1.本技术属于医药领域,具体地,涉及一种布鲁顿酪氨酸激酶btk抑制剂的用途,特别是该类化合物在制备治疗btk相关肿瘤疾病的药物中的用途。
背景技术:
2.b细胞抗原受体(b cell receptor,bcr)信号传导对正常b细胞发育和适应性免疫有着至关重要的作用,该信号传导通路的激活有助于b细胞恶性肿瘤和自身免疫疾病的发生和发展。布鲁顿酪氨酸激酶(bruton’s tyrosine kinase,btk)是隶属于tec酪氨酸激酶家族的一种非受体酪氨酸激酶,是b细胞受体(bcr)信号通路中的关键调节因子,主要在b淋巴细胞的各个发育阶段表达(b淋巴细胞发育终末的浆细胞除外),对b细胞的增殖、分化和凋亡有重要影响。
3.在与b细胞相关的恶性肿瘤中,bcr信号通路过度活跃,从而抑制b细胞的正常分化和凋亡,促进异常增殖,已知多种b细胞类型的恶性肿瘤中经常存在bcr通路的异常调节。目前,已有4个btk抑制剂获批上市,依鲁替尼(ibrutinib,商品名imbruvica)是首个上市的btk小分子抑制剂,属于一代btk抑制剂,于2013年被美国fda批准用于套细胞淋巴瘤(mantle cell lymphoma,mcl)和慢性淋巴细胞白血病(chronic lymphocytic leukemia,cll)等的临床治疗,其后依鲁替尼不断扩充适应症,目前获批的适应症还包括携带17p删除的慢性淋巴细胞白血病/小淋巴细胞淋巴瘤(cll/small lymphocytic lymphoma,sll)、华氏巨球蛋白血症(waldenstrom macroglobulinemia,wm)、边缘区淋巴瘤(marginal zone lymphoma,mzl)、慢性移植物抗宿主病(chronic graft-versus-host disease,cgvhd)。依鲁替尼可以与btk激酶atp结合域中第481位半胱氨酸(cys481)形成共价结合,不可逆性抑制btk活化,阻断btk信号通路,从而抑制b淋巴瘤细胞的增殖和存活,达到肿瘤治疗的目的。依鲁替尼在临床治疗中取得了实质性的疗效。但由于依鲁替尼靶点选择性较差,临床存在一定毒副作用。阿卡替尼(acalabrutinib,acp-196,商品名calquence)于2017年被批准用于mcl和cll的治疗,属于第二代btk靶向药。相对于依鲁替尼,阿卡替尼对btk的选择性更高,脱靶毒性较低。另外,百济神州生物科技有限公司开发的zanubrutinib(泽布替尼,bgb-3111)于2019年11月经fda批准用于经治的成年mcl患者,成为首获fda突破性疗法认定的中国本土抗癌药。日本小野制药有限公司研发的tirabrutinib(ono-4059)于2020年3月经日本医药品医疗器械综合机构(pmda)批准上市,用于复发或难治性的原发性中枢神经系统淋巴瘤(pcnsl)和淋巴浆细胞性淋巴瘤(lpl)的治疗。总的来讲,第一代抑制剂对btk的抑制活性高,但靶点选择和生物利用度差;第二代抑制剂选择性好,但对btk的抑制率低于第一代抑制剂。结构上,目前上市的btk药物的核心骨架主要是双环体系。
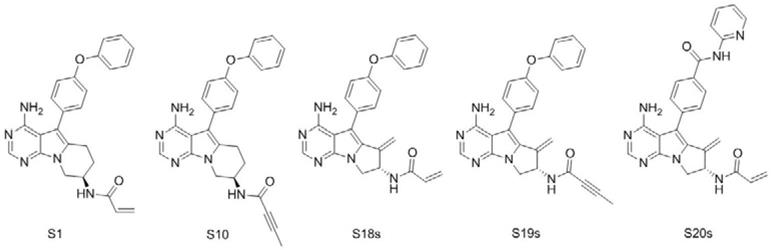
4.为了获得兼具第一代抑制剂的高活性和第二代抑制剂的良好选择性的新一代btk抑制剂,中国科学院上海药物研究所在cn108101905a专利中公开了一系列具有嘧啶并[5,4-b]吲嗪或嘧啶并[5,4-b]吡呤结构的化合物。其中,嘧啶并[5,4-b]吲嗪类化合物s1和s10及嘧啶并[5,4-b]吡呤类化合物s18、s19和s20显示了较高的btk抑制活性,在进一步的工作
中,还合成了s18、s19和s20的s构型的化合物(即s18s、s19s和s20s)[yu xue,et al.discovery of4,7-diamino-5-(4-phenoxyphenyl)-6-methylenepyrimido[5,4-b]pyrrolizines as novel bruton’s tyrosine kinase inhibitors.j.med.chem.,2018,61,4608-4627.],但进一步研究发现s1、s10、s18s和s19s这些化合物代谢过程中不稳定(末端苯环对位为活性代谢位点之一),末端苯基的4-位易于氧化;而化合物s20s的口服生物利用度不理想。
[0005][0006]
基于上述问题,进一步研发具有优良的btk抑制活性和选择性、体内抗肿瘤活性高、并具有优良的口服给药性能和代谢稳定性的btk激酶抑制剂,对于治疗btk相关肿瘤疾病十分重要。
技术实现要素:
[0007]
本发明的目的在于提供一种布鲁顿酪氨酸激酶btk抑制剂化合物a或其药学上可接受的盐在制备治疗btk相关肿瘤疾病的药物中的用途,所述化合物a具有如下结构:
[0008][0009]
上述用途,所述btk相关肿瘤疾病包括血液肿瘤和实体瘤。
[0010]
优选地,上述血液肿瘤为淋巴瘤和白血病。
[0011]
进一步优选地,上述淋巴瘤为b细胞淋巴瘤。
[0012]
上述用途,所述btk相关肿瘤疾病包括组织细胞性淋巴瘤、套细胞淋巴瘤、弥漫性大b细胞淋巴瘤、慢性淋巴细胞白血病、小淋巴细胞淋巴瘤、边缘区淋巴瘤、滤泡性淋巴瘤、burkitt淋巴瘤或华氏巨球蛋白血症。
[0013]
上述用途,所述弥漫性大b细胞淋巴瘤选自非特殊类型的弥漫性大b细胞淋巴瘤、其他大b细胞淋巴瘤、伴myc和/或bcl基因异常的高级别b细胞淋巴瘤和非特殊类型的高级别b细胞淋巴瘤组成的组中的1种或多种;优选地,所述弥漫性大b细胞淋巴瘤选自其他大b细胞淋巴瘤、伴myc和/或bcl基因异常的高级别b细胞淋巴瘤和非特殊类型的高级别b细胞淋巴瘤组成的组中的1种或多种。
[0014]
上述用途,所述其他大b细胞淋巴瘤包括富于t细胞/组织细胞的dlbcl、原发中枢神经系统dlbcl、原发皮肤dlbcl(腿型)、ebv阳性的dlbcl,非特指型dlbcl、慢性炎症相关的大b细胞淋巴瘤、淋巴瘤样肉芽肿、伴有irf4重排的大b细胞淋巴瘤、原发纵隔(胸腺)大b细胞淋巴瘤、血管内大b细胞淋巴瘤、alk阳性大b细胞淋巴瘤、浆母细胞性淋巴瘤、hhv8阳性dlbcl、原发性渗出性淋巴瘤等;
