少一种过渡金属催化剂或过渡金属催化剂前体。
[0053]
本发明提供了一种可逆氢气装载和排放系统,所述系统包括:至少一种n
‑
杂环;以及一种过渡金属催化剂或过渡金属催化剂前体。
[0054]
本发明进一步提供了一种可逆氢气装载和排放方法,所述方法包括以下步骤:
[0055]
(a)氢气释放过程:使至少一种经取代的n
‑
杂环富h2化合物与至少一种过渡金属催化剂在允许经取代的n
‑
杂环富h2化合物脱氢的条件下接触,从而形成三个氢分子和至少一种经取代的n
‑
杂环贫h2化合物;
[0056]
(b)氢气装载过程:使所述至少一种经取代的n
‑
杂环贫h2化合物与所述至少一种过渡金属催化剂和三个氢分子在所述经取代的n
‑
杂环贫h2化合物氢化的条件下接触,由此形成至少一种经取代的n
‑
杂环h2化合物。
[0057]
本发明进一步提供了一种可逆氢气装载和排放方法,所述方法包括以下步骤:
[0058]
(c)氢气释放过程:使至少一种经取代的/未经取代的哌啶与至少一种过渡金属催化剂在允许经取代的/未经取代的哌啶脱氢的条件下接触,从而形成三个氢分子和至少一种经取代的吡啶;
[0059]
(d)氢气装载过程:使所述至少一种经取代的/未经取代的吡啶与所述至少一种过渡金属催化剂和三个氢分子在所述经取代的/未经取代的吡啶氢化的条件下接触,从而形成至少一种经取代的哌啶。
[0060]
在一些实施例中,根据本发明的系统、过程和/或方法中的n
‑
杂环是富h2化合物。在其它实施例中,富h2化合物是经取代的/未经取代的哌啶。在其它实施例中,富h2化合物是经取代的哌啶。在其它实施例中,n
‑
杂环是贫h2化合物。在其它实施例中,贫h2化合物是经取代的/未经取代的吡啶。在其它实施例中,贫h2化合物是经取代的吡啶。在其它实施例中,n
‑
杂环是富h2化合物和贫h2化合物的组合。在其它实施例中,n
‑
杂环是经取代的/未经取代的
吡啶和经取代的/未经取代的哌啶的组合。
[0061]
在一些实施例中,术语“经取代的哌啶”和/或“经取代的吡啶”是指具有环取代的哌啶或吡啶衍生物,所述环取代包含但不限于选自以下的至少一种:c1‑
c3烷基(例如,甲基、乙基、丙基)、c1卤代烷基(例如,cf3、chf2、ch2f)、c1‑
c2烷氧基(och3、och2ch3)、f、cl、oh、nh2、n(ch3)2、cn、芳基、苯基。在其它实施例中,所述取代为选自以下的至少一种:ch3、oh、och3、nh2。在其它实施例中,取代为至少一个ch3。在其它实施例中,取代为两个ch3基团。
[0062]
在一些实施例中,n
‑
杂环富h2化合物选自:哌啶、2
‑
甲基哌啶、3
‑
甲基哌啶、4
‑
甲基哌啶、2,6
‑
二甲基哌啶、2,4
‑
二甲基哌啶、2,3
‑
二甲基哌啶、2,5
‑
二甲基哌啶、3,4
‑
二甲基哌啶、3,5
‑
二甲基哌啶、2,5
‑
二甲基哌啶、2,4,6
‑
三甲基哌啶或其组合;每一个表示根据本发明的单独实施例。在一些实施例中,n
‑
杂环富h2化合物是哌啶。在一些实施例中,n
‑
杂环富h2化合物是2
‑
甲基哌啶。在一些实施例中,n
‑
杂环富h2化合物是2,6
‑
二甲基哌啶。在一些实施例中,n
‑
杂环富h2化合物是哌啶。在一些实施例中,n
‑
杂环富h2化合物是3
‑
甲基哌啶。在一些实施例中,n
‑
杂环富h2化合物是4
‑
甲基哌啶。在一些实施例中,n
‑
杂环富h2化合物是2,4
‑
二甲基哌啶。在一些实施例中,n
‑
杂环富h2化合物是2,3
‑
二甲基哌啶。在一些实施例中,n
‑
杂环富h2化合物是2,5
‑
二甲基哌啶。在一些实施例中,n
‑
杂环富h2化合物是3,4
‑
二甲基哌啶。在一些实施例中,n
‑
杂环富h2化合物是3,5
‑
二甲基哌啶。在一些实施例中,n
‑
杂环富h2化合物是2,5
‑
二甲基哌啶。在一些实施例中,n
‑
杂环富h2化合物是2,4,6
‑
三甲基哌啶。
[0063]
在一些实施例中,n
‑
杂环贫h2化合物选自:吡啶、2
‑
甲基吡啶(2
‑
皮考啉)、3
‑
甲基吡啶、4
‑
甲基吡啶、2,6
‑
二甲基吡啶(2,6
‑
二甲基吡啶)、2,4
‑
二甲基吡啶、2,3
‑
二甲基吡啶、2,5
‑
二甲基吡啶、3,4
‑
二甲基吡啶、3,5
‑
二甲基哌啶、2,5
‑
二甲基吡啶、2,4,6
‑
三甲基吡啶或其组合;每一个表示根据本发明的单独实施例。在一些实施例中,n
‑
杂环贫h2化合物为吡啶。在一些实施例中,n
‑
杂环贫h2化合物为2
‑
甲基吡啶(2
‑
皮考啉)。在一些实施例中,n
‑
杂环贫h2化合物为2,6
‑
二甲基吡啶(2,6
‑
二甲基吡啶(lutidine))。在一些实施例中,n
‑
杂环贫h2化合物为吡啶。在一些实施例中,n
‑
杂环贫h2化合物为3
‑
甲基吡啶。在一些实施例中,n
‑
杂环贫h2化合物为4
‑
甲基吡啶。在一些实施例中,n
‑
杂环贫h2化合物为2,4
‑
二甲基吡啶。在一些实施例中,n
‑
杂环贫h2化合物为2,3
‑
二甲基吡啶。在一些实施例中,n
‑
杂环贫h2化合物为2,5
‑
二甲基吡啶。在一些实施例中,n
‑
杂环贫h2化合物为3,4
‑
二甲基吡啶。在一些实施例中,n
‑
杂环贫h2化合物为3,5
‑
二甲基哌啶。在一些实施例中,n
‑
杂环贫h2化合物为2,5
‑
二甲基吡啶、2,4,6
‑
三甲基吡啶。
[0064]
在一些实施例中,n
‑
杂环的液体范围宽。
[0065]
根据本发明,物质的“液体范围”是指允许液体存在的温度的可能范围。其液体范围的值是通过从其沸点减去熔点而获得的。如果沸点高于其熔点,则液体范围据说为正;如果沸点低于其熔点,则液体范围为负,这意味着液体不可能形成,除非施加在物质上的压力上升。在根据本发明的一些实施例中,“宽液体范围”是指温度范围在物质的熔点与沸点之间为至少80度。在其它实施例中,物质的熔点与沸点之间为至少90、100、110、120、130、140、150、160、170、180、190、200、220、250、300度;每一个表示根据本发明的单独实施例。
[0066]
在一些实施例中,n
‑
杂环的熔点低于
‑
10℃、低于
‑
