1.本发明属于化学合成领域,具体涉及一种噻托溴铵的制备方法。
背景技术:
2.慢性阻塞性肺疾病(copd)是一种致死率和致残率很高的疾病,由于其患病人数多较,社会经济负担重,已成为世界各国的公共卫生问题。目前临床上用于治疗copd一线药物是异丙托溴铵,而由德国boehringeringelheim公司首先开发并在荷兰和菲律宾上市销售的新型长效药物噻托溴铵(tiotropium bromide),舒张支气管的作用优于异丙托溴铵,且在有效治疗剂量下未出现明显副作用,因而噻托溴铵在copd治疗中表现出良好的应用前景。
3.噻托溴铵是一种白色固体的化学品,无臭无味,在二甲基甲酰胺中溶解,在甲醇和水中微溶,在氯仿中几乎不溶,化学名称为(1α,2β,4β,5α,7β)3-恶-9氮翁三环[3,3,1,0
2,4
]壬烷,7-[羟基-二-2-噻吩乙酰)氧基]-9,9-二甲基,溴,一水合物,具有如下结构式:
[0004][0005]
cn1861598a公开的噻托溴铵合成方法中,先以氢溴酸东莨菪碱为重要原料合成东莨菪醇,然后通过格式试剂制备2,2-二噻吩基乙醇酸甲酯,之后再合成二(2-噻吩基)乙醇酸东莨菪酯,最终经过乙腈-甲醇以及活性炭重结晶制备出噻托溴铵,反应过程如下。
[0006]
[0007][0008]
该路线操作过程过于复杂,引入过多有机小分子也会带来大量的后处理流程,不适合工业上大规模生产。
技术实现要素:
[0009]
本发明的目的是通过一种工艺流程时间短、操作简单、成本低、质量好、适合工业化生产的方法制备噻托溴铵。
[0010]
本发明中噻托溴铵的制备方法包括以下步骤:
[0011]
(1)在盐酸盐反应罐中加入氢溴酸东莨菪碱、有机溶剂,搅拌降温后,于10~15℃温度下每隔10~15min加入还原剂,分3~5次加入,密闭保温反应,然后降温至5℃以下,通入干燥的氯化氢气体调节ph,加入乙醚搅拌后静置,过滤真空干燥得东莨菪醇盐酸盐;
[0012]
(2)将东莨菪醇盐酸盐加入到碱化反应罐中,加入二氯甲烷,回流通入氨气调节ph,过滤至东莨菪醇浓缩罐,减压浓缩至无溶剂蒸出,得无色至略黄色微粘液体东莨菪醇,用甲苯淋洗得东莨菪醇洗涤液;
[0013]
(3)将东莨菪醇洗涤液加入到噻托溴铵前体反应罐,加入二(2-溴噻吩)羟乙酸甲酯,开真空,升温至60~65℃,每隔15~20min加入钠屑,分2~4次加入,抽真空后在60~65℃下保温搅拌,然后升温搅拌反应;
[0014]
(4)搅拌反应结束后,在惰性气氛保护下往前体反应罐中加入甲苯,然后加入到装有稀盐酸的前体物洗涤罐中,搅拌,静置分取酸水层,有机层再用稀盐酸提取;前体反应罐中未溶解的固体在氮气保护下用二氯甲烷溶解,加入到装有稀盐酸的前体物洗涤罐中,搅拌,静置分取酸水层,有机层再用稀盐酸提取;合并酸水层,用乙醚洗涤后,酸水层转入前体物析料罐中冷却,用碱性溶液调节ph,过滤后用二氯甲烷溶解水洗,用无水硫酸钠脱水干燥,过滤浓缩至干,加入乙腈回流溶解,再加入活性炭回流脱色,过滤浓缩至糊状,冷却过
滤,真空干燥得二(2-噻吩基)乙醇酸东莨菪酯;
[0015]