[0015]
上述用途,所述伴myc基因异常的高级别b细胞淋巴瘤选自myc基因扩增的高级别b细胞淋巴瘤、myc基因融合的高级别b细胞淋巴瘤组成的组中的1种或多种;所述bcl基因异常的高级别b细胞淋巴瘤选自bcl2和/或bcl6基因异常的高级别b细胞淋巴瘤,所述bcl2和/或bcl6基因异常的高级别b细胞淋巴瘤选自bcl2基因扩增的高级别b细胞淋巴瘤、bcl2基因融合的高级别b细胞淋巴瘤、bcl6基因扩增的高级别b细胞淋巴瘤和bcl6基因融合的高级别b细胞淋巴瘤组成的组中的1中或多种。
[0016]
上述用途,本发明涉及的弥漫性大b细胞淋巴瘤也可以是携带异常染色体的弥漫性大b细胞淋巴瘤。
[0017]
上述用途,所述套细胞淋巴瘤选自经典型套细胞淋巴瘤、白血病样非淋疤结性套细胞淋巴瘤和cyclin d1阳性的套细胞淋巴瘤组成的组中的1种或多种。
[0018]
上述用途,所述套细胞淋巴瘤选自bcl基因异常的套细胞淋巴瘤;优选为bcl1和/或bcl2基因异常的套细胞淋巴瘤;进一步优选为bcl1基因融合或重排的套细胞淋巴瘤或bcl2基因融合或重排的套细胞淋巴瘤。
[0019]
上述的用途,本发明涉及的套细胞淋巴瘤也可以是携带异常染色体的套细胞淋巴瘤。
[0020]
上述用途,所述burkitt淋巴瘤是携带异常染色体的burkitt淋巴瘤。
[0021]
上述用途,所述基因异常是指基因突变、基因融合或重排和/或基因扩增。
[0022]
上述用途,所述异常染色体是指发生了扩增、缺失、断裂、重排和/或易位的染色体。
[0023]
上述用途,所述药物中还可含有一种或多种其他靶向药物或化疗药物。所述的其他靶向药物或化疗药物是指临床用于治疗肿瘤相关疾病的靶向药物或化疗药物。
[0024]
上述的用途,所述药物制成临床接受的制剂,例如口服制剂、注射制剂、外用制剂等。
[0025]
上述用途,所述药物中含有治疗有效量的化合物a或其药学上可接受的盐,所述治疗有效量优选每天给药剂量为0.001-1000mg,进一步优选每天给药剂量为0.01-500mg,更进一步优选每天给药剂量为0.1-200mg,更进一步优选每天给药剂量为0.5-100mg,更进一步优选每天给药剂量为0.5-50mg,更进一步优选每天给药剂量为0.5-30mg,更进一步优选每天给药剂量为0.5-20mg。可以单剂量施用或分剂量施用。
[0026]
另一方面,本发明还提供一种治疗btk相关肿瘤疾病的方法,其特征在于,给予受试者或患者含有治疗有效量的化合物a或其药学上可接受的盐的药物。
[0027]
上述方法,所述btk相关肿瘤疾病包括血液肿瘤和实体瘤。
[0028]
优选地,上述血液肿瘤为淋巴瘤和白血病。
[0029]
进一步优选地,上述淋巴瘤为b细胞淋巴瘤。
[0030]
上述方法,所述btk相关肿瘤疾病包括组织细胞性淋巴瘤、套细胞淋巴瘤、弥漫性
大b细胞淋巴瘤、慢性淋巴细胞白血病、小淋巴细胞淋巴瘤、边缘区淋巴瘤、滤泡性淋巴瘤、burkitt淋巴瘤或华氏巨球蛋白血症。
[0031]
上述用途,所述弥漫性大b细胞淋巴瘤选自非特殊类型的弥漫性大b细胞淋巴瘤、其他大b细胞淋巴瘤、伴myc和/或bcl基因异常的高级别b细胞淋巴瘤和非特殊类型的高级别b细胞淋巴瘤组成的组中的1种或多种;优选地,所述弥漫性大b细胞淋巴瘤选自其他大b细胞淋巴瘤、伴myc和/或bcl基因异常的高级别b细胞淋巴瘤和非特殊类型的高级别b细胞淋巴瘤组成的组中的1种或多种。
[0032]
上述用途,所述其他大b细胞淋巴瘤包括富于t细胞/组织细胞的dlbcl、原发中枢神经系统dlbcl、原发皮肤dlbcl(腿型)、ebv阳性的dlbcl,非特指型dlbcl、慢性炎症相关的大b细胞淋巴瘤、淋巴瘤样肉芽肿、伴有irf4重排的大b细胞淋巴瘤、原发纵隔(胸腺)大b细胞淋巴瘤、血管内大b细胞淋巴瘤、alk阳性大b细胞淋巴瘤、浆母细胞性淋巴瘤、hhv8阳性dlbcl、原发性渗出性淋巴瘤等;
[0033]
上述用途,所述伴myc基因异常的高级别b细胞淋巴瘤选自myc基因扩增的高级别b细胞淋巴瘤、myc基因融合的高级别b细胞淋巴瘤组成的组中的1种或多种;所述bcl基因异常的高级别b细胞淋巴瘤选自bcl2和/或bcl6基因异常的高级别b细胞淋巴瘤,所述bcl2和/或bcl6基因异常的高级别b细胞淋巴瘤选自bcl2基因扩增的高级别b细胞淋巴瘤、bcl2基因融合的高级别b细胞淋巴瘤、bcl6基因扩增的高级别b细胞淋巴瘤和bcl6基因融合的高级别b细胞淋巴瘤组成的组中的1种或多种。
[0034]
上述用途,本发明涉及的弥漫性大b细胞淋巴瘤也可以是携带异常染色体的弥漫性大b细胞淋巴瘤。
[0035]
上述的用途,所述套细胞淋巴瘤选自经典型套细胞淋巴瘤、白血病样非淋疤结性套细胞淋巴瘤和cyclin d1阳性的套细胞淋巴瘤组成的组中的1种或多种。
[0036]
上述的用途,所述套细胞淋巴瘤选自bcl基因异常的套细胞淋巴瘤;优选为bcl1和/或bcl2基因异常的套细胞淋巴瘤;进一步优选为bcl1基因融合或重排的套细胞淋巴瘤或bcl2基因融合或重排的套细胞淋巴瘤。
[0037]
上述的用途,本发明涉及的套细胞淋巴瘤也可以是携带异常染色体的套细胞淋巴瘤。
[0038]
上述用途,所述burkitt淋巴瘤是携带异常染色体的burkitt淋巴瘤。
[0039]
上述用途,所述基因异常是指基因突变、基因融合或重排和/或基因扩增。
[0040]
上述用途,所述异常染色体是指发生了扩增、缺失、断裂、重排和/或易位的染色体。
[0041]
上述方法,所述给予可以是口服给予、注射给予、局部给予或体外给予,优选为口服给予或注射给予。
[0042]
上述方法,化合物a或其药学上可接受的盐的施用剂量和剂量频率可以通过常规方法诸如建模、剂量递增研究或临床试验的常规方法和通过考虑诸如待治疗疾病的特性和严重程度、患者的年龄、一般情况和体重,以及所施用的具体化合物,它的药物代谢动力学特性,以及施用途径等因素进行确定。化合物a或其药学上可接受的盐合适的剂量范围为每天从约0.001mg/kg至约1000mg/kg;优选的,从约0.01mg/kg至约100mg/kg;进一步优选的,从约0.02mg/kg至约50mg/kg;更进一步优选的,从约0.03mg/kg至约20mg/kg。优选的,化合
