用于无抗生素发酵制备低分子量物质和蛋白质的微生物菌株和方法
1.本技术是2018年6月11日提交的题为“用于无抗生素发酵制备低分子量物质和蛋白质的微生物菌株和方法”的中国专利申请201580085273.3 的分案申请。
2.本发明涉及用于无抗生素发酵生产低分子量物质和蛋白质的微生物菌株和方法。
3.首要地,借助于微生物的低分子量物质的自然生产已经增多,重组蛋白药物(“生物制剂”)的市场近年来也呈现强劲增长。由于发酵生产(特别是对于基于蛋白质的活性药物成分)中的高成本压力,正在寻求更有效且因此更具成本效益的用于其生产的方法和系统。各种微生物,如细菌、酵母、丝状真菌或其它植物细胞或动物细胞均可用作生产者。在此,从经济角度来看至关重要的是经济有效的发酵、高产物产量及在蛋白质的情况中的获得功能性蛋白质的正确折叠和/或修饰。由于其经广泛研究的遗传学和生理学、短的传代时间和简便处理,革兰氏阴性肠杆菌大肠杆菌(escherichia coli,e. coli)是目前最常用于生产低分子量物质和蛋白质的生物体。
4.基本上,使用两种不同的微生物:天然产生所述物质的微生物及已经遗传修饰的微生物。遗传修饰所需的技术方法在现有技术中已久为人所知。在此的目标是将靶蛋白质需要的或者合成低分子量物质需要的基因导入宿主细胞。所述基因由宿主细胞转录、翻译、必要时修饰,且可能向外转移到培养基中。
5.生物技术方法的经济可行性关键取决于所得产物的产量。所述产量可以通过表达系统(宿主细胞,遗传元件等)、发酵参数和培养基而优化。
6.基本上,宿主细胞可以通过两种不同的方式进行修饰。从而,可以将新的遗传信息整合进基因组(丝状真菌,酵母等)和/或导入染色体外元件上 (例如质粒)(原核生物,酵母等)。在将基因遗传整合进基因组中的情况中,即使没有选择压力,所述基因在宿主细胞中仍然良好保持。然而,缺点是在原核生物情况中,宿主中只有一个基因拷贝,且由于序列特异性重组事件,整合相同基因的进一步的拷贝,以通过基因
‑
剂量效应增加产物形成是非常具有挑战性的(ep 0284126 b1)。
7.当使用染色体外dna时,通常将质粒形式的靶蛋白的遗传信息转化进大肠杆菌生产菌株中。由于基因
‑
剂量效应在此也产生作用,因此争取尽可能最高的每个细胞的质粒拷贝数。由于这种遗传因子因胁迫而容易从细胞丢失(归因于质粒的复制以及靶蛋白的产生),因此有必要对整个培养施加选择压力。标准的是使用抗生素抗性基因作为选择标记,这使得包含这种元件的细胞在抗生素存在下能够生长。因此,只有携带质粒的细胞可以繁殖。由于产生靶物质或靶蛋白质的能力也由于质粒丢失而丧失,这对于在发酵中可以达到的产量有直接影响。
8.近年来,使用抗生素抗性作为选择标记越来越受到关注。首先,抗生素的使用相当昂贵,尤其是当抗性基于抗生素降解酶时,因为需要不断提供额外剂量的抗生素。其次,在医学和其它领域的广泛使用促成抗性基因扩散到其它菌株,在某些情况中是致病性菌株。这对疾病的治疗具有负面影响。
9.同时,现有技术中也开发了无抗生素选择系统。已经开发了多种不同的无抗生素
系统。这些可以分为三种不同的基本系统。使用营养缺陷型、毒素
‑
抗毒素系统及其它方法。
10.其它方法的类别涵盖不遵循一般原理的机制,例如抑制因子trna、fabi/三氯生(fa合成)、操纵子/阻抑物滴定(peubez et al.microbial cell fac,2010, 9:65)。然而,这些方法通常用于合成用于基因治疗的dna和dna片段,未优化用于产生高产量的靶物质。在一些情况中,代替抗生素用于选择的是其它物质,例如三氯生、除草剂和重金属,但也会引起健康问题。例如 herrero et al.(1990,j.bacteriol.172,6557
–
6567)描述了作为选择标记的除草剂和重金属抗性基因。
11.毒素
‑
抗毒素系统(hok
‑
sok,ccda/b等)由两个遗传元件组成,其可能均在质粒上编码或者在染色体上和质粒上编码(gerdes et al.proc natl acadsci usa,1986,83:3116
–
20)。在此,抗毒素对毒素具有中和作用。在质粒丢失的情况中,该机制丧失且由于染色体编码的且长久的毒素无质粒细胞死亡。