(5)开溴甲烷钢瓶阀门,使溴甲烷液化气经缓冲罐气化后进入放有乙腈的配制罐,放出一定量溴甲烷后关闭阀门,乙腈吸收完溴甲烷后,即得溴甲烷-乙腈溶液。将二(2-噻吩基)乙醇酸东莨菪酯加入到噻托溴铵粗品反应罐中,二氯甲烷溶解后加入配制好的溴甲烷-乙腈溶液,搅拌均匀后反应,过滤干燥得到类黄色固体,用甲醇及丙酮加热溶解,减压浓缩至糊状,冷却过滤干燥得噻托溴铵粗品;
[0016]
(6)在成品溶解脱色罐中加入噻托溴铵粗品,加纯化水及丙酮加热回流溶解,加活性炭搅拌脱色过滤,加热溶解澄清后降温过滤,用纯水、丙酮淋洗罐壁、活性炭和物料,抽干出料,真空干燥得噻托溴铵。
[0017]
反应反应式如下:
[0018][0019][0020]
现有技术中多用已有的东莨菪醇来合成噻托溴铵中间体,但是东莨菪醇是一种敏感化合物,在空气中不稳定,倾向于液化,会影响反应过程的实施。本发明中该反应在反应罐中进行,可有效避免东莨菪醇不稳定性带来的负面影响。反应过程中使用氯化氢气体、氨气、碱性溶液来调节ph,严格控制反应过程中的酸碱度,以得到最好的合成效果和产品收率。
[0021]
进一步地,步骤(1)中还原剂为草酸(h2c2o4),硼氢化钾(kbh4),硼氢化钠(nabh4)和亚硫酸钠(na2so3)中的一种或多种,优选为硼氢化钠,硼氢化钠加入体系会升温并产生氢气和硼烷气体,分次加入有利于控制反应温度和反应罐内压力。
[0022]
进一步地,步骤(1)中有机溶剂为乙醚、无水乙醇、丙酮、正丙醚、正丁醚中的一种或多种,优选为乙醚。
[0023]
进一步地,步骤(1)中密闭保温反应的温度为18~22℃,时间为3~5h,真空干燥的温度为25~35℃,时间为9~11h。
[0024]
进一步地,步骤(1)中氯化氢气体调节ph值为1~1.5。
[0025]
进一步地,步骤(2)中氨气调节ph值为11.2~12,反应体系为非均相反应,ph值11.2~12能保证东莨菪醇盐酸盐反应完全,提高东莨菪醇收率。
[0026]
进一步地,步骤(3)中升温搅拌反应的温度是80~90℃,时间为3~5h。
[0027]
进一步地,步骤(4)中酸水层转入前体物析料罐中冷却至0~10℃,真空干燥的温度为50~60℃,时间为13~15h。
[0028]
进一步地,步骤(4)中调节ph的碱性溶液为碳酸钠溶液、碳酸氢钠、醋酸钠或者氢氧化钠溶液中的一种,优选饱和碳酸钠溶液,调节ph值为11~12。
[0029]
进一步地,步骤(4)中惰性气氛为氮气、氦气、氩气中的一种,优选为氮气。
[0030]
进一步地,步骤(5)中反应温度为20~30℃,反应时间为20~24h,干燥温度为95~105℃,干燥时间为2~5h。
[0031]
相比现有技术,本发明具有以下优点:
[0032]
(1)本发明方法多次调节反应的ph值,控制好反应过程的酸碱度,可以有效控制反应产物的纯度和收率;
[0033]
(2)本发明方法合成的东莨菪醇纯度达到94.23%,异构率较小;
[0034]
(3)本发明方法所得噻托溴铵纯度达到99.87%,最大杂质0.07%,符合ep标准;
[0035]
(4)本方法中硼氢化钠和钠屑分次加入,有利于控制反应温度和反应罐内压力,可以保证反应的安全,所得噻托溴铵的纯度大于99%,收率大于93%,相比一次性加入都有提高;
[0036]
(5)本发明方法合成中间体操作简单,成本低、质量好、适合工业化生产。