物a或其药学上可接受的盐每天给药剂量为0.001mg-1000mg,进一步优选的,化合物a或其药学上可接受的盐每天给药剂量为0.01-500mg;更进一步优选的,化合物a或其药学上可接受的盐每天给药剂量为0.1-200mg;更进一步优选的,化合物a或其药学上可接受的盐每天给药剂量为0.5-100mg;更进一步优选的,化合物a或其药学上可接受的盐每天给药剂量为0.5-50mg;更进一步优选的,化合物a或其药学上可接受的盐每天给药剂量为0.5-30mg;更进一步优选的,化合物a或其药学上可接受的盐每天给药剂量为0.5-20mg;以单剂量或分剂量施用。
[0043]
另一方面,本发明还提供一种治疗患者的病症的方法,通过向患者施用含有治疗有效量的化合物a或其药学上可接受的盐的药物,所述患者的病症为btk相关肿瘤疾病。
[0044]
上述方法,所述btk相关肿瘤疾病包括血液肿瘤和实体瘤。
[0045]
优选地,上述血液肿瘤为淋巴瘤和白血病。
[0046]
进一步优选地,上述淋巴瘤为b细胞淋巴瘤。
[0047]
上述方法,所述btk相关肿瘤疾病包括组织细胞性淋巴瘤、套细胞淋巴瘤、弥漫性大b细胞淋巴瘤、慢性淋巴细胞白血病、小淋巴细胞淋巴瘤、边缘区淋巴瘤、滤泡性淋巴瘤、burkitt淋巴瘤或华氏巨球蛋白血症。
[0048]
上述用途,所述弥漫性大b细胞淋巴瘤选自非特殊类型的弥漫性大b细胞淋巴瘤、其他大b细胞淋巴瘤、伴myc和/或bcl基因异常的高级别b细胞淋巴瘤和非特殊类型的高级别b细胞淋巴瘤组成的组中的1种或多种;优选地,所述弥漫性大b细胞淋巴瘤选自其他大b细胞淋巴瘤、伴myc和/或bcl基因异常的高级别b细胞淋巴瘤和非特殊类型的高级别b细胞淋巴瘤组成的组中的1种或多种。
[0049]
上述用途,所述其他大b细胞淋巴瘤包括富于t细胞/组织细胞的dlbcl、原发中枢神经系统dlbcl、原发皮肤dlbcl(腿型)、ebv阳性的dlbcl,非特指型dlbcl、慢性炎症相关的大b细胞淋巴瘤、淋巴瘤样肉芽肿、伴有irf4重排的大b细胞淋巴瘤、原发纵隔(胸腺)大b细胞淋巴瘤、血管内大b细胞淋巴瘤、alk阳性大b细胞淋巴瘤、浆母细胞性淋巴瘤、hhv8阳性dlbcl、原发性渗出性淋巴瘤等;
[0050]
上述用途,所述伴myc基因异常的高级别b细胞淋巴瘤选自myc基因扩增的高级别b细胞淋巴瘤、myc基因融合的高级别b细胞淋巴瘤组成的组中的1种或多种;所述bcl基因异常的高级别b细胞淋巴瘤选自bcl2和/或bcl6基因异常的高级别b细胞淋巴瘤,所述bcl2和/或bcl6基因异常的高级别b细胞淋巴瘤选自bcl2基因扩增的高级别b细胞淋巴瘤、bcl2基因融合的高级别b细胞淋巴瘤、bcl6基因扩增的高级别b细胞淋巴瘤和bcl6基因融合的高级别b细胞淋巴瘤组成的组中的1种或多种。
[0051]
上述用途,本发明涉及的弥漫性大b细胞淋巴瘤也可以是携带异常染色体的弥漫性大b细胞淋巴瘤。
[0052]
上述的用途,所述套细胞淋巴瘤选自经典型套细胞淋巴瘤、白血病样非淋疤结性套细胞淋巴瘤和cyclin d1阳性的套细胞淋巴瘤组成的组中的1种或多种。
[0053]
上述的用途,所述套细胞淋巴瘤选自bcl基因异常的套细胞淋巴瘤;优选为bcl1和/或bcl2基因异常的套细胞淋巴瘤;进一步优选为bcl1基因融合或重排的套细胞淋巴瘤或bcl2基因融合或重排的套细胞淋巴瘤。
[0054]
上述的用途,本发明涉及的套细胞淋巴瘤也可以是携带异常染色体的套细胞淋巴
瘤。
[0055]
上述用途,所述burkitt淋巴瘤是携带异常染色体的burkitt淋巴瘤。
[0056]
上述用途,所述基因异常是指基因突变、基因融合或重排和/或基因扩增。
[0057]
上述用途,所述异常染色体是指发生了扩增、缺失、断裂、重排和/或易位的染色体。
[0058]
本发明所述化合物a或其药学上可接受的盐的治疗有效量、施用剂量或给药剂量,均以化合物a计。
[0059]
根据造血与淋巴组织肿瘤2017年版(第4版修订版),将弥漫大b细胞淋巴瘤(dlbcl)分为四类:非特殊类型的dlbcl、其他大b细胞淋巴瘤、高级别b细胞淋巴瘤及介于dlbcl和经典霍奇金淋巴瘤之间不能分类的b细胞淋巴瘤。其中,非特殊类型的dlbcl包括形态学上包括中心母细胞变型、免疫母细胞变型、间变变型以及其它少见的变型(如梭形细胞变型、印戒细胞样变型)等;其他大b细胞淋巴瘤包括富于t细胞/组织细胞的dlbcl、原发中枢神经系统dlbcl、原发皮肤dlbcl(腿型)、ebv阳性的dlbcl,非特指型dlbcl、慢性炎症相关的大b细胞淋巴瘤、淋巴瘤样肉芽肿、伴有irf4重排的大b细胞淋巴瘤、原发纵隔(胸腺)大b细胞淋巴瘤、血管内大b细胞淋巴瘤、alk阳性大b细胞淋巴瘤、浆母细胞性淋巴瘤、hhv8阳性dlbcl、原发性渗出性淋巴瘤等;高级别b细胞淋巴瘤包括伴myc、bcl2和/或bcl6异常(例如,基因重排或融合、基因扩增、基因突变等)的高级别b细胞淋巴瘤、非特殊类型的高级别b细胞淋巴瘤等,例如本技术中提及的u-2932细胞(伴bcl2基因扩增)、will-2细胞(伴bcl2基因融合)等。(来源:atcc官网信息和dsmz官网信息)
[0060]
本技术中提及的“携带异常染色体的弥漫性大b细胞淋巴瘤”是指携带有异常染色体(例如,染色体扩增、缺失、断裂、重排和/或易位等)的弥漫性大b细胞淋巴瘤,例如本技术中提及的pfeiffer细胞(存在多处染色体异常,包括染色体t(14;18)(q32;q21)易位)等。(来源:atcc官网信息和dsmz官网信息)
[0061]
本技术中提及的“携带异常染色体的套细胞淋巴瘤”是指携带有异常染色体(例如,染色体易位、染色体融合、染色体缺失等)的套细胞淋巴瘤,例如本技术中提及的z-138细胞(存在染色体异常,例如染色体t(11;14)(q13;q32)易位和/或染色体del(5)(p15)缺失等)、mino细胞(存在染色体异常,例如染色体del(6)(q16)缺失等)、rec-1细胞(存在染色体异常,例如染色体t(11;14)(q13;q32)易位等)。(来源:atcc官网信息和dsmz官网信息)