5℃、低于0℃、低于5℃、低于10℃、低于15℃、低于20℃、低于25℃、低于30℃、低于35℃、低于40℃、低于45℃、低于50℃、低于55℃;每一个表示根据本发明的单独实施例。
[0067]
在一些实施例中,n
‑
杂环的沸点高于50℃、高于70℃、高于90℃、高于100℃、高于120℃、高于130℃、高于150℃、高于170℃、高于200℃、高于230℃、高于250℃、高于300℃;每一个表示根据本发明的单独实施例。
[0068]
在一些实施例中,n
‑
杂环在至少以下温度下为液体:介于15℃与200℃之间;介于0℃与250℃之间;介于10℃与180℃之间;介于25℃与250℃之间;介于0℃与300℃之间;介于5℃与200℃之间;介于5℃与250℃之间;介于15℃与150℃之间;介于5℃与150℃之间;介于5℃与170℃之间;介于
‑
10℃与250℃之间;每一个表示根据本发明的单独实施例。
[0069]
在一些实施例中,贫h
2 n
‑
杂环和富h
2 n
‑
杂环在室温下为液体。在一些实施例中,经取代的/未经取代的吡啶和经取代的/未经取代的哌啶两者在室温下均为液体。
[0070]
在一些实施例中,根据本发明的系统、过程或方法在以下温度范围下起作用:介于约130℃与约180℃之间、介于约50℃与约180℃之间、介于约100℃与约180℃之间、介于约100℃与约250℃之间、介于约140℃与约180℃之间、介于约100℃与约200℃之间、介于约130℃与约220℃之间、介于约150℃与约170℃之间、介于约130℃与约200℃;每一个是根据本发明的单独实施例。在一些实施例中,根据本发明的系统、过程或方法在以下温度下起作用:170℃、150℃、119℃、117℃、or 130℃;每一个是根据本发明的单独实施例。
[0071]
在一些实施例中,在根据本发明的系统、过程或方法中执行的氢化过程在低氢气压力下发生。在一些实施例中,在根据本发明的系统、过程或方法中执行的氢化过程在以下氢气压力下发生:介于约1巴与约80巴之间、介于约15巴与约60巴之间、介于约1.5巴与约59巴之间、介于约1巴与约59巴之间、介于约4巴与约50巴之间、介于约1.3巴与约50巴之间、介于约1.5巴与约20巴之间、介于约1.5巴与约10巴之间、介于约1.5巴与约8巴之间、介于约1.2巴与约6巴之间、介于约1巴与约8巴之间、介于约1.6巴与约5巴之间、介于约2.6巴与约5巴之间、介于约1.2与约8巴之间、介于约30巴与约50巴之间、介于约3.5巴与约5巴之间、介于约2.5巴与约5巴之间、介于约1.5巴与约5巴之间、介于约1.5与约7巴之间;每一个表示根据本发明的单独实施例。在一些实施例中,在根据本发明的系统、过程或方法中执行的氢化过程在以下氢气压力下执行:1.0巴、1.1巴、1.2巴、1.3巴、1.4巴、1.5巴、1.6巴、1.7巴、1.8巴、1.9巴、2.0巴、2.5巴、3.0巴、3.5巴、4.0巴、4.5巴、5.0巴、5.5巴、6.0巴、6.5巴、7.0巴、7.5巴或8.0巴;每一个表示根据本发明的单独实施例。
[0072]
在一些实施例中,过渡金属催化剂在本发明的系统、过程或方法中在氢气释放和氢气装载反应中相同。在一些实施例中,对氢气装载(氢化)和氢气排放(脱氢)过程两者使用同一催化剂。
[0073]
在其它实施例中,在用于储存并释放氢气(h2)的可逆方法中重复使用同一催化剂(照现状)。在其它实施例中,在氢气(h2)的储存和/或释放过程中再循环并使用同一催化剂。
[0074]
在一些实施例中,过渡金属选自mn、fe、co、ni、ru、rh、pd、cu、ag。在一些实施例中,过渡金属选自ru、pd和pt。在一些实施例中,过渡金属催化剂是非均相的。在一些实施例中,过渡金属催化剂是均相的。在一些实施例中,所述过渡金属催化剂是钯/活性炭(pd/c)。在一些实施例中,过渡金属催化剂是pd(oac)2。在一些实施例中,过渡金属催化剂是ru/al2o3。在一些实施例中,催化剂可商购获得。可商购获得的催化剂的实例包含但不限于:5wt%pt/c、10wt%pt/c、1wt%pt/c、3wt%pt/c、30wt%pt/c、0.5wt%pt/al2o3、1wt%pt/al2o3、
4wt%pd/mcm
‑
48、5wt%pd/sio2、0.5wt%pd/al2o3、1wt%pd/al2o3、5wt%pd/al2o3、10wt%pd/al2o3、0.6wt%pd/c、1wt%pd/c、3wt%pd/c、5wt%pd/c、10wt%pd/c、20wt%pd/c、30wt%pd/c、5wt%pd/baso4、10wt%pd/baso4、5wt%pd/baco3、5wt%pd/caco3、10wt%pd/caco3、5wt%pd/srco3、5wt%pd/tio2、5wt%ru/c、5wt%ru/al2o3等。在一些实施例中,催化剂由催化剂前体原位产生。催化剂前体的实例包含但不限于:pd(oac)2、pdcl2、pd(tfa)2、pd(acac)2和pd2(dba)3。在一些实施例中,催化剂负载在如无机氧化物(例如,氧化铝或二氧化硅,任选地通过系链连接)等不溶性基质(或固体载体,如以下所述)或有机不溶性聚合物(例如交联聚苯乙烯)上。不溶性基质的更多实例包含但不限于:活性炭、干式酸性活性炭、sio2、baso4、bn、γ
‑
al2o3或ceo2。在一些实施例中,本发明的系统、方法和/或过程中的活性催化剂是钯/碳或pd/c,其中钯金属负载在活性炭上,以最大化其表面积和活性。
[0075]
在一些实施例中,按富h2化合物计,各自表示根据本发明的单独实施例的催化剂或催化剂前体以以下量存在:介于0.05w/w%与5w/w%之间、介于0.01w/w%与1w/w%之间、介于0.1w/w%与1w/w%之间、介于0.15w/w%与0.5w/w%之间、介于0.1w/w%与0.7w/w%之间、介于0.1w/w%与0.5w/w%之间、介于0.05w/w%与0.5w/w%、介于0.15w/w%与0.3w/w%之间、0.15w/w%、0.2w/w%、0.3w/w%;每一个表示根据本发明的单独实施例。
[0076]