12.另一种已知的选择方法是营养缺陷型菌株的补足。在此,生产菌株基因组中具有基本代谢功能的基因被去除或失活。因此,这些基因被称为必需基因。只有当代谢功能被规避或重新建立时,所得到的营养缺陷型菌株能生长、繁殖或存活。这可以通过给予适当的前体或代谢终端产物(氨基酸,碱基等)或通过导入在宿主基因组中失活的基因而实现。专利ep 0284126 b1 命名了氨基酸代谢的营养缺陷标记。由于通过向培养基中加入必需的代谢产物可以容易地补充营养缺陷,所以这些菌株可以容易地产生。这是因为即使在不存在质粒的情况中也可以在存在代谢产物的条件下培养细胞。通过转化,将氨基酸/碱基的合成信息重新导入细胞中,即使在没有补加条件下细胞也可以生长。因此,选择必须在没有补加的基本培养基上完成。
13.实际上,由于具有复合组成成分的培养基通常用于工业发酵,因此迄今还不可能实施使用这种营养缺陷作为选择标记。在此,出于成本的原因,废产物通常是发酵培养基的组分,例如谷类(乙醇生产)、玉米(淀粉生产)、马铃薯(淀粉回收)或酵母提取物的残留物。这些既用作碳源又用作氮源。在某些情况中,这些组分没有精确定义,但含有氨基酸、碱基、维生素等,其可自培养基摄取。因此,如果有可能的话在工业发酵中也很难用营养缺陷型菌株建立足够的选择压力。
14.如wo2008/135113a1所述,在葡萄糖代谢中使用营养缺陷型在工业应用中的培养基中也具有不足的选择性。尽管微生物可以在葡萄糖上生长特别良好且比含有其它碳源的培养基更快速地繁殖,但由于质粒或靶物质的产生所致较高的胁迫,使得这种优点作废。其它碳源可用于复合培养基并为细胞使用。
15.这也适用于使用pyrc基因(二氢乳清酸酶)作为营养缺陷型标记,如 wo07039632a1中所述。这种酶存在于嘧啶碱基合成起始,失活也导致抑制重新合成。但是,这些碱基可以得自培养基并利用,在工业培养基中不能建立适当的选择压力。
16.例外是对于必要的胸腺嘧啶核苷和d
‑
丙氨酸的营养缺陷,即使在复合组分中在发酵培养基也只有微量或根本不存在(ep 0251579 a1;ep 0185512 b1)。但是这些系统也不适合在通常争取的高细胞密度方法中有效生产。因为在所述方法中有一些细胞死亡和裂解,因此释放胸腺嘧啶核苷和d丙氨酸或其它氨基酸等,其可以转而补充营养缺陷。
17.总体而言,必须指出的是尽管有发酵生产低分子量物质和蛋白质的多年经验,但是除了通过昂贵或者表现健康和/或环境危险的物质如抗生素之外,迄今为止还没有开发
出可普遍使用的系统。由于工业中使用的复合生长培养基,因此通过营养缺陷型标记进行选择的各种方法迄今也仅带来不理想的结果。
18.这主要适用于微生物,但尤其适用于较不强力的生产菌株,例如由于其特殊性质(释放蛋白质至培养基中)使用的渗漏菌株。在工业规模上工业性应用所述菌株的情况中,这种应用通常在培养基中使用复合成分。
19.为了生产工业上使用的产物,出于成本原因使用由纯化组分组成的确定成分培养基是不经济的。
20.本发明的一个目的是提供用于生产低分子量物质或蛋白质的微生物菌株,该菌株即使在含有复合成分的培养基中也保持稳定,在该菌株中生产质粒不是由抗生素/抗性标记系统而在细胞中稳定的。
21.通过在其基因组中含有基因突变的微生物菌株实现这个目的,所述突变在菌株中引起营养缺陷,所述菌株还含有生产质粒,所述生产质粒编码用于生产低分子量物质的至少一种酶或者至少一种重组蛋白,以及基因的功能性拷贝,其染色体失活导致营养缺陷,特征在于所述营养缺陷是不可补足的营养缺陷。
22.在本发明上下文中,不可补足的营养缺陷应理解为指所述营养缺陷降低微生物细胞的生长或者导致微生物细胞死亡,且不能通过在生长培养基中加入代谢特异性前体、中间体和/或终产物而补足。
23.在本发明上下文中,当基因突变后菌株在发酵中的生长速率与基因突变前菌株的生长速率相比降低至≤10%时,优选不再生长时,有降低的生长。
24.特别优选地,突变导致基因失活,以这种方式导致不可补足的营养缺陷及因此导致微生物细胞的死亡。
25.因此,在根据本发明的微生物菌株的细胞基因组中,有对于细胞生长或存活必需的合成代谢途径重要的基因的失活,通过在生长培养基中加入代谢特异性前体、中间体和/或终产物不可能再次实现细胞的生长或存活。
26.本发明上下文中不可补充的营养缺陷基因是微生物细胞基因组中的基因,其失活不能通过在生长培养基加入代谢特异性前体、中间体和/或终产物而补足,其失活导致生长速率降低或微生物细胞死亡。
27.优选地,导致菌株中不可补足的营养缺陷型的基因突变导致所述基因的失活或者由该基因编码的基因产物的活性失活。