[0062]
本技术中提及的“bcl基因异常的套细胞淋巴瘤”是指存在bcl基因异常(例如,基因突变、基因扩增、基因重排或/融合、基因异常活化等)的套细胞淋巴瘤,可导致bcl相关蛋白过表达,例如本技术中提及的jeko-1细胞(存在bcl-1基因重排)等。(来源:atcc官网信息)
[0063]
本技术中提及的“携带异常染色体的burkitt淋巴瘤”是指携带有异常染色体(例如,染色体大小和/或数量的异常、染色体易位、染色体融合、染色体缺失等)的burkitt淋巴瘤,例如本技术中提及的raji细胞(1号或4号染色体大小和/数量异常)。(来源:atcc官网信息)
[0064]
化合物的药学上可接受的盐可以为所述化合物与无机酸、有机酸、无机碱或有机碱反应形成的常规的无毒盐。
[0065]
在该制备方法以及本发明中,使用的术语如下所述:
[0066]
dcm:二氯甲烷;diad:偶氮二甲酸二异丙酯;dipea:二异丙基乙胺;dmf:n,n-二甲基甲酰胺;ea:乙酸乙酯;hatu:2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯;nbs:n-溴代丁二酰亚胺;nis:n-碘代丁二酰亚胺;pdcl2(dppf):[1,1'-双(二苯基膦基)二茂铁]二氯化钯;pd(pph3)4:四(三苯基膦)钯;pdcl2:二氯化钯;pd(oac)2:醋酸钯;pd(pph3)2cl2:双三苯基磷二氯化钯;pe:石油醚;thf:四氢呋喃;dmso:二甲基亚砜。
[0067]
本发明取得了以下一种或多种有益的技术效果:
[0068]
(1)体内外药效试验结果显示,化合物a对btk和体外培养淋巴瘤细胞的抑制活性不仅优于前期化合物s1、s10、s18s、s19s和s20s,也优于目前已上市的第一代btk抑制剂依鲁替尼和第二代btk抑制剂阿卡替尼。在体内移植瘤模型中,化合物a也显示出了比s18s、依鲁替尼明显更优的抑制肿瘤生长的效果。
[0069]
(2)体外药效试验结果显示,化合物a对多种类型肿瘤细胞均具有良好的抑制效果,而且,对同种细胞类型的不同具体细胞株也具有良好的抑制效果,即化合物a对于同种肿瘤细胞类型、不同来源细胞株能实现较好的抑制效果,具有较好的临床应用前景。
[0070]
(3)药代动力学研究显示,化合物a的体内清除率显著低于依鲁替尼及其结构类似物s18s、s19s、s20s,仅为后者的1/10~1/20;化合物a口服后血浆中药物暴露量(auc)比依鲁替尼高70倍,绝对生物利用度明显增高。
[0071]
(4)s1、s10、s18s和s19s因末端苯基的4-位易于氧化,代谢产物种类多,且代谢稳定性差,s18s经大鼠肝微粒体孵育后仅能保留的原型药物51%。化合物a通过结构改造有效克服了上述化合物中二苯醚结构的末端苯环羟基化,在不同种属肝微粒体(hlm:人肝微粒体;rlm:大鼠肝微粒体;mlm:小鼠肝微粒体)作用下,代谢产物种类和占比少,基本以原型药物为主(60min:84%-98%),代谢稳定性更好。
[0072]
综上,化合物a具有良好的btk抑制活性和抑瘤作用,并且体内清除率低,口服生物利用度好,代谢稳定,具有重要的临床应用价值。
附图说明
[0073]
图1:rec-1异种移植瘤模型的实验结果示意图。
[0074]
图2:tmd8异种移植瘤模型的实验结果示意图。
具体实施方式
[0075]
以下进一步提供实施实例,这些实施实例有助于理解本发明,仅用作说明而不限制本发明的应用范围。
[0076]
实施例1:化合物a的合成
[0077]
1、中间体3的合成
[0078][0079]
在250ml圆底烧瓶中加入4-氯-5-碘-7h-吡咯并[2,3-d]嘧啶(原料2,17.28g,1eq)
和无水碳酸钾(2eq),真空干燥除水。加入干燥的dmf作为溶剂,粉碎的(s)-甲磺酸2-((叔丁氧基羰基)氨基)-丁-3-烯-1-基酯(原料1,24.6g,1.5eq),置换氮气。于55℃下加热搅拌12小时,时间可适当延长以确保反应完全。
[0080]
反应完成后,加入水和乙酸乙酯萃取三次,合并酯层,用水反萃一次,用饱和食盐水洗。无水硫酸钠干燥。干法过柱(洗脱液:chcl3:meoh=100:1)得产品(s)-(1-(4-氯-5-碘-7h-吡咯并[2,3-d]嘧啶-7-基)丁-3-烯-2-基)氨基甲酸叔丁酯(中间体3,19.28g),产率为69.5%。
[0081]1h nmr(300mhz,cdcl3)δ8.60(s,1h),7.39(s,1h),5.82(ddd,j=17.1,10.5,5.5hz,1h),5.33-5.14(m,2h),4.80(s,1h),4.63-4.51(m,1h),4.51-4.42(m,1h),4.35(s,1h),1.33(s,9h)。ee》99.5%。
[0082]
2、中间体4的合成
[0083][0084]
在350ml耐压管中加入(s)-(1-(4-氯-5-碘-7h-吡咯并[2,3-d]嘧啶-7-基)丁-3-烯-2-基)氨基甲酸叔丁酯(中间体3,9.2g),加入1,4-二氧六环(40ml)作为溶剂,加入氨水(40ml)。于120℃下密封反应2.5小时。
[0085]
反应完成后,冷却至室温,加入水和乙酸乙酯萃取,合并酯层,用饱和食盐水洗。无水硫酸钠干燥。干法过柱(洗脱液:chcl3:meoh=30:1)得产品(s)-(1-(4-氨基-5-碘-7h-吡咯并[2,3-d]嘧啶-7-基)丁-3-烯-2-基)氨基甲酸叔丁酯(中间体4,6.86g),产率为78.0%。
[0086]1h nmr(300mhz,cdcl3)δ8.25(s,1h),7.05(s,1h),5.87-5.74(m,1h),5.72(s,2h),5.34-5.13(m,3h),4.56-4.43(m,1h),4.34(dd,j=14.8,4.9hz,1h),4.30-4.15(m,1h),1.35(s,9h)。ee》99.5%。
[0087]
3、中间体6的合成
[0088][0089]