在一些实施例中,按贫h2化合物计,各自表示根据本发明的单独实施例的催化剂或催化剂前体以以下量存在:介于0.05w/w%与5w/w%之间、介于0.01w/w%与1w/w%之间、介于0.1w/w%与1w/w%之间、介于0.15w/w%与0.5w/w%之间、介于0.1w/w%与0.7w/w%之间、介于0.1w/w%与0.5w/w%之间、介于0.05w/w%与0.5w/w%之间、介于0.15w/w%与0.3w/w%之间、0.15w/w%、0.2w/w%、0.3w/w%;每一个表示根据本发明的单独实施例。
[0077]
在一些实施例中,如本文所描述的本发明的实施例中的任何实施例的过程/方法在纯条件下进行。在一些实施例中,本发明的所述系统、过程和/或方法不包括任何溶剂。在一些实施例中,本发明的所述系统和方法进一步包括至少一种有机溶剂。在一些实施例中,所述至少一种有机溶剂选自苯、甲苯、邻二甲苯、间二甲苯或对二甲苯、均三甲苯(1,3,5
‑
三甲基苯)、二噁烷、thf、dme、dmso、二甘醇二甲醚、dmf(二甲基甲酰胺)、戊腈、dmac(二甲基乙酰胺)、nmm(n
‑
甲基吗啉)、吡啶、n
‑
bucn、苯甲醚、环己烷和其组合。在一些实施例中,本发明的所述系统和方法进一步包括一种有机溶剂。在其它实施例中,本发明的所述系统和方法进一步包括至少两种有机溶剂的混合物。
[0078]
在另一个实施例中,催化剂被吸收在固体载体上,并且在没有溶剂的情况下完成氢气的储存/装载和释放/排放。
[0079]
在一些实施例中,本发明的所述系统和/或方法进一步包括至少一种酸。可以包含在本发明的系统、方法和/或方法中的酸的实例包含但不限于:乙酸(hoac)、苯甲酸(ba)、羧基聚苯乙烯(cps)、聚丙烯酸(paa)、4
‑
甲基苯磺酸(p
‑
tsoh)和其混合物。在一些实施例中,酸以催化量存在。在一些实施例中,按n
‑
杂环(例如,经取代的/未经取代的吡啶或经取代的/未经取代的哌啶)的量计,酸以以下量存在:0.01w/w%、0.05w/w%、0.1w/w%、0.2w/w%、0.3w/w%、0.4w/w%、0.5w/w%、0.6w/w%、0.7w/w%、0.8w/w%、0.9w/w%、1w/w%、5w/w%、10w/w%、15w/w%、25w/w%、35w/w%;每一个表示根据本发明的单独实施例。在一些实施例中,酸是弱酸。
[0080]
在一些实施例中,氢气的排放通过以下实现:使经取代的/未经取代的哌啶与过渡
金属催化剂反应;由此形成三个氢分子和经取代的吡啶。
[0081]
在一些实施例中,氢气的装载通过以下实现:使经取代的/未经取代的吡啶与三个氢分子和过渡金属催化剂反应;由此形成经取代的/未经取代的哌啶。
[0082]
在一些实施例中,本发明的可逆氢气装载和排放系统、方法和/或方法的氢气储存容量为至少4%、至少5%、至少5.2%、至少5.3%、至少5.5%、至少6%、至少6.1%;每一个表示根据本发明的单独实施例。在其它实施例中,本发明的可逆氢气装载和排放系统、方法和/或方法的氢气储存容量介于约4%到约6.5%之间、介于约4%到约6.3%之间、介于约5%到约6.5%之间、介于约5.3%到约6.1%之间、介于约4%到约6.1%之间;每一个表示根据本发明的单独实施例。
[0083]
利用本发明的经取代的/未经取代的哌啶作为lohc的反应途径在图1d中概述,所述图描述了基于n
‑
杂环(经取代的/未经取代的哌啶/吡啶)的pd催化的脱氢和氢化的lohc系统。发现两种贫h2/富h2化合物对是供未来使用的有前途的lohc。一种是2
‑
皮考啉/2
‑
甲基哌啶系统,其理论氢气储存容量为6.1wt%,这很好地满足了us doe板载氢气储存目标2020。另一种是2,6
‑
二甲基吡啶/2,6
‑
二甲基哌啶系统,去理论氢气储存容量为5.3wt%,接近us doe目标。所有化合物具有宽液体范围和低于0℃的熔点。脱氢和氢化两者均是在温和条件下使用同一催化剂以优异产率实现的。具体地,对于2,6
‑
二甲基吡啶/2,6
‑
二甲基哌啶系统,2,6
‑
二甲基哌啶的完全脱氢可以在170℃下以较快且可靠的h2释放速率执行。机理研究揭示了酸和酸性基团在活性炭表面上的特殊作用。值得注意的是,反向氢化仅需要低h2压力(对于2
‑
皮考啉为2
‑
7巴,并且对于2,6
‑
二甲基吡啶为1.6
‑
5巴),这是已知最低的。催化剂再循环和相互转化实验表明催化剂2,6
‑
二甲基哌啶和2,6
‑
二甲基吡啶具有良好的稳定性。
[0084]
系统
[0085]
本发明的可逆氢气装载和排放/释放系统是指能够保持在所述系统中执行的反应的反应物的任何类型的布置,其中氢分子的排放和装载是使用n
‑
杂环和如上所述的至少一种过渡金属催化剂执行的,优选地n
‑
杂环是经取代的/未经取代的哌啶(对于氢气排放)和经取代的/未经取代的吡啶(对于氢气装载)。
[0086]
在经取代的/未经取代的哌啶与所述至少一种过渡金属催化剂反应时,氢分子释放以形成对应的经取代的/未经取代的吡啶和氢分子。
[0087]
在装载氢分子时,经取代的/未经取代的吡啶与氢分子反应以形成经取代的/未经取代哌啶。
[0088]
在一个实施例中,本发明涉及一种用于按需储存/装载并释放/排放氢气(h2)的lohc系统,所述系统包括n
‑
杂环;以及至少一种过渡金属催化剂。
[0089]
在一些实施例中,本发明涉及一种用于储存氢气(h2)的液体有机氢气载体(lohc)系统,所述系统包括(i)经取代的/未经取代的吡啶,以及(ii)过渡金属催化剂,其中在存在所述催化剂的情况下,所述经取代的/未经取代的吡啶能够与氢气(h2)在足以产生对应的经取代的/未经取代的哌啶作为氢气储存系统的条件下反应。
[0090]
在一些实施例中,本发明涉及一种用于释放氢气(h2)的液体有机氢气载体(lohc)系统,所述系统包括(i)经取代的/未经取代的哌啶;以及(ii)过渡金属催化剂,其中所述经取代的/未经取代的哌啶能够在存在所述催化剂的情况下,在足以产生经取代的/未经取代
的吡啶和分子氢气的条件下脱氢。
[0091]
在一些实施例中,本发明涉及一种用于按需储存氢气(h2)并释放氢气的液体有机氢气载体(lohc)系统,所述系统包括(i)经取代的/未经取代的吡啶;(ii)经取代的/未经取代的哌啶;以及(iii)如上文所述的第一催化剂和第二催化剂,其中所述第一催化剂能够与经取代的/未经取代的吡啶在足以储存氢气的条件下反应,并且其中所述第二催化剂能够与经取代的/未经取代的哌啶在足以如期望的按需释放氢气的条件下反应,并且其中所述第一催化剂和所述第二催化剂可以相同或不同。