28.不可补足的营养缺陷型基因(致死基因)的实例在baba et al.(2006,mol. syst.biol.2:2006.0008)中描述。优选地,所述基因是基因map、pyrh、ftsl、 rpsb、tsf、plsc或其同源基因。
29.特别优选地,所述基因是pyrh基因或plsc基因或具有相同功能或活性的同源基因。
30.本发明上下文中的基因不仅包含被转录的dna区段,而且还包含参与调节这个复制过程的dna区段,即该基因的调控元件,例如优选启动子和终止子。
31.同源基因优选被理解为是指编码与指定基因编码的蛋白质具有相同活性的蛋白质的基因,及与考虑中的微生物中指定基因的序列具有大于30%、特别优选大于70%的序列相同性,所述序列在各种情况中从数据库获知。
32.pyrh基因编码酶ump激酶(ec:2.7.4.22;uk,尿苷单磷酸激酶,尿苷酸激酶,尿苷5'
‑
单磷酸(ump)激酶)。所述酶使用atp激活ump形成udp。在下一步中,udp被另一激酶(udp激酶)激活形成utp,其可用于合成rna 以及合成ctp(胞嘧啶三磷酸)和ttp(胸腺嘧啶三磷酸)。ctp和ttp转而又是rna(ctp)和dna(dctp和dttp)的组成部分。因此,ump是rna和 dna的嘧啶结构模块的起源。由于尿嘧啶转变为胞嘧啶或胸腺嘧啶仅在三磷酸水平发生,因此ump激酶的失活导致这个合成途径完全阻断。
33.基本上,细胞只能从培养基中摄取三种碱基的单磷酸盐。对于ump转变为utp,ump激酶是必不可少的。这意味着这种营养缺陷不能通过补加来补足,因此不依赖于培养基成分。
34.pyrh基因由seq id no.1表征。pyrh基因产物(pyrh)由seq id no.2 表征。
35.在本发明中,pyrh同源物是编码具有pyrh活性的蛋白质并与seq idno.1具有大于30%的序列相同性的基因。特别优选与seq id no.1具有大于70%的序列相同性。pyrh基因是特别优选的。
36.pyrh同系物是与seq id no.2具有大于30%的序列相同性的蛋白质,该蛋白质具有根据ec号2.7.4.22的ump激酶活性。特别优选地,pyrh同系物具有根据ec编号2.7.4.22的ump激酶活性及与seq id no.2具有大于70%的序列相同性。pyrh蛋白是特别优选的。
37.细胞中的pyrh活性可以根据bucurenci et al.(1998,j.bact.180:473
–
77) 描述的测定法确定。
38.plsc基因编码酶1
‑
酰基甘油
‑3‑
磷酸o
‑
酰基转移酶(ec:2.3.1.51)。所述酶将一个脂肪酸从酰基
‑
coa转移至一个酰基甘油
‑3‑
磷酸并释放一个 coa
‑
sh。所得二酰甘油3
‑
磷酸用于合成必要的膜组分,如甘油三酯和甘油磷脂。这意味着这一步对于产生大肠杆菌膜系统是必需的。在大肠杆菌中,虽然可以摄取脂肪酸,但甘油三酯和甘油磷脂必须在细胞或膜中合成。
39.plsc基因由seq id no.3表征。plsc基因产物(plsc)由seq id no.4表征。
40.在本发明中,plsc同系物是编码具有plsc活性的蛋白质且与seq idno.3具有大于30%的序列相同性的基因。特别优选与seq id no.3具有大于70%的序列相同性。plsc基因是特别优选的。
41.plsc同系物是与seq id no.4具有大于30%序列相同性的蛋白质,该蛋白质具有ec编号2.3.1.51的1
‑
酰基甘油
‑3‑
磷酸o
‑
酰基转移酶活性。特别优选地,plsc同系物具有根据ec编号2.3.1.51的1
‑
酰基甘油
‑3‑
磷酸o
‑ꢀ
酰基转移酶活性且与seq id no.4具有大于70%的序列相同性。plsc蛋白是特别优选的。
42.细胞中的plsc活性可以根据monrand et al.(1998,biochem.biophys. res.commun.244:79
–
84)描述的测定法确定。
43.dna相同性的程度是通过程序“nucleotide blast”确定,该程序可以在网页http://blast.ncbi.nlm.nih.gov/上找到,且基于blastn算法。默认参数用作两个或多个核苷酸序列比对的算法参数。默认的通用参数是:max targetsequences=100;short queries=“automatically adjust parameters for shortinput sequences”;expect threshold=10;word size=28;automatically adjustparameters for short input sequences=0。相应的默认打分参数是: match/mismatch scores=1,