在1l圆底烧瓶中加入(s)-(1-(4-氨基-5-碘-7h-吡咯并[2,3-d]嘧啶-7-基)丁-3-烯-2-基)氨基甲酸叔丁酯(中间体4,32.9g,1eq),n-(吡啶-2-基)-4-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)苯甲酰胺(原料5,34.8g,1.4eq)和pd(pph3)4(17.7g,0.2eq)。加入1,4-二氧六环(383ml)作为溶剂,并置换n2。在搅拌下加入2m碳酸钠溶液(76.6ml)。于
90℃下回流搅拌5小时。
[0090]
加入水与乙酸乙酯萃取,合并酯层,用饱和食盐水洗。无水硫酸钠干燥。干法过柱,先用ea作为洗脱液除去大部分杂质,然后用chcl3:meoh=30:1的混合物作为洗脱液。产品可能含有少量杂质,可用pe重结晶析出纯品。得产品(s)-(1-(4-氨基-5-(4-(吡啶-2-基氨基甲酰基)苯基)-7h-吡咯并[2,3-d]嘧啶-7-基)丁-3-烯-2-基)氨基甲酸叔丁酯(中间体6,28.4g),产率为74.3%。ee》99.5%。
[0091]
4、中间体7的合成
[0092][0093]
在1l圆底烧瓶中加入(s)-(1-(4-氨基-5-(4-(吡啶-2-基氨基甲酰基)苯基)-7h-吡咯并[2,3-d]嘧啶-7-基)丁-3-烯-2-基)氨基甲酸叔丁酯(中间体6,32.7g,1eq),加入600ml dmf作为溶剂。在搅拌下缓慢加入nbs(12.8g,1.1eq),于室温下搅拌过夜。
[0094]
反应完成后,加入水与乙酸乙酯萃取,合并酯层,用水反萃一次,饱和食盐水洗。无水硫酸钠干燥。干法过柱,首先用chcl3:meoh=50:1的混合物作为洗脱液,然后换chcl3:meoh=30:1的混合物作为洗脱液。得产品(s)-(1-(4-氨基-6-溴-5-(4-(吡啶-2-基氨基甲酰基)苯基)-7h-吡咯并[2,3-d]嘧啶-7-基)丁-3-烯-2-基)氨基甲酸叔丁酯(中间体7,25.8g),产率为68.2%。ee》99.5%。
[0095]
5、中间体8的合成
[0096][0097]
在250ml圆底烧瓶中加入(s)-(1-(4-氨基-6-溴-5-(4-(吡啶-2-基氨基甲酰基)苯基)-7h-吡咯并[2,3-d]嘧啶-7-基)丁-3-烯-2-基)氨基甲酸叔丁酯(中间体7,11.9g,1eq)和pdcl2(dppf)(1.66g,0.11eq),加入51ml thf作为溶剂,并置换氮气,多置换几次以确保完全。在搅拌下加入4m氢氧化钠溶液(8.2ml)。于85℃下回流搅拌15小时。
[0098]
反应完全后,加入水与乙酸乙酯萃取,合并酯层,用饱和食盐水洗。无水硫酸钠干燥。干法过柱(洗脱液:chcl3:meoh=30:1)得产品(s)-(4-氨基-6-亚甲基-5-(4-(吡啶-2-基氨基甲酰基)苯基)-7,8-二氢-6h-嘧啶并[5,4-b]吡呤-7-基)氨基甲酸叔丁酯(中间体8,
8.79g),产率为85.9%。ee》99.5%。
[0099]
6、化合物a的合成
[0100][0101]
在250ml圆底烧瓶中加入(s)-(4-氨基-6-亚甲基-5-(4-(吡啶-2-基氨基甲酰基)苯基)-7,8-二氢-6h-嘧啶并[5,4-b]吡呤-7-基)氨基甲酸叔丁酯(中间体8,2.75g,1eq),加入110ml dcm作为溶剂。于搅拌下逐滴加入三氟乙酸(10.5ml)。于室温下搅拌3小时。反应完成后,直接将反应液旋干,多用甲醇带几次将三氟乙酸带出,旋干后得到氨基脱boc保护粗品,直接投下一步。
[0102]
将上一步产物移至250ml圆底烧瓶中,再加入三乙胺(1eq),搅拌五分钟后再加入2-丁炔酸(0.511g,1.1eq)和hatu(2.31g,1.1eq),加入100ml dcm作为溶剂。冰水浴降温至0℃,逐滴加入三乙胺(1.54ml 0.77ml)。逐渐升温至室温,于室温下搅拌1.5小时。反应液呈微黄色。加入水和dcm萃取,合并有机相,用饱和食盐水洗。无水硫酸钠干燥,柱层析(chcl3:meoh=30:1)后得终产物a(1.88g),产率为73.3%。ee》99.5%。
[0103]
1h nmr(400mhz,cdcl3)δ8.98(s,1h),8.43(dt,j=8.3,1.0hz,1h),8.34(ddd,j=5.0,1.9,0.9hz,1h),8.22(s,1h),8.09-8.03(m,2h),7.81(ddd,j=8.4,7.4,1.9hz,1h),7.69-7.63(m,2h),7.13(ddd,j=7.4,4.9,1.0hz,1h),6.55(d,j=8.2hz,1h),5.67(m,j=8.1,5.7,2.6hz,1h),5.56(d,j=2.3hz,1h),5.40(s,2h),5.27(d,j=2.3hz,1h),4.70(dd,j=11.7,8.1hz,1h),4.09-3.99(m,1h),1.99(s,3h)。
[0104]
实验例1:布鲁顿激酶(btk)分子水平酶活抑制活性评价
[0105]
将酶反应底物poly(glu,tyr)
4:1
用无钾离子的pbs(10mm磷酸钠缓冲液,150mm nacl,ph 7.2-7.4)稀释成20μg/ml包被酶标板,在37℃下反应12-16小时后,用200μl/孔的t-pbs(含0.1%tween-20的pbs)洗板三次,于37℃烘箱中干燥酶标板1-2小时。在以上包被底物的酶标板中,首先加入用反应缓冲液(50mm hepes ph 7.4,50mm mgcl2,0.5mm mncl2,0.2mm na3vo4,1mm dtt)稀释的atp溶液49μl/孔(终浓度为5μm)。每孔中加入1μl待测试化合物(化合物孔)或含相应浓度的dmso(阴性对照孔),每次实验需设无酶对照孔。再加入50μl以反应缓冲液稀释的btk酪氨酸激酶蛋白启动反应。
[0106]
将上述反应体系置于37℃摇床(100rpm)中1小时,然后t-pbs洗板三次,加入一抗py99100μl/孔(santa cruz),37℃摇床反应0.5小时。t-pbs洗板后,加入辣根过氧化物酶标记的羊抗鼠二抗稀释液100μl/孔,37℃摇床反应0.5小时。t-pbs洗板后,加入2mg/ml的opd显色液100μl/孔,25℃避光反应1-10分钟。然后加入2m h2so