[0092]
在一个实施例中,氢气的排放/释放是通过以下实现的:使经取代的/未经取代的与所述至少一种过渡金属催化剂反应;由此形成三个氢分子和经取代的/未经取代的吡啶。
[0093]
在一个实施例中,氢气的装载/储存是通过以下实现的:在存在所述过渡金属催化剂的情况下,使所述经取代的/未经取代的吡啶与三个氢分子反应;由此形成经取代的/未经取代的哌啶。
[0094]
在一个实施例中,本发明涉及一种lohc系统。在另一个实施例中,lohc系统用于氢燃料电池。在另一个实施例中,lohc系统用于为内燃机提供燃料。本发明的lohc将氢气板载释放在由氢燃料电池供电的车辆中,以用于内燃机,或lohc系统将氢气储存并释放在服务站、仓库、中央车队加油站以及居民个人住宅或其它使用点处。氢气的释放是现场产生的;并可以在个人住宅或其它使用地点产生。在释放氢气之后,将脱氢的化合物带到专门氢化设施,并在用加压的氢气和催化剂处理后回收lohc。
[0095]
在一个实施例中,本发明的lohc系统用于分配和监测车辆中基于氢气的燃料。系统被配置成在车辆中储存、释放和分配氢气。系统还包含车辆上的被配置成将氢气递送到发动机的燃料递送系统,以及被配置成控制产生系统并监测车辆对氢气的使用的控制系统。
[0096]
本发明提供了一种用于从本发明的lohc中释放氢气并使用氢气气体以供由氢气燃料电池供电的车辆和/或以供内燃机的方法。
[0097]
在一个实施例中,lohc可以被泵送或倾倒以用于分配到容纳罐和储存容器。使用常规的液体运输和分配方法(管道、轨道车、油罐卡车)轻松地运输液体。氢气在车辆中现场产生或通过脱氢反应器系统产生,所述脱氢反应器系统递送氢气并在氢化反应器位点中回收脱氢的底物。
[0098]
在一个实施例中,本发明的用于在车辆中使用的lohc系统包括:反应室,所述反应室被配置成收集本发明的lohc和催化剂;加热元件,所述加热元件被配置成加热lohc和催化剂以释放氢气;与所述反应室流体连通的缓冲罐,所述缓冲罐被配置成收集并临时储存所述氢气;与所述缓冲罐流体连通的压缩机系统,所述压缩机系统被配置成将所述氢气加压到选定压力;与所述压缩机系统流体连通的储存系统,所述储存系统被配置成储存选定量的选定压力的所述氢气;与所述储存系统流体连通的分配系统,所述分配系统被配置成将所述氢气分配给氢燃料电池或内燃机。与所述反应室流体连通的第二分配系统,所述第二分配系统被配置成将反应的废料分配给废料罐,其中在存在加压的氢气的情况下回收脱氢的底物。脱氢的底物的回收可以在车上或车外进行。
[0099]
方法
[0100]
本发明进一步涉及一种按需储存氢气(h2)并释放氢气的方法,所述方法包括以下
步骤:
[0101]
(a)当期望储存氢气时,在存在第一催化剂的情况下,使经取代的/未经取代的吡啶与氢气(h2)在足以产生对应的经取代的/未经取代的哌啶的条件下反应;以及
[0102]
(b)当期望释放氢气时,使经取代的/未经取代的哌啶与第二催化剂在足以产生对应的经取代的/未经取代的吡啶和氢气(h2)的条件下反应,其中所述第一催化剂和所述第二催化剂可以相同或不同。
[0103]
在一些实施例中,所述第一催化剂和所述第二催化剂相同。在一些实施例中,催化剂是pd/c。在一些实施例中,催化剂是pd(oac)2。在一些实施例中,催化剂是ru/al2o3。在一些实施例中,所述方法不包括任何溶剂。在一些实施例中,氢气储存在温和温度(例如,介于50
‑
180℃之间)下执行。在一些实施例中,氢气储存在温和氢气压力(例如,介于1.5巴与8巴之间)下执行。在一些实施例中,氢气释放在温和压力(例如,大气压)下执行。在一些实施例中,氢气释放在温和温度(例如介于100
‑
180℃之间)下执行。
[0104]
在一些实施例中,催化剂附接到固体载体,或在其它实施例中,催化剂嵌入在固体载体上,或在其它实施例中,位于固体载体的表面上。
[0105]
在一些实施例中,固体载体包括无机材料。无机材料的实例包含但不限于:二氧化硅、氧化铝、氧化镁、二氧化钛、氧化锆、蒙脱石、页硅酸盐、沸石、滑石、粘土、层状双氢氧化物、磷灰石、硅胶、玻璃、玻璃纤维、氧化镍和其混合物。在其它实施例中,固体载体包括有机聚合物。有机聚合物的实例包含但不限于:聚烯烃、聚酰胺、聚对苯二甲酸乙二醇酯、聚氯乙烯、聚偏二氯乙烯、聚苯乙烯、聚甲基丙烯酸酯(polymethracrylate)、天然橡胶、聚异戊二烯、丁二烯
‑
苯乙烯无规共聚物、丁二烯丙烯腈共聚物、聚碳酸酯、聚缩醛、聚苯硫醚、环烯烃共聚物、苯乙烯
‑
丙烯腈共聚物、abs、苯乙烯
‑
马来酸酐共聚物、氯丁二烯聚合物、异丁烯共聚物、聚丙烯、聚四氟乙烯、聚丙烯酸甲酯、聚甲基丙烯酸甲酯、聚碳酸酯、聚乙二醇、聚(有机)硅氧烷和其混合物。
[0106]
化学定义
[0107]
如本文所使用的,在一个实施例中,单独或作为另一基团的一部分使用的术语烷基是指“c1到c8烷基”、“c1到c3烷基”或“c1到c
10
烷基”表示直链和支链基团。非限制性实例是含有1到3个碳原子的烷基(c1到c3烷基)或含有1到4个碳原子的烷基(c1到c4烷基)。饱和烷基的实例包含但不限于甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、戊基、叔戊基和己基。
[0108]
烷基可以是未经取代的,或者被选自由以下组成的组的一个或多个取代基取代:卤素、羟基、烷氧基、芳氧基、烷基芳氧基、杂芳氧基、氧代、环烷基、苯基、杂芳基、杂环基、萘基、氨基、烷基氨基、芳基氨基、杂芳基氨基、二烷基氨基、二芳基氨基、烷基芳基氨基、烷基杂芳基氨基、芳基杂芳基氨基、酰基、酰氧基、硝基、羧基、氨基甲酰基、羧酰胺基、氰基、磺酰基、磺酰基氨基、亚磺酰基、亚磺酰基氨基、硫醇、烷硫基、芳硫基或烷基磺酰基。任何取代基可以是未经取代的或进一步被这些前述取代基中的任何一个取代。通过说明,“烷氧基烷基”是被烷氧基取代的烷基。
[0109]
描述以下实例是为了更全面地展示本发明的优选实施例。然而,所述实例决不应该被解释为限制本发明的广泛范围。
[0110]
实例
[0111]
基本信息