‑
2;gap costs=
linear。
44.对于蛋白质序列对比,使用网页http://blast.ncbi.nlm.nih.gov/上的程序“protein blast”。所述程序基于blastp算法。默认参数用作两或多个蛋白质序列比对的算法参数。默认的一般参数是:max target sequences=100;shortqueries=“automatically adjust parameters for short input sequences”;expectthreshold=10;word size=3;automatically adjust parameters for short inputsequences=0。默认打分参数是:matrix=blosum62;gap costs=existence: 11extension:1;compositional adjustments=conditional compositional scorematrix adjustment。
45.为了产生本发明的菌株,将具有突变或缺失的不可补足的营养缺陷基因的功能性拷贝的温度敏感型质粒导入合适微生物菌株中。随后,失活所述菌株基因组中相应的不可补足的营养缺陷基因。之后,温度敏感型质粒在非允许温度下在所述菌株中换成生产质粒,其中编码至少一种用于生产低分子量物质或至少一种重组蛋白的酶的生产质粒还含有不可补足的营养缺陷基因的功能性拷贝。
46.这种方法确保了即使在复合培养基中,生产质粒在生产低分子量化合物或重组蛋白质的发酵过程期间也稳定地保持在细胞中。因此,根据本发明的菌株可以在没有添加的选择剂或补足营养缺陷型的添加剂的情况中培养而不丢失质粒。
47.基本上,具有基因(其失活导致菌株的不可补足的营养缺陷)的任何微生物菌株均适合作为用于生产本发明的菌株的起始菌株。
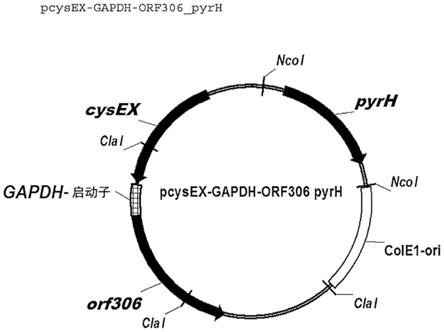
48.优选地,用于产生本发明菌株的起始菌株是肠杆菌科菌株,特别优选物种大肠杆菌的菌株。
49.优选的大肠杆菌菌株是那些具有“渗漏”突变的菌株。“渗漏突变”应理解为是指外层细胞膜(outer cell membrane)或细胞壁的结构元件的基因中的突变,所述基因选自omp基因、tol基因、excd基因、excc基因、lpp基因、 pal基因、env基因和lky基因,所述突变导致细胞将周质蛋白释放进培养基中的程度增加(shokri et al.,appl.microbiol.biotechnol.60(2003), 654
–
664)。优选地,“渗漏”突变是lpp基因中的突变,特别优选选自lpp1 突变、lpp缺失突变和lpp蛋白质14位甘氨酸残基突变(编号系统包括信号肽),例如,lpp
‑
3突变。lpp1突变是lpp基因中的突变,其导致位置77的精氨酸残基由半胱氨酸残基取代,lpp3突变是lpp基因中的突变,其导致位置14的甘氨酸残基由天冬氨酸残基取代。这些突变在us2008254511中详细描述。lpp缺失突变优选是lpp基因自身或lpp基因的启动子区域中的至少一个核苷酸缺失,导致细胞呈现出增加的周质蛋白渗漏。
50.在本发明上下文中,增加的渗漏应理解为是指在细胞发酵后,在培养基中的周质蛋白质例如碱性磷酸酶的浓度高于在相同的条件下发酵大肠杆菌菌株w3110(atcc 27325)的培养基中的浓度。
51.失活微生物菌株中的基因的方法为现有技术领域已知。下文详细描述两个优选基因(pyrh基因和plsc基因)的突变及其基因产物。也可以以相似方式将这些方法应用于其失活引起微生物菌株的不可补足营养缺陷型的其它基因。
52.温度敏感型质粒含有温度敏感型复制起点和不可补足的营养缺陷基因的功能性拷贝。另外,所述质粒含有用于选择转化体的选择标记基因。选择标记是例如抗生素抗性。
53.温度敏感型复制起点的优选实例是“orir101&repa101
‑
ts”,质粒 psc101
(hashimoto
‑