4 50μl/孔中止反应,用可调波长式微孔板酶标仪spectra max plus384读数,波长为490nm。
[0107]
以化合物s1、s10、依鲁替尼、阿卡替尼、s18s、s19s和s20s作为阳性对照化合物,其中,化合物s1、s10、s18s、s19s和s20s采用现有技术(例如cn108101905a)中公开的方法或类似方法制备,依鲁替尼和阿卡替尼购自selleck公司。
[0108]
各化合物的抑制率通过下列公式求得:
[0109][0110]
ic
50
值采用酶标仪随机附带软件以四参数法回归求得。结果列于下表1中。
[0111]
表1不同化合物对btk的抑制作用
[0112]
化合物ic
50
(nm)s1~1s10《10依鲁替尼~1阿卡替尼~10s18s~1s19s~1s20s~1化合物a0.5
[0113]
以上结果表明,化合物a对btk的抑制活性优于前期化合物s1、s10、s18s、s19s和s20s,也优于目前已上市的第一代btk抑制剂依鲁替尼和第二代btk抑制剂阿卡替尼。
[0114]
实验例2:化合物对人b淋巴瘤细胞的体外增殖抑制活性检测
[0115]
实验细胞
[0116][0117][0118]
注:dlbcl:弥漫性大b细胞淋巴瘤;fbs:胎牛血清;2-mercaptoethanol:2-巯基乙醇
[0119]
试验方法:
[0120]
将细胞悬液(ramos:10000细胞/孔;tmd8:12000细胞/孔)接种于96孔板中,于37℃培养箱静置2小时待细胞状态稳定后,每孔加入不同浓度的受试化合物(每个浓度设3个复孔),并同时设置空白对照(仅包含培养液,不含细胞的孔)、阴性对照(仅加细胞,不加化合物的孔)及阳性化合物对照。加药处理72h后,每孔加入20μl mtt(5mg/ml)于37℃孵育4h,加入100μl三联液(10%sds,5%异丁醇,0.01m hcl),37℃放置过夜。用可调波长式微孔板酶标仪spectramax plus384在570nm波长条件下测定od值。
[0121]
化合物的抑制率通过下列公式求得:
[0122][0123]
ic
50
值采用酶标仪随机附带软件以四参数法回归求得。实验独立重复3次,其结果列于下表2中。
[0124]
同样以上文所述化合物s1、s10、依鲁替尼、阿卡替尼、s18s、s19s和s20s作为阳性对照化合物。
[0125]
表2不同化合物对ramos细胞和tmd8细胞的增殖抑制活性
[0126]
化合物ramos细胞ic
50
tmd8细胞ic
50
s194.73μm0.006μms108.72μm0.030μm依鲁替尼12.91μm0.005μm阿卡替尼38.16μm0.023μm化合物a3.15μm0.003μms18s5.04μm0.016μms19s—0.017μms20s14.3μm0.004μm
[0127]
以上结果表明,在细胞水平,化合物a对b细胞淋巴瘤的增殖抑制能力优于前期化合物s1、s10、s18s、s19s和s20s,也优于目前已上市的第一代btk抑制剂依鲁替尼和第二代btk抑制剂阿卡替尼。进一步需要说明的是,与其他化合物相比,本发明的化合物a对于ramos细胞具有较高的增殖抑制活性,且对于tmd8细胞具有更高的增殖抑制活性。
[0128]
实验细胞
[0129][0130][0131]
注:dlbcl:弥漫性大b细胞淋巴瘤;fl:滤泡性淋巴瘤;mcl:套细胞淋巴瘤;pmbcl:原发性纵隔b细胞淋巴瘤
[0132]
试验方法
[0133]
采用cck8(cell counting kit-8,#d3100l4057,上海李记生物科技有限公司)染色法检测化合物对细胞的增殖抑制活性。
[0134]
(1)处于生长对数期的肿瘤细胞根据其不同的生长速度,将适当密度的细胞悬液接种于96孔板中,每孔95μl。37℃培养箱静置2-4h待细胞状态稳定后,依次加入梯度稀释的所需浓度化合物10μl,每剂量设3个复孔,同时设置溶剂对照和无细胞的空白对照孔。于37℃二氧化碳培养箱培养72h。
[0135]
(2)加入cck8染色液,10μl/孔。培养箱孵育2-4h,用酶标仪spectramax plus 384读数,测定波长为450nm。化合物对细胞的生长抑制率计算公式为:
[0136]
抑制率%=(对照组od值-给药组od值)/对照组od值
×
100%。
[0137]
半数抑制量ic
50
值采用四参数法计算。每个实验独立重复3次,求出各次实验的平均ic
50
值作为抑制能力的最终指标。
[0138]
表3化合物a对b淋巴瘤细胞(btk抑制剂敏感细胞株)的增殖抑制活性
[0139][0140]
以上结果表明,化合物a对2株btk抑制剂敏感细胞株的抑制活性较强,且优于阳性对照药依鲁替尼和阿卡替尼。
[0141]
表4化合物a对b淋巴瘤细胞的增殖抑制活性
[0142][0143][0144]
以上结果表明,化合物a对其他b淋巴瘤细胞的抑制活性,或者与依鲁替尼相当,或者介于依鲁替尼和阿卡替尼之间,均为微摩尔水平。表3和表4的结果共同表明,相比阳性对照药,化合物a对btk抑制剂敏感细胞株抑制作用更明显,而对其他b淋巴瘤细胞也能具有一定的抑制活性,即具有更好的btk抑制选择性。
[0145]
由以上表3和表4的结果表明,化合物a对u-2932、will-2的抑制作用提示本发明的化合物a预期具有治疗弥漫性大b细胞淋巴瘤中伴bcl基因异常的高级别b细胞淋巴瘤的作用;化合物a对pfeiffer的抑制作用提示本发明的化合物a预期具有治疗弥漫性大b细胞淋巴瘤中携带异常染色体的弥漫性大b细胞淋巴瘤的作用;化合物a对rl的抑制作用提示本发明的化合物a预期具有治疗滤泡性淋巴瘤的作用;化合物a对raji、namalwa的抑制作用提示本发明的化合物a预期具有治疗burkitt淋巴瘤的作用;化合物a对raji的抑制作用提示本发明化合物a预期具有治疗携带异常染色体的burkitt淋巴瘤的作用;化合物a对z-138、mino、rec-1的抑制作用提示本发明的化合物a预期具有治疗套细胞淋巴瘤中携带异常染色体的套细胞淋巴瘤的作用;化合物a对jeko-1的抑制作用提示本发明的化合物a预期具有治疗套细胞淋巴瘤中携带异常染色体的套细胞淋巴瘤的作用;化合物a对karpas-1106p的抑制作用提示本发明的化合物a预期具有治疗弥漫性大b细胞淋巴瘤中原发性纵隔b细胞淋巴瘤的作用。