[0112]
在配备有mo 40
‑
2惰性气体纯化器或使用标准施兰克(schlenk)技术的vacuum atmospheres手套箱中的净化氮的气氛下执行用金属复合物进行的所有实验。所有溶剂均为试剂等级或更好。在氩气气氛下根据标准程序对所有非氘化溶剂进行纯化。如所接收到的使用氘化溶剂。用n2对所有溶剂进行脱气并将其保存在手套箱中。根据标准程序对催化反应中使用的化学品中的大部分化学品进行纯化。
[0113]
在300mhz下使用bruker amx
‑
300nmr光谱仪记录1h nmr光谱。在环境温度下执行测量,如每个实验所指出的。1h nmr化学位移参考氘化溶剂的残留氢信号。对具有ms检测器的agilent 7820a/5975c gcms系统执行gsms分析,并且将氦气作为载体气体。在eds bruker xflash/60mm下使用zeiss ultra 55扫描电子显微镜记录eds。在jem
‑
2100电子显微镜下记录透射电子显微镜(tem)。
[0114]
实例1
[0115]2‑
甲基哌啶脱氢为2
‑
皮考啉
[0116]
选择2
‑
甲基哌啶作为模型富h2化合物,因为其潜在氢气储存容量为6.1wt%,并且2位处的供电子甲基可以提供关于电子和空间效应两者的一些益处。在作为溶剂的对二甲苯中筛选几个种类的非均相催化剂(更多细节见下文)。发现钯/活性炭(pd/c,4wt%)是将2
‑
甲基哌啶无受体脱氢为2
‑
皮考啉的最有效催化剂。然后,对作为2
‑
甲基哌啶的无溶剂脱氢催化剂的pd/c进行研究。应用商业pd/c(5wt%)产生了42
‑
48%产率的2
‑
皮考啉和9
‑
10%副产物,这取决于供应商(表2,条目1和2)。重要的是,添加催化量的乙酸(条目3)提高了反应的产率(60%)和选择性(形成了仅约3%的副产物)两者。
[0117]
表2. 2
‑
甲基哌啶脱氢的优化
a
[0118][0119][0120]
a
条件:2
‑
甲基哌啶(10mmol)、催化剂(0.2mol%的[pd])170℃(油浴温度,内部温度为119℃)、在冷凝器的顶部的氩气流下的开放系统、用冷水循环。通过gc使用正庚烷作为内部标准物确定产率和转化率。
[0121]
b hoac(0.04mmol)。
[0122]
c
载体(50mg)。pd(tfa)2=三氟乙酸钯(ii),pd2(dba)3=三(二亚苄基丙酮)二钯(0),pd(acac)2=乙酰丙酮钯(ii),ac=活性炭,bn=氮化硼。
[0123]
进一步地,规划了原位催化剂产生策略。选择pd(oac)2作为钯前体,干式酸性活性炭(darco@kb,表面积:1500m2/g、ph
pzc
=4.25:零电荷的点处的ph值)作为载体以进行初步尝试,其中可能潜在地产生pd/c和2当量的乙酸(相对于pd)。此原位产生的钯催化剂比预先制备的商用催化剂更活跃,产生了72%产率的2
‑
皮考啉(条目4)。包含pdcl2、pd(tfa)2、pd2(dba)3和pd(acac)2的其它钯源的催化剂活性比pd(oac)2更低(条目5
‑
8)。接下来,选择pd(oac)2作为用于研究载体的作用的最佳钯前体(表2,条目9
‑
12)。在由sio2、bn、γ
‑
al2o3或ceo2代替活性炭的情况下,获得了非常低产率的2
‑
皮考啉。在不存在载体的情况下,检测到仅3%的2
‑
皮考啉,并且在施兰克管的底部观察到钯镜(palladium mirror),这示出在反应系统中产生了pd0物种。因此,pd(oac)2和活性炭的组合是2
‑
甲基哌啶脱氢的最有效的催化剂。
[0124]
接下来,将反应连接到气体收集系统(图3),以记录时间依赖性h2释放曲线。如图2a
‑
2b所述,在pd(oac)2/活性炭(pd(oac)2=0.03mmol,ac=40mg)催化下,在170℃下(浴温,内部温度为119℃)在51小时后获得27.1mmol(90%产率)的h2(条目1,三角形
▲
)。还在较低浴温下实现了脱氢;在150℃下(内部温度也为119℃),在117小时之后收集到84%产率的h2(条目2,正方形
■
)。此外,用相同量的催化剂,将2
‑
甲基哌啶的比例加倍,脱氢(条目3,点
●
)甚至更快,并且在94小时之后产生了54.5mmol(91%产率)的h2。可喜地,反向氢化可以通过直接用h2对同一反应混合物进行加压来实现。在2
‑
7巴的h2下,混合物中的2
‑
皮考啉完全转化为90%产率的2
‑
甲基哌啶(图4)。这些结果表明脱氢和氢化是由同一单个金属非均相催化剂催化的,并且副产物的总量少于10%。
[0125]
为了了解副反应的过程,通过gc
‑
ms对副产物的混合物进行了分析。这些副产品的保留时间相似(其中一些部分重叠,见下文)且分子量相似,可能是py
‑
py和py
‑
pi,这大概是通过将烯胺钯催化加成为亚胺而形成的(图5a)。因此,抑制烯胺加成为亚胺可能是提高选择性的有效方法。策略是增加对亲核和亲电中间体的空间位阻(图5b)。
[0126]
实验细节:
[0127]
在溶剂中将2
‑
甲基哌啶脱氢为2
‑
皮考啉:在手套箱中,将催化剂、t
‑
buok(0.2mmol)、2
‑
甲基哌啶(1mmol)和溶剂(1ml)添加到施兰克管中。施兰克管装配有冷凝器,并且在开放系统中在冷凝器顶部的氩气流下利用搅拌使溶液回流持续指定时间。冷却到室温之后,使用正庚烷作为内部标准物通过gc确定转化率和产率。
[0128]
表3.用于溶剂中的2
‑
甲基哌啶脱氢的非均相催化剂
[0129]
[0130][0131]
条件:2
‑
甲基哌啶(1mmol)、催化剂(50mg)、添加剂和对二甲苯(1ml)、170℃(油浴)、开放系统、在冷凝器顶部的氩气流下,使用正庚烷作为内部标准物通过gc确定产率和转化率。p
‑
tsoh=4
‑
甲基苯磺酸。
[0132]2‑
甲基哌啶的无溶剂脱氢:在手套箱中,将2
‑
甲基哌啶(10mmol)、43mg的5wt%pd/c或(4.5mg的pd(oac)2和50mg的载体)添加到施兰克管中。施兰克管装配有冷凝器,并且在开放系统中在氩气流下利用搅拌使溶液回流持续指定时间。冷却到室温之后,使用正庚烷作为内部标准物通过gc确定转化率和产率(结果在表2和表4中给出)。
[0133]