gotoh et al.;gene,2000,241:1:185
–
191)的复制起点的衍生物,其位于尤其是质粒pkd20和pkd46上(datsenko and wanner,2000, p.n.a.s.97:6640
–
6645)。
54.使用本领域技术人员已知的转化技术将温度敏感型质粒导入细胞,例如tss法、cacl/rbcl法、电穿孔法。
55.通过存在于温度敏感质粒上的选择标记对包含温度敏感型质粒的细胞进行选择,细胞在许可(permissiver)该质粒的温度下温育。
56.通过在对温度敏感型质粒非许可的温度下用生产质粒转化后培养微生物菌株,可以将这种温度敏感型质粒置换生产质粒。优选的非许可温度范围是37
‑
45℃,特别优选39
‑
43℃。
57.使用本领域技术人员已知的转化技术将生产质粒导入细胞,例如tss 方法、cacl/rbcl方法、电穿孔方法。
58.转化体可以在琼脂平板上和液体培养物中培养。在优选的实施方案中,微生物菌株在用生产质粒转化后立即暴露于47
‑
55℃、优选52℃的温度休克30
‑
90分钟,优选60分钟。然后在非许可温度下进一步温育。结果,温度敏感型质粒一步置换生产质粒,及产生用于无抗生素生产低分子量物质或重组蛋白质的本发明生产菌株。
59.例如,使用以下已知表达载体之一作为生产质粒:pjf118eh,pkk223
‑
3,puc18,pbr322,pacyc184,pask
‑
iba3或pet。
60.除了不可补足的营养缺陷基因的功能性拷贝之外,生产质粒还包含一或多个靶基因以及表达所述靶基因所必需的表达信号,例如启动子序列、操纵子序列和终止子序列。靶基因编码至少一种用于生产低分子量物质的酶或至少一种重组蛋白质。低分子量物质是生物体能够合成的基本结构模块(碱基,氨基酸,脂肪酸等)以及次生代谢产物(维生素,抗氧化剂等)。优选氨基酸,特别优选氨基酸l
‑
半胱氨酸及从其衍生的化合物。
61.重组蛋白质优选是异源蛋白质。
62.异源蛋白质应理解为是指不属于宿主生物体的蛋白质组即全部天然蛋白质组成的蛋白质。
63.优选地,异源蛋白质是真核蛋白质,特别优选含有一或多个二硫键或者以其功能形式以二聚体或多聚体形式存在的蛋白质,即所述蛋白质具有四级结构,且由多个相同的(同源的)或不同的(异源的)亚基构建。
64.由多个蛋白质亚基组成的蛋白质的优选类别是抗体或抗体片段。特别优选功能性fab抗体片段。
65.下文描述了生产根据本发明的微生物菌株,其中不可补足的营养缺陷基因pyrh已被突变
66.基本上,具有ump激酶pyrh的基因的任何宿主生物均适合作为用于生产根据本发明的具有基因组pyrh失活的微生物菌株的起始菌株
67.现有技术领域中已知用于使微生物菌株中pyrh基因失活的方法。例如, pyrh基因可通过在pyrh基因的读框中导入突变(取代、插入或缺失一或多个核苷酸)而失活,该突变导致pyrh的特异性活性失活。本领域技术人员已知产生这种pyrh等位基因的方法。例如,可以通过link et al.(1997,j. bacteriol.179:6228
–
37)所述方法,通过同源重组机制将染色体突变插入基因。例如,借助于λ红重组酶系统根据datsenko and wanner(2000,
proc.natl. acad.sci.u s a.97:6640
–
5)所描述的方法,可以对全部或部分pyrh基因进行染色体缺失。pyrh等位基因也可以通过p1噬菌体转导或者与具有pyrh 突变的菌株接合而转移至pyrh野生型菌株,染色体中的pyrh野生型基因被相应的pyrh等位基因置换。
68.此外,细胞的pyrh基因也可以通过取代、插入或缺失一或多个核苷酸而突变至少一个调控表达必需的元件(例如启动子,增强子,核糖体结合位点)而失活。在本发明上下文中,当发酵细胞的生长速率由于基因失活而与突变前的细胞相比降低至≤10%、优选不再生长时,则有失活。
69.特别优选的是所述突变导致微生物细胞死亡。
70.由于pyrh基因的失活导致不可补足的营养缺陷,pyrh基因的功能性拷贝在染色体失活前必须已经存在于细胞中。这可以通过瞬时导入含有pyrh 基因功能性拷贝的温度敏感型质粒实现。
71.在含有温度敏感型质粒的这种细胞中,通过所述方法可以使基因组中的pyrh基因失活。
72.在进一步的步骤中,将所述温度敏感型质粒换成生产质粒,其也含有 pyrh基因的功能性拷贝。优选以已经描述的方式完成。