[0146]
另外,鉴于化合物a对oci-ly10、rec-1(btk抑制剂敏感细胞株)的抑制活性优于阳性对照药依鲁替尼和阿卡替尼的抑制活性,并且对其他b淋巴瘤细胞的抑制活性与依鲁替尼相当,或者介于依鲁替尼和阿卡替尼之间的结果提示:本发明的化合物a预期能够治疗依鲁替尼和阿卡替尼所治疗的疾病,例如慢性淋巴细胞白血病、携带17p删除的慢性淋巴细胞白血病/小淋巴细胞淋巴瘤、华氏巨球蛋白血症、边缘区淋巴瘤、慢性移植物抗宿主病。
[0147]
实验例3:体内抗肿瘤活性评价
[0148]
实验动物:
[0149]
tmd8异种移植瘤模型
[0150]
1)种属:小鼠
[0151]
2)品系:cb-17scid
[0152]
3)周龄及体重:6-8周;18-22g
[0153]
4)性别:雌性
[0154]
5)供应商:北京维通利华实验动物技术有限公司
[0155]
rec-1异种移植瘤模型
[0156]
1)种属:小鼠
[0157]
2)品系:balb/c裸小鼠
[0158]
3)周龄及体重:6-8周;17-20g
[0159]
4)性别:雌性
[0160]
5)供应商:上海灵畅生物科技有限公司
[0161]
细胞培养:人淋巴癌tmd8细胞体外悬浮培养,培养条件为rpmi 1640培养基(供应商:gibco;货号:22400-089;生产批号:4868546)中加10%胎牛血清,100u/ml青霉素和100μg/ml链霉素,37℃5%co2培养。一周两次进行常规处理传代。当细胞饱和度为80%-90%时,收取细胞,计数,接种。
[0162]
人套细胞淋巴癌rec-1细胞体外悬浮培养,培养条件为rpmi 1640培养基(供应商:gibco;货号:22400-089;生产批号:1868795)中加10%胎牛血清,100u/ml青霉素和100μg/ml链霉素,37℃5%co2培养。一周两次进行常规处理传代。当细胞饱和度为80%-90%时,收取细胞,计数,接种。
[0163]
肿瘤细胞接种:将0.2ml 10
×
106个人淋巴癌tmd8细胞皮下接种于每只裸小鼠的右后背(pbs:matrigel=1:1)。肿瘤平均体积达到104mm3时开始分组给药。根据动物肿瘤体积通过一个基于excel随机分组软件进行分组,每组6只小鼠。
[0164]
将0.2ml 5
×
106个rec-1细胞皮下接种于每只裸小鼠的右后背(pbs:matrigel=1:1)。肿瘤平均体积达到100mm3时开始分组给药。根据动物肿瘤体积通过一个基于excel随机分组软件进行分组,每组6只小鼠。
[0165]
受试物的配制:
[0166]
受试物配制方法参见下表5和表6:
[0167]
表5 tmd8异种移植瘤模型受试物配制方法
[0168][0169]
注:样品现配现用,配置好的样品于4℃条件下保存,并在给动物给药前需要轻轻将药物充分混匀;给药方式:灌胃;给药体积10μl/g
[0170]
表6 rec-1异种移植瘤模型受试物配制方法
[0171][0172][0173]
注:样品现配现用,配置好的样品于4℃条件下保存,并在给动物给药前需要轻轻将药物充分混匀;给药方式:灌胃;给药体积10μl/g。
[0174]
实验动物日常观察:本实验方案的拟定及任何修改均通过了苏州药明康德新药开发股份有限公司实验动物管理与使用委员会(iacuc)的评估核准。实验动物的使用及福利遵照国际实验动物评估和认可委员会(aaalac)的规定执行。每天监测动物的健康状况及死亡情况,例行检查包括观察肿瘤生长和药物治疗对动物日常行为表现的影响如行为活动,摄食摄水量(仅目测),体重变化(每周测量三次体重),外观体征或其它不正常情况。基于各组动物数量记录了组内动物死亡数和副作用。
[0175]
肿瘤测量和实验指标:实验指标是考察肿瘤生长是否被抑制、延缓或治愈。每周三次用游标卡尺测量肿瘤直径。
[0176]
肿瘤体积的计算公式为:
[0177]
v=0.5a
×
b2,
[0178]
a和b分别表示肿瘤的长径和短径。
[0179]
化合物的抑瘤疗效用tgi(%)或相对肿瘤增殖率t/c(%)评价。tgi(%),反映肿瘤生长抑制率。
[0180]
tgi(%)的计算:
[0181]
tgi(%)=【1-(某处理组给药结束时平均瘤体积-该处理组开始给药时平均瘤体积)/(溶剂对照组治疗结束时平均瘤体积-溶剂对照组开始治疗时平均瘤体积)】
×
100%。
[0182]
相对肿瘤增殖率t/c(%):计算公式如下:
[0183]
t/c%=t
rtv
/c
rtv
×
100%(t
rtv
:治疗组相对肿瘤体积;c
rtv
:阴性对照组相对肿瘤体
积)。根据肿瘤测量的结果计算出相对肿瘤体积(rtv),计算公式为rtv=v
t
/v0,其中v0是分组给药时(即d0)测量所得平均肿瘤体积,v
t
为某一次测量时的平均肿瘤体积,t
rtv
与c
rtv
取同一天数据。
[0184]
统计分析:统计分析,包括每个组的每个时间点的肿瘤体积的平均值和标准误(sem)。治疗组在给药后分别在第15天(rec-1异种移植瘤模型)和第17天(tmd8异种移植瘤模型)表现出最好的治疗效果,因此基于此数据进行统计学分析评估组间差异。三组或多组间比较用one-way anova进行分析,如果f值有显著性差异,应用games-howell法进行检验。用spss 17.0进行所有数据分析。p《0.05认为有显著性差异。
[0185]
化合物a在人套细胞淋巴癌rec-1异种移植瘤模型中的体内药效如表7和图1所示。开始给药后15天,溶剂对照组荷瘤鼠的瘤体积达到3501mm3,依鲁替尼25mg/kg组瘤体积为1323mm3,与溶剂对照组相比具有显著的抑瘤作用(t/c=38%,tgi=64%,p=0.008)。化合物a15mg/kg和30mg/kg组的瘤体积分别为1034和680mm3,与溶剂对照组相比具有显著的抑瘤作用(t/c值分别为30%和19%,tgi值分别为73%和83%,p值均小于0.01),与阳性药依鲁替尼25mg/kg组相比,化合物a15mg/kg组的抑瘤作用更优。