表4.由其它金属催化的2
‑
甲基哌啶的无溶剂脱氢
[0134][0135]2‑
甲基哌啶的无溶剂脱氢(连接到气体收集系统):在手套箱中,将2
‑
甲基哌啶和催化剂添加到施兰克管中(参见表5)。施兰克管装配有冷凝器,所述冷凝器连接到气体收集系统(图3)。由所述收集系统记录h2的体积,并且时间依赖性h2释放曲线在图10中示出。基于峰面积通过gc确定产率和转化率。在h2体积的基础上计算关于2
‑
甲基哌啶完全转化为2
‑
皮考啉的h2产率(10mmol的2
‑
甲基哌啶在28℃下可以产生30mmol的h2,740ml)。
[0136]
表5.利用pd催化剂进行的2
‑
甲基哌啶的脱氢
[0137][0138][0139]
注意:
a
环境温度=27
‑
30℃(30mmol的h2为约740ml)。
[0140]
b
ba=苯甲酸(0.06mmol)
[0141]
ac=活性炭,pd/c
hs
=用于氢气储存的钯/活性炭。
[0142]
表6. 2
‑
甲基哌啶/正庚烷和2
‑
皮考啉/正庚烷的相对gc响应因子的测量
[0143][0144]
gc条件(对于有机化合物):hp 6890或agilent 7890b系列gc系统;柱:hp
‑
5、30m、320μm,入口:280℃;检测器:fid 280℃;载体气体:he;流量:1毫升/分钟;烤箱:50℃,保持8分钟;15℃/分钟到280℃,保持2分钟。
[0145]
实例2
[0146]
基于n
‑
杂环的无溶剂液体到液体的lohc系统的优化
[0147]
取代对脱氢过程的影响
[0148]
本着增加空间位阻的想法,对取代基的影响进行了研究。对哌啶、3
‑
甲基哌啶、4
‑
甲基哌啶和2,6
‑
二甲基哌啶进行了研究(表7)。使用哌啶或3
‑
甲基哌啶作为富h2化合物产生了良好的选择性,但产生了低产率的贫h2产物(条目1和2)。使用4
‑
甲基哌啶产生了46%产率的4
‑
皮考啉和约7%的副产物(条目3)。
[0149]
表7.哌啶和甲基哌啶的脱氢
[0150][0151][0152]
a
条件:哌啶(10mmol)、催化剂(0.2mol%的[pd])、活性炭(darco@kb,50mg)、170℃(油浴温度)、在冷凝器的顶部的氩气流下开放系统、用冷水循环。通过gc使用正庚烷作为内部标准物确定产率和转化率。
[0153]
b
通过在1h nmr上使用均三甲苯作为内部标准物来确定。
[0154]
c
基于峰面积通过gc来确定。
[0155]
d
开放系统,氩气气氛,冷凝器连接到气体收集系统。
[0156]
使用2,6
‑
二甲基哌啶产生了多于99%产率和100%选择性(条目4)。重要的是,2,6
‑
二甲基哌啶的定量脱氢也可以在没有氩气流的情况下实现,并得到100%产率的纯h2气体(通过具有热导检测器的gc确认,h2纯度>99.99%,未观察到杂质)。因此,在2,6
‑
二甲基吡啶/2,6
‑
二甲基哌啶的基础上,以同一催化剂系统建立lohc系统也是有前景的。
[0157]
基于2,6
‑
二甲基吡啶/2,6
‑
二甲基哌啶的lohc系统的最大重量容量为5.3wt%,高于欧盟目标并且非常接近us doe目标。另外,2,6
‑
二甲基吡啶和2,6
‑
二甲基哌啶的理化性质满足理想lohc的所有要求。因此,基于lohc系统的2,6
‑
二甲基吡啶/2,6
‑
二甲基哌啶基的lohc系统是有吸引力的。
[0158]
通过气体收集系统记录了2,6
‑
二甲基哌啶脱氢的时间依赖性h2释放曲线(图6a
‑
6b)。在pd(pd(oac)2=6.7mg,0.3mol%,ac=40mg)的催化下,2,6
‑
二甲基哌啶的脱氢工作良好,13小时之后得到97%的h2,并且23小时候得到100%的h2(图6a)。有趣的是,在12小时内,在达到95%产率的h2之前,h2释放速率恒定(图6a,内插页)。这些结果表明,由于催化剂的表面被2,6
‑
二甲基哌啶饱和,在2,6
‑
二甲基哌啶中发生了零级反应。此外,通过使用不同催化剂负载量测量h2释放速率,可能存在pd的一阶速率依赖性。
[0159]
此外,通过在150℃下用1.6
‑
5巴的h2对混合物加压18小时实现了2,6
‑
二甲基吡啶到2,6
‑
二甲基哌啶再生的反向氢化,产率为100%(图6b,右列1)。所产生的混合物可以重复用于第二轮脱氢(93%产率,图6b,左列2)和氢化(100%产率,图6b,右列2),并且无2,6
‑
二甲基哌啶和2,6
‑
二甲基吡啶分解发生。另外,利用0.3mol%的催化剂,在30
‑
50巴的h2压力下,在150℃下,2,6
‑
二甲基吡啶的氢化在1.5小时、2小时和3小时之后分别得到产率为87%、95%和98%的2,6
‑
二甲基哌啶,这可以实现快速h2装载(详情见下文)。
[0160]
因此,建立了基于n
‑
杂环的无溶剂液体到液体lohc系统,所述系统由单一催化剂在相对温和的条件下催化,以用于脱氢和氢化两者。
[0161]
实验数据:
[0162]
2,6
‑
二甲基哌啶到2,6
‑
二甲基吡啶的无溶剂脱氢(与气体收集系统连接):在手套箱中,将2,6
‑
二甲基哌啶(10mmol)和催化剂添加到配备有冷凝器的施兰克管中,所述冷凝器与气体收集系统连接(图3)。在开放系统中利用搅拌使溶液回流持续指定时间。在形成的h2体积的基础上计算关于2,6
‑
二甲基哌啶完全转化为2,6
‑
二甲基吡啶的h2的产率(30mmol的h2,在20℃下为721ml,在28℃下为740ml)。在冷却到室温之后,使用均三甲苯作为内部标准物通过1h nmr,或基于峰面积通过gc确定转化率和产率。
[0163]
gc条件(对于有机化合物):hp 6890或agilent 7890b系列gc系统;柱:hp
‑
5、30m、320μm,入口:280℃;检测器:fid 280℃;载体气体:he;流量:1毫升/分钟;烤箱:50℃,保持8分钟;15℃/分钟到280℃,保持2分钟。
[0164]
表8.用于2,6
‑
二甲基哌啶的脱氢的非均相催化剂
[0165][0166][0167]
条件:2,6
‑
二甲基哌啶(10mmol)和催化剂,油浴温度,开放系统,冷凝器连接到气体收集系统,基于峰面积通过gc确定2,6
‑
二甲基吡啶的产率。
[0168]