73.可以以相似的方式产生具有失活的plsc基因或一些其它突变的不可补足的营养缺陷基因的本发明的微生物菌株。
74.在重组蛋白质的生产中,细胞质生产和分泌性生产之间是有区别的。在细胞质生产的情况中,靶蛋白积聚在细胞的细胞质中,而在分泌性生产的情况中,靶蛋白转移至周质或培养基中。在本发明上下文中优选分泌性生产。特别优选靶蛋白分泌性生产至培养基中。
75.为了分泌性生产蛋白质,即蛋白质从细胞质转移至周质或培养基中,有必要将符合读框产生的蛋白质基因的5'末端与信号序列的3'末端连接以便蛋白质输出。适用于此目的的主要是在大肠杆菌中导致靶蛋白转移至周质中的所有信号序列的基因。在大肠杆菌中,已知三种主要的转移途径: sec途径,tat途径和srp途径。优选那些允许通过sec装置易位的信号序列。现有技术中描述了这种类型的各个信号序列,例如以下基因的信号序列:phoa,ompa,pelb,ompf,ompt,lamb,male,dsba,葡萄球菌蛋白a,stii等(choi and lee,appl.microbiol.biotechnol.64(2004), 625
–
635)。
76.根据本发明优选的是大肠杆菌的phoa基因或ompa基因的信号序列,或者肺炎克雷伯菌(klebsiella pneumoniae)m5a1的环糊精糖基转移酶 (cgtase)的信号序列,或者衍生自这个信号序列的序列,其在us2008076157 中公开。
77.含有生产质粒的细胞的培养(发酵)是根据本领域技术人员已知的常规发酵方法在不加入抗生素的生物反应器(发酵罐)中完成的。
78.本发明还提供通过微生物菌株产生低分子量物质或重组蛋白质的方法,该方法的特征在于使用根据本发明的微生物菌株及使用不含抗生素的发酵培养基。
79.在常规生物反应器中进行发酵,例如搅拌罐、泡罩式发酵罐或气升式发酵罐。在工业规模优选搅拌罐式发酵罐,因此大小>100l。
80.在发酵中,本发明的菌株的细胞在液体培养基中培养,持续监测并精确控制各个参数,例如营养素的供应、氧分压、ph和培养温度。培养期优选16
‑
150小时,特别优选24
‑
72小时。
81.可能的发酵培养基是本领域技术人员已知的用于培养微生物的所有常见培养基。然而,所述发酵培养基不含任何抗生素。
82.可以使用复合培养基或最低盐培养基,向其中加入确定比例的复合组分,例如蛋白胨、胰蛋白胨、酵母提取物、糖蜜或玉米浆。优选含有复合培养基成分的培养基。
83.作为发酵的主要碳源,可以使用所有细胞可利用的糖、糖醇或者有机酸或其盐。优选使用葡萄糖、乳糖或甘油。特别优选葡萄糖和乳糖。也可以组合给予多种不同碳源。碳源可以在发酵开始时最初充入发酵培养基中,或者最初什么都不加入或者在开始时首先只加入部分碳源,并在整个发酵过程中加入碳源。特别优选的实施方案是最初加入部分碳源并补加部分。特别优选地,碳源最初以10
‑
30g/l的浓度加入,当浓度降至低于5g/l时开始补加,由此保持低于5g/l的浓度。
84.培养物中的氧分压(po2)优选为10
‑
70%饱和。优选20
‑
60%的po2;po2特别优选在45
‑
55%饱和之间。
85.培养ph优选在ph 6与ph 8之间。优选ph在6.5
‑
7.5之间;培养ph 特别优选为6.8
‑
7.2。
86.培养温度在15
‑
45℃之间。优选18
‑
40℃的温度范围,特别优选25
‑
35℃的温度范围,非常特别优选30℃。
87.在优选的安排中,发酵是高细胞密度发酵。高细胞密度发酵应理解为是指其过程中细胞干重达到50g/l以上的发酵。特别优选细胞干重大于 70g/l。
88.图1示出实施例2的质粒pkd46的限制和功能图。
89.图2示出在实施例2中产生的质粒paf
‑
ts
‑
pyrh的限制和功能图。
90.图3示出在实施例2中产生的质粒paf
‑
ts
‑
plsc的限制和功能图。
91.图4示出在实施例5中使用的质粒pmt1的限制和功能图。
92.图5示出在实施例5中产生的表达质粒pcysex
‑
gapdh
‑
orf306_tetr 的限制和功能图。
93.图6示出在实施例6中产生的表达质粒pcgt_tetr的限制和功能图。
94.图7示出在实施例7中产生的表达质粒pfab
‑
anti
‑
lysozym_tetr的限制和功能图。
95.图8示出在实施例8中产生的表达质粒 pcysex
‑
gapdh