附图说明
[0037]
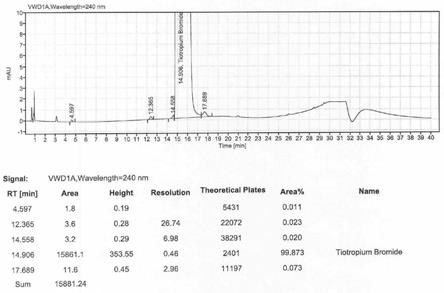
图1是本发明实施例1的噻托溴铵液相图谱;
[0038]
图2是本发明实施例1的噻托溴铵红外图谱;
[0039]
图3是本发明实施例1的东莨菪醇液相图谱;
[0040]
图4是本发明实施例1的东莨菪醇红外图谱。
具体实施方式
[0041]
下面通过具体实施例和附图,对本发明的技术方案作进一步描述说明,应当理解的是,此处所描述的具体实施例仅用于帮助理解本发明,不用于本发明的具体限制。且本文中所使用的附图,仅仅是为了更好地说明本发明所公开内容,对保护范围并不具有限制作用。如果无特殊说明,本发明的实施例中所采用的原料均为本领域常用的原料,实施例中所采用的方法,均为本领域的常规方法。
[0042]
实施例1
[0043]
(1)在盐酸盐反应罐中加入4.0kg氢溴酸东莨菪碱,加入无水乙醇搅拌降温到10℃,缓慢分次加入2.56kg硼氢化钠,每隔10min加一次,分5次加完,升温至20℃密闭保温反应4h,然后降温至5℃,通入干燥的氯化氢气体至ph值为1.5,加入40l乙醚,搅拌10min,静置5min,过滤,30℃真空干燥10h得东莨菪醇盐酸盐;
[0044]
(2)将东莨菪醇盐酸盐加入到碱化反应罐中,加入96l二氯甲烷,回流下通入氨气至ph为12,过滤至东莨菪醇浓缩罐,控制外温50℃以下减压浓缩至无溶剂蒸出,得无色至略
黄色微粘液体东莨菪醇,用甲苯淋洗得东莨菪醇洗涤液;
[0045]
(3)将含有1.6kg东莨菪醇的东莨菪醇洗涤液加入到噻托溴铵前体反应罐中,加入2.60kg二(2-溴噻吩)羟乙酸甲酯,开真空升温至60℃,分3次加入0.06kg钠屑,每隔20min加一次,升温至80℃搅拌反应3.5h。
[0046]
(4)氮气保护下加入8l甲苯,搅拌8min,然后加入到装有稀盐酸的前体物洗涤罐中,搅拌10min,静置分取酸水层,有机层用9l稀盐酸,分二次提取。前体反应罐中未溶解的固体在氮气保护下用8l二氯甲烷搅拌8min溶解,加入到装有稀盐酸的前体物洗涤罐中,搅拌8min,静置分取酸水层,有机层用9l稀盐酸,分二次提取。合并酸水层,用32l乙醚洗涤三次,酸水层转入前体物析料罐中,冷却至5℃,用饱和碳酸钠溶液调节ph至11,5℃析料,次日过滤,用8l二氯甲烷溶解水洗,2kg无水硫酸钠脱水干燥2h,过滤浓缩至干,加入16l乙腈回流溶解,再加入0.16kg活性炭回流脱色30min,过滤浓缩至糊状,冷却至5℃过滤,60℃真空干燥15h得二(2-噻吩基)乙醇酸东莨菪酯。
[0047]
(5)开溴甲烷钢瓶阀门,使溴甲烷液化气经缓冲罐气化后进入放有8.3l乙腈的配制罐,放出17.1kg溴甲烷后关闭阀门,乙腈吸收完溴甲烷后,既得溴甲烷-乙腈溶液。将1.5kg二(2-噻吩基)乙醇酸东莨菪酯,加入到噻托溴铵粗品反应罐中,加入5l二氯甲烷溶解,加入配制好的溴甲烷-乙腈溶液,搅拌均匀,于25℃反应24h,过滤,100℃干燥2h,得类黄色固体。用7l甲醇,7l丙酮加热溶解类黄色固体,减压浓缩至糊状,冷却至5℃,过滤,100℃干燥4.5h,得噻托溴铵粗品。
[0048]