表9.[pd]与h2释放速率之间的关系
[0169][0170]
k
obs
=92.066
[0171]
gc条件(对于气体分析):hp 6890系列gc系统;柱:supelco 1
‑
2382,5ft
×
1/8in s.s.support 45/60carboxentm 1000,填充柱。入口:87℃;检测器:tcd 250℃;载体气体:he;流量:29.1毫升/分钟;烤箱:35℃,保持2分钟;10℃/分钟到60℃,保持0分钟;30℃/分钟到200℃。
[0172]
对于90%产率内的平均周转频率(atof
90
)的计算:在手套箱中,将2,6
‑
二甲基哌啶(10mmol)、pd/c
hs
([pd]=0.03mmol)和酸(0.06mmol)添加到配备有冷凝器的施兰克管中,所述冷凝器与气体收集系统连接(图3)。在开放系统中利用搅拌使溶液回流持续指定时间。在形成的h2体积的基础上计算关于2,6
‑
二甲基哌啶完全转化为2,6
‑
二甲基吡啶的h2的产率(30mmol的h2,在20℃下为721ml,在28℃下为740ml)。冷却到室温之后,通过gc确定转化率和产率。atof
90
的计算基于h2释放速率(未示出)。
[0173]
实例3
[0174]
用于吡啶的氢化的一般程序
[0175]
在手套箱中,将催化剂(pd或ru)和2,6
‑
二甲基吡啶装入衬有teflon管(或90ml fisher porter管)的50ml不锈钢高压釜中,所述管内含有搅拌棒。用h2(10atm
×
2)吹扫之后,用h2对高压釜加压(压力参见表10)。在150℃下搅拌混合物,h2降低到指定压力,然后冷却到室温并且再次用h2加压(重复几次)。表11提供了氢化的5次重复的结果。当压力未降低时,将高压釜冷却到室温,并且小心地释放h2。然后离心以分离催化剂和产物抽出50μl澄清溶液并在cdcl3和gc中通过by 1
h nmr测量。蒸馏之后,获得了纯的2,6
‑
二甲基哌啶以供进一步使用。
[0176]
表10. 2,6
‑
二甲基吡啶到2,6
‑
二甲基哌啶的氢化
a
[0177][0178][0179]
a
条件:催化剂装载,h2压力,浴温,并且反应时间在表中示出,基于峰面积通过gc确定2,6
‑
二甲基哌啶的产率。
[0180]
b 2,6
‑
二甲基吡啶(54mmol)。
[0181]
c 2,6
‑
二甲基吡啶(72mmol)。
[0182]
d 2,6
‑
二甲基吡啶(10mmol)。
[0183]
e
在具有teflon管的50ml不锈钢高压釜中。
[0184]
f
在90ml fisher
‑
porter管中
[0185]
表11.催化剂重复使用(在每个氢化步骤之后,将催化剂分离并再循环),以用于将2,6
‑
二甲基吡啶氢化为2,6
‑
二甲基哌啶。
[0186][0187][0188]
实例4
[0189]
催化过程的机理研究
–
对照实验
[0190]
执行几个对照实验以查明催化脱氢是否涉及均相或非均相催化。当将0.3mol%的pd(oac)2、40mg的活性炭和10mmol的2,6
‑
二甲基哌啶在170℃(浴温,内部温度为128℃)下加热5小时时,收集到46%产率的h2,这与形成的2,6
‑
二甲基吡啶的产率(46.2%)非常匹配(图7a)。使用teflon注射器过滤器(0.22μm孔径的ptfe)对混合物进行过滤,以提供2,6
‑
二甲基哌啶和2,6二甲基吡啶的无色混合物。将获得的混合物在170℃下加热另外20小时,未形成气体,并且2,6
‑
二甲基吡啶的量也未改变(45.9%,图7b)。因此,均相催化过程是不可能的。通过icp
‑
ms分析最终混合物示出了仅0.172ppm的pd,这表明实际上pd浸出到反应溶液中未发生。第三,在没有载体的情况下进行的反应提供了仅5%产率的2,6
‑
二甲基吡啶并且产生pd镜(图7c)。综上所述,这些结果示出可以排除均相过程。
[0191]
实验细节:
[0192]
对照实验:在手套箱中,将2,6
‑
二甲基哌啶(1.13g,10mmol)、pd(oac)2(6.7mg,0.03mmol)和活性炭(40mg)添加到施兰克管中。施兰克管配备有冷凝器,所述冷凝器连接到气体收集系统。将溶液在开放系统中在搅拌下回流5小时。收集h2(331ml,46%产率)。在冷却到室温之后,使用teflon注射器过滤器(0.22μm孔径的ptfe)对反应混合物进行过滤,以提供2,6
‑
二甲基哌啶(53.8%)和2,6二甲基吡啶(46.2%)的无色混合物。然后将2,6
‑
二甲基哌啶和2,6
‑
二甲基吡啶的混合物转移到新施兰克管中,并且回流另一20小时,但没有收集到气体。在冷却到室温之后,提取样品以用于gc和icp
‑
ms分析。
[0193]
实例5
[0194]
机理研究
‑
酸对脱氢过程的影响
[0195]
由pd(oac)2和酸性活性炭(darco@kb)制备pd/c
hs
(pd/c
hs
=用于氢气储存的钯/活性炭)以研究酸的影响。通过傅里叶转变红外光谱(fourier
‑
transform infrared spectroscopy,ftir)、扫描电子显微镜(sem)和透射电子显微镜(tem)对制备的pd/c
hs
进行
表征。发现与活性炭表面的羧基结合的钯纳米粒子均匀分布,pd纳米粒子的平均直径约为1.93
±
0.44nm。
[0196]
然后,测试pd/c
hs
对2,6
‑
二甲基哌啶脱氢的催化活性;时间依赖性h2释放曲线和90%产率内的平均周转频率(atof
90
=85,mol h2每mol pd每小时)在图8中示出。使用pd/c
hs
作为催化剂的atof
90
与pd(oac)2/活性炭系统(atof
90
=91)接近。
[0197]
通过将2当量(相对于pd)的乙酸(pk
a
=4.76)、苯甲酸(pk
a
=4.20)、4
‑
甲基苯磺酸(pk
a
=1.99)、羧基聚苯乙烯(单体pk
a
=4.35)或聚丙烯酸(pk
a
=4.75)添加到反应混合物中对酸的影响进行了研究(图8,条目2
‑
6)。乙酸、苯甲酸和聚合物羧基聚苯乙烯对脱氢过程示出积极影响,分别产生了103、128和107的atof
90
。使用聚丙烯酸作为添加剂,atof
90
略微降低到80,并且使用强酸4