‑
orf306_pyrh1_tetr的限制和功能图。
96.图9示出在实施例8中产生的表达质粒pcgt_pyrh1_tetr的限制和功能图。
97.图10示出在实施例8中产生的表达质粒pfab
‑
anti
‑
lysozym_pyrh1_tetr 的限制和功能图。
98.图11示出在实施例8中产生的表达质粒 pcysex
‑
gapdh
‑
orf306_plsc1_tetr的限制和功能图。
99.图12示出在实施例8中产生的表达质粒pcgt_plsc1_tetr的限制和功能图。
100.图13示出在实施例8中产生的表达质粒pfab
‑
anti
‑
lysozym_plsc1_tetr 的限制和功能图。
101.图14示出在实施例9中产生的本发明的生产质粒 pcysex
‑
gapdh
‑
orf306_pyrh的限制和功能图。
102.图15示出在实施例9中产生的本发明的生产质粒pcgt_pyrh的限制和功能图。
103.图16示出在实施例9中产生的本发明的生产质粒 pfab
‑
anti
‑
lysozym_pyrh的限制和功能图。
104.图17示出在实施例9中产生的本发明的生产质粒 pcysex
‑
gapdh
‑
orf306_plsc的限制和功能图。
105.图18示出在实施例9中产生的本发明的生产质粒pcgt_plsc的限制和功能图。
106.图19示出在实施例9中产生的本发明的生产质粒 pfab
‑
anti
‑
lysozym_plsc的限制和功能图。
107.如下实施例用于进一步阐明本发明。使用的所有分子生物学和微生物学方法如聚合酶链式反应(pcr)、基因合成、dna分离和纯化、限制酶修饰dna、klenow片段和连接酶、转化、p1转导等,均以本领域技术人员已知的、在文献中描述的或由各个厂商推荐的方式进行。
108.实施例1:扩增具有其自身启动子的标记基因(pyrh或plsc)
109.使用引物pyrh
‑
ncoi
‑
fw(seq id no.5)和pyrh
‑
ncoi
‑
rev(seq id no.6) 扩增大约1.0kb大小的dna片段,其编码包括天然启动子区域的pyrh基因。用于pcr反应的模板是来自大肠杆菌菌株w3110(atcc 27325)的染色体dna。
110.将1.0kb大小的pcr片段通过琼脂糖凝胶电泳纯化并从琼脂糖凝胶中分离,使用“qiaquick gel extraction kit”(qiagen gmbh,hilden,germany) 根据来自厂商的信息进行。之后,使用限制酶ncoi消化纯化的pcr片段,并储存于
‑
20℃。以类似的方式扩增、纯化、消化及贮存编码包括天然启动子区域的plsc基因的大约1.0kb大小的dna片段。为了扩增,使用引物 plsc
‑
ncoi
‑
fw(seq id no.7)和plsc
‑
ncoi
‑
rev(seq id no.8)。
111.实施例2:用温度敏感型复制起点产生质粒paf
‑
ts
‑
pyrh和paf
‑
ts
‑
plsc
112.用于构建具有温度敏感型复制起点的质粒paf
‑
ts
‑
pyrh和paf
‑
ts
‑
plsc 的起始质粒是质粒pkd46(datsenko and wanner,2000,p.n.a.s.97: 6640
–
6645)。图1示出质粒pkd46的限制和功能图。将在实施例1中描述的并使用ncoi消化的pcr产物(该pcr产物编码具有其自身启动子的pyrh 基因或plsc基因)克隆进pkd46的ncoi限制位点。将连接制备物转化进“dh5α
tm
‑
t1r大肠杆菌细胞”(life technologies gmbh),在所述细胞中扩增,并通过测序验证分离的质粒的dna序列。以这种方式产生的总共4个可能构建体中的2个命名为paf
‑
ts
‑
pyrh和paf
‑
ts
‑
plsc(参见图2和3)。
113.实施例3:使用paf
‑
ts
‑
pyrh or paf
‑
ts
‑
plsc转化选择的大肠杆菌菌株
114.将实施例2中描述的具有温度敏感型复制起点的质粒paf
‑
ts
‑
pyrh和 paf
‑
ts
‑
plsc转化进两个大肠杆菌菌株w3110(atcc 27325)和w3110lpp3 (在us2008076158a1中描述为“渗漏”菌株),使用本领域技术人员已知的 cacl2方法进行。转化的细胞在含有100mg/l氨苄青霉素的lb琼脂平板上选择。以这种方式产生的菌株命名为w3110/paf