[0186]
表7化合物a对rec-1异种移植瘤模型的抑瘤药效评价(基于给药后第15天肿瘤体积计算得出)
[0187][0188]
注:a.平均值
±
sem;b.肿瘤生长抑制由t/c和tgi(tgi(%)=[1-(t
15-t0)/(v
15-v0)]
×
100)计算;c.给药方式:每天一次;**:p《0.01。
[0189]
化合物a在人淋巴癌tmd8异种移植瘤模型中的体内药效如表8和图2所示。开始给药后17天,溶剂对照组荷瘤鼠的瘤体积达到1852mm3,依鲁替尼25mg/kg组瘤体积为661mm3与,溶剂对照组相比具有显著的抑瘤作用(t/c=35.68%,tgi=68.18%,p《0.001)。化合物a 5mg/kg和10mg/kg组的瘤体积分别为912mm3和553mm3,与溶剂对照组相比具有显著的抑瘤作用(t/c值分别为49.27%和29.85%,tgi值分别为53.78%和74.35%,p值均小于0.01),与阳性药依鲁替尼25mg/kg组相比,化合物a10mg/kg组的抑瘤作用更优。
[0190]
表8化合物a对tmd8异种移植瘤模型的抑瘤药效评价(基于给药后第17天肿瘤体积计算得出)
[0191][0192]
注:a.平均值
±
sem;b.肿瘤生长抑制由t/c和tgi(tgi(%)=[1-(t
17-t0)/(v
17-v0)]
×
100)计算;c.给药方式:每天一次;**:p《0.01。
[0193]
结果表明,在两种btk敏感的小鼠移植瘤模型中,化合物a具有显著的肿瘤生长抑制活性,明显优于目前已上市的第一代btk抑制剂依鲁替尼。
[0194]
另外,使用化合物s18s,重复上述在人淋巴癌tmd8异种移植瘤模型中的实验,t/c(%)结果列于下表中。在下表中,还列出了化合物a的t/c(%)结果作为对比。
[0195]
表9化合物a及s18对tmd8异种移植瘤模型的抑瘤效果
[0196][0197][0198]
注:给药方式:每天一次。
[0199]
由上述数据可以看出,在更低剂量下(10mg/kg),化合物a表现出较化合物s18s(15mg/kg)更好的肿瘤生长抑制效果。
[0200]
实验例4:大鼠药代动力学性质评价
[0201]
sd大鼠14只,雄性,体重200-220g,随机分成4组,每组4/3只,分别灌胃和静脉给予受试化合物,具体安排见下表10:
[0202]
表10受试化合物给药方法
[0203]
组别动物数化合物给药途径给药剂量(mg/kg)14化合物a灌胃(po)323化合物a静脉(iv)134依鲁替尼灌胃(po)343依鲁替尼静脉(iv)1
[0204]
注:灌胃给药以含1%吐温80的0.5%羧甲基纤维素钠(cmc-na)配制,配置药物浓
度为0.3mg/ml;静脉给药以5%dmso/5%吐温80/90%生理盐水配制成溶液,配置给药浓度为0.2mg/ml。
[0205]
试验前禁食12小时,自由饮水。给药后2小时统一进食。
[0206]
采血时间点及样品处理:
[0207]
灌胃给药:给药后0.25,0.5,1.0,2.0,4.0,6.0,8.0和24小时;
[0208]
静脉给药:给药后5分钟,0.25,0.5,1.0,2.0,4.0,6.0,8.0和24小时;
[0209]
在以上设定时间点经大鼠眼球后静脉丛取静脉血0.3ml,置肝素化试管中,11000rpm离心5分钟,分离血浆,于-20℃冰箱中冷冻。
[0210]
样品测试和数据分析
[0211]
采用lc/ms/ms法测定大鼠血浆中化合物a的浓度。
[0212]
采用winnonlin 5.3软件(美国pharsight公司)的非房室模型计算给药后的药代动力学参数。
[0213]
达峰浓度c
max
和达峰时间t
max
为实测值;
[0214]
药时曲线下面积auc
0-t
值:采用梯形法计算;
[0215]
auc
0-∞
=auc
0-t
c
t
/ke,
[0216]ct
为最后一个可测得时间点的血药浓度,
[0217]
ke为消除速率常数;
[0218]
消除半衰期t
1/2
=0.693/ke;
[0219]
平均滞留时间mrt=aumc/auc。
[0220]
清除率cl=d/auc
0-∞
;稳态分布容积vss=cl
×
mrt
[0221]
绝对生物利用度f=(auc
灌胃
×d静脉
)/(auc
静脉
×d灌胃
)
×
100%
[0222]
试验结果见下表11:
[0223]
表11不同化合物的药代动力学实验结果
[0224][0225]
注:以上s18s、s19s和s20s的药代数据摘自“yu xue,et al.discovery of4,7-diamino-5-(4-phenoxyphenyl)-6-methylenepyrimido[5,4-b]pyrrolizines as novel bruton’s tyrosine kinase inhibitors.j.med.chem.,2018,61,4608-4627.”[0226]
以上结果表明,化合物a在大鼠体内清除率显著低于依鲁替尼(20倍),口服后血浆中药物暴露量也比依鲁替尼高达70倍;化合物a的体内清除率也显著高于s18s、s19s和s20s,口服后血浆中药物暴露量显著高于s18s、s19s和s20s。即同等剂量下,相比依鲁替尼、s18s、s19s和s20s,化合物a的口服给药性能更好,且具有良好的口服生物利用度。
[0227]
因此,化合物a是一个结构新颖的、可口服的、高选择性的、高活性的btk抑制剂,体内外活性明显优于目前国外已上市的btk抑制剂,在同等剂量下,对肿瘤生长抑制活性显著优于阳性对照药依鲁替尼,极具开发价值。
[0228]
以上实施方式本质上仅为辅助说明,且并不欲用以限制申请目标的实施例或这些实施例的应用或用途。在本文中,用语“例示性”代表“作为一个实例、范例或说明”。本文中任一种例示性的实施形态并不必然可解读为相对于其他实施形态而言为优选或较有利者。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。