‑
甲基苯磺酸具有负面影响,导致atof
90
降低到45。然而,在使用前用t
‑
buok对pd/c
hs
进行处理,此时羧基和pd纳米粒子的相互作用可能会被碱破坏,催化剂变得完全失活(图8,条目8)。这些结果表明,羧酸和活性炭表面上的羧基加快了钯催化的2,6
‑
二甲基哌啶脱氢。此外,pd/c
hs
和羧基聚苯乙烯可以通过离心轻松回收而不会损失催化活性(atof
90
=101,图8,条目9)。
[0198]
基于对照实验、初步机理研究和文献,提出了2
‑
皮考啉和2,6
‑
二甲基吡啶形成的可信的脱氢机制(图9a)和途径(图9b)。n
‑
杂环与pd(ii)纳米粒子的配位引起n
‑
h基团的酸度增加,所述基团可以通过pd
n2
的反羧酸根阴离子去质子化,以产生酰胺
‑
钯物种和羧酸(图9a)。然后β
‑
氢化物消除发生以产生亚胺和钯氢化物物种。最后,在羧酸的辅助下,h2释放并且活性催化剂pd
n2
重新产生。羧酸在反应系统中的另一个作用是加速亚胺互变异构化为关键中间体烯胺(图9b)。脱氢和互变异构化的级联步骤产生最终的贫h2产物2
‑
皮考啉或2,6
‑
二甲基吡啶和h2。
[0199]
实验细节:
[0200]
用于用t
‑
buok对pd/c
hs
进行处理的程序:在手套箱中,将pd/c
hs
(43mg,[pd]=0.03mmol)、t
‑
buok(16.8mg,0.15mmol)和thf(1ml)添加到施兰克管中。在室温下搅动12小时之后,通过离心将催化剂分离,用thf进行洗涤(1ml
×
2),并且在真空下干燥。
[0201]
实例6
[0202]2‑
甲基哌啶作为lohc系统
[0203]2‑
甲基哌啶和2
‑
皮考啉的可逆相互转化
[0204][0205]
脱氢:在手套箱中,将2
‑
甲基哌啶(20mmol)、pd(oac)2(6.7mg,0.03mmol)和活性炭(40mg)添加到施兰克管中。施兰克管装配有冷凝器,所述冷凝器连接到气体收集系统(图3)。在开放系统中利用搅拌使溶液回流。当不再产生气体时,停止反应。基于h2体积(1344ml)计算关于2
‑
甲基哌啶完全转化为2
‑
皮考啉的h2的产率(91%)(表11)。冷却到室温之后,使施兰克管进入手套箱中,并且将混合物转移到90ml的fisher
‑
porter管中以进行氢
化。
[0206]
氢化:从手套箱中取出fisher
‑
porter管,用7巴的h2进行加压并且在150℃下加热。6小时之后,h2压力降低到2巴,并且反应混合物冷却到室温,然后用h2(7巴)第二次进行加压,并且在150℃下加热另外6小时。用h2(7巴)第三次对fisher
‑
porter管进行加压并且在150℃下加热12小时。在第四次加热之后(7巴),将fisher
‑
porter管在150℃下加热最后12小时。然后,将fisher
‑
porter管冷却到室温,并且小心释放氢气,添加950mg的正庚烷作为内部标准物,提取100μl的样品(用et2o稀释)以供gc分析(表12)。
[0207]
表12. 2
‑
甲基哌啶和2
‑
皮考啉的相互转化
[0208][0209]
a gc产率,使用正庚烷作为内部标准物。
[0210]
b
基于峰面积通过gc确定。
[0211]
c
在h2体积的基础上计算关于2
‑
甲基哌啶完全转化为2
‑
皮考啉的h2产率(60mmol的h2,在28℃下为1480ml)。
[0212]
实例7
[0213]
2,6
‑
二甲基哌啶作为lohc系统
[0214]
2,6
‑
二甲基哌啶和2,6
‑
二甲基吡啶的可逆相互转化以及催化剂重复使用
[0215][0216]
脱氢:将2,6
‑
甲基哌啶(50mmol)、pd(oac)2(33.6mg,0.15mmol)和活性炭(200mg)添加到施兰克管中。施兰克管装配有冷凝器,所述冷凝器连接到气体收集系统(图3)。在开放系统中利用搅拌使溶液回流23小时。冷却到室温之后,使施兰克管进入手套箱中,并且将混合物转移到90ml的fisher
‑
porter管中以进行氢化。
[0217]
氢化:从手套箱中取出fisher
‑
porter管,用7巴的h2进行加压(第一次)并且在150℃下加热。6小时之后,h2压力降低到1.6巴,并且fisher
‑
porter管冷却到室温,然后用h2(7巴)第二次进行加压。8次加压和加热之后,将fisher
‑
porter管冷却到室温,小心释放氢气,然后将具有反应混合物的管放入到手套箱中,并且将混合物转移到施兰克管中以进行脱氢。
[0218]
催化剂重复使用:在三次脱氢和两次氢化之后,通过离心将固体催化剂分离(4000rpm,持续40分钟),并且用正戊烷进行洗涤(4ml
×
4),然后在真空下干燥8小时。使用回收的催化剂以进行第四次和第五次脱氢。
[0219]
图12展示了在可逆方法步骤脱氢
→
氢化
→
脱氢
‑→
氢化
→
脱氢之后,催化剂的100%转化,其中未再循环催化剂并且照现状使用。在脱氢的最后步骤之后,将催化剂再循环并再次用于另外的两个脱氢步骤(催化剂在最后的脱氢步骤中的每个脱氢步骤之间再循
环)。在所有方法中,催化剂都表现出100%转化率。
[0220]
实例8
[0221]
催化剂pd/c
hs
的制备
[0222][0223]
在手套箱中i,将pd(oac)2(100.8mg)、活性炭(600mg,darco@kb,表面积:1500m2/g,ph
pzc
=4.25)、2,6
‑
二甲基哌啶(170mg)和i
‑
proh(15ml)添加到90ml fisher
‑
porter管中。用5巴的h2对fisher
‑
porter管加压,并且将混合物在120℃下加压5小时,然后在室温下搅拌另外5天。在真空下去除溶剂并且用h2流(40毫升/分钟)在200℃下对残余物进行处理,持续2小时。获得的催化剂pd/c
hs
必须保持在惰性气氛下。
[0224]
虽然本文已经说明并描述了本发明的某些特征,但本领域普通技术人员现在将想到许多修改、取代、变化及等效物。因此,应当理解,所附权利要求书旨在覆盖如落入本发明的真正精神内的所有这种修改和改变。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。