(6)在成品溶解脱色罐中加入1.36kg噻托溴铵粗品,加入13.6l纯化水,2.72l丙酮,加热回流溶解,加入0.136kg活性炭搅拌脱色30min,过滤,加入1.36l水、0.28l丙酮淋洗罐壁和活性炭,加热溶解澄清,降温至5℃,再次用1.36l水、0.28l丙酮淋洗罐壁和物料,再用0.4l丙酮淋洗,抽干出料,30℃真空干燥6h得噻托溴铵。
[0049]
实施例2
[0050]
实施例2与实施例1的区别仅在于步骤(1)硼氢化钠每隔15min加一次,分4次加完。
[0051]
实施例3
[0052]
实施例3与实施例1的区别仅在于步骤(3)分4次加入钠屑,每隔15min加一次。
[0053]
对比例1
[0054]
对比例1与实施例1的区别仅在于步骤(1)中加入硼氢化钠的温度为35℃。
[0055]
对比例2
[0056]
对比例2与实施例1的区别仅在于步骤(1)中密闭保温反应时间为10h。
[0057]
对比例3
[0058]
对比例3与实施例1的区别仅在于步骤(3)中钠屑一次加入。
[0059]
对比例4
[0060]
对比例4与实施例1的区别仅在于步骤(3)中钠屑分7次加入。
[0061]
对比例5
[0062]
对比例5与实施例1的区别仅在于步骤(2)中氨气调节ph值为8。
[0063]
对比例6
[0064]
对比例6与实施例1的区别仅在于步骤(5)用溴甲烷替代原本的溴甲烷-乙腈溶液。
[0065]
附图是实施例1所得噻托溴铵及东莨菪醇的特征谱图,图1是噻托溴铵液相图谱,从图谱中可以看出,本方法所得噻托溴铵纯度达到99.87%,最大杂质0.07%,符合ep标准,图2是噻托溴铵红外图谱,在3191cm-1
、1734cm-1
,、1201cm-1
处都出现特征峰,图3是东莨菪醇液相图谱,本方法所得东莨菪醇纯度达到94.23%,异东莨菪醇2.87%,原料氢溴酸东莨菪碱2.15%,图4是东莨菪醇红外图谱,在2930cm-1
、1079cm-1
,、870cm-1
处有特征峰。
[0066]
实施例1、实施例2、实施例3以及对比例1、对比例2、对比例3、对比例4、对比例5、对比例6所制备的噻托溴铵最终的纯度分别为99.9%、99.4%、99.2%、98.2%、98.0%、97.9%、98.3%、98.5%、97.5%,收率分别为95.0%、94.8%、93.9%、90.5%、91.0%、90.2%、90.7%、90.3%、89.8%,其中按照实施例1的方法操作得到的噻托溴铵纯度和收率均最高,对比例6得到噻托溴铵纯度和收率均最低,原因在于溴甲烷未被完全吸收,用量不足。本实验方法所制备的噻托溴铵有较好的纯度和收率。
[0067]
最后应说明的是,本文中所描述的具体实施例仅仅是对本发明精神作举例说明,而并非对本发明的实施方式的限定。本发明所属技术领域的技术人员可以对所描述的具有实施例做各种各样的修改或补充或采用类似的方式替代,这里无需也无法对所有的实施方式予以全例。而这些属于本发明的实质精神所引申出的显而易见的变化或变动仍属于本发明的保护范围,把它们解释成任何一种附加的限制都是与本发明精神相违背的。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。