‑
ts
‑
pyrh、 w3110lpp3/paf
‑
ts
‑
pyrh、w3110/paf
‑
ts
‑
plsc和w3110lpp3/paf
‑
ts
‑
plsc。
115.实施例4:在大肠杆菌中缺失基因pyrh(尿苷酸激酶失活)或者基因plsc (1
‑
酰基甘油
‑3‑
磷酸酯o
‑
酰基转移酶失活)
116.a)基因pyrh的缺失
117.根据由datsenko and wanner(datsenko and wanner,2000,p.n.a.s.97: 6640
–
6645)开发的“λ红方法”,在大肠杆菌菌株w3110/paf
‑
ts
‑
pyrh和 w3110lpp3/paf
‑
ts
‑
pyrh
中缺失编码大肠杆菌中尿苷酸激酶(pyrh)的基因 pyrh。使用引物pyrh
‑
fw(seq id no.9)和pyrh
‑
rev(seq id no.10)扩增编码卡那霉素抗性标记基因(kanr)的dna片段。引物pyrh
‑
fw编码由与pyrh 基因的5'
‑
末端同源的30个核苷酸组成的序列及与编码质粒pkd13(coligenetic stock center(cgsc)no.7633)上两个frt位点(flp识别靶位)之一的dna序列互补的包含20个核苷酸的序列。引物pyrh
‑
rev编码由与pyrh 基因的3'
‑
末端同源的30个核苷酸组成的序列及与编码质粒pkd13上的第二个frt位点的dna序列互补的包含20个核苷酸的序列。
118.通过电穿孔将扩增的pcr产物导入大肠杆菌菌株w3110/pafts
‑
pyrh 和w3110lpp3/paf
‑
ts
‑
pyrh(参见实施例3)。在含有50mg/l卡那霉素和 100mg/l氨苄青霉素的lb琼脂平板上选择具有染色体整合的卡那霉素抗性标记基因(kanr)的细胞。使用在质粒pcp20(cgsc no.7629)上编码的酶flp 重组酶实现染色体导入的卡那霉素抗性标记基因(kanr)的去除。在含有 100mg/l氨苄青霉素(选择paf
‑
ts
‑
pyrh)和34mg/l氯霉素(选择pcp20)的lb 琼脂平板上选择pcp20的细胞。由于温度敏感的复制起点(ori),在进行转化后质粒pcp20通过在非许可,即升高的温度,如在42℃,培养而除去。
119.在含有100mg/l氨苄青霉素(选择paf
‑
ts
‑
pyrh)的lb琼脂平板上进行丢失温度敏感型质粒pcp20及同时维持温度敏感型质粒paf
‑
ts
‑
pyrh的首次选择。之后,检测预选的氨苄青霉素抗性细菌克隆的卡那霉素敏感性,即丢失染色体导入的卡那霉素标记基因,以及氯霉素敏感性,即丢失温度敏感型质粒pcp20。
120.使用引物pyrh
‑
check
‑
for(seq id no.11)和pyrh
‑
check
‑
rev(seq idno.12),最终检测仅卡那霉素和氯霉素敏感但氨苄青霉素抗性克隆的pyrh 基因的染色体缺失。用于通过pcr检测染色体pyrh缺失的模板是来自选择的氨苄青霉素抗性、氯霉素和卡那霉素敏感性克隆的染色体dna。
121.由此产生并检测具有染色体pyrh缺失和质粒编码的pyrh表达的氨苄青霉素抗性大肠杆菌菌株命名为w3110δpyrh/paf
‑
ts
‑
pyrh和 w3110lpp3δpyrh/paf
‑
ts
‑
pyrh。
122.b)plsc基因的缺失
123.与pyrh基因相似,在大肠杆菌菌株w3110/paf
‑
ts
‑
plsc和 w3110lpp3/paf
‑
ts
‑
plsc(datsenko and wanner,2000,p.n.a.s.97:6640
‑
6645) 中缺失编码大肠杆菌中1
‑
酰基甘油
‑3‑
磷酸o
‑
酰基转移酶(plsc)的基因plsc。使用引物plsc
‑
fw(seq id no.13)和plsc
‑
rev(seq id no.14)扩增编码卡那霉素抗性标记基因(kanr)的dna片段。引物plsc
‑
fw编码由与plsc基因的 5'末端同源的30个核苷酸组成的序列及与编码在质粒pkd13(coli geneticstock center(cgsc)no.76333)上两个frt位点(flp识别靶位)之一的dna 序列互补的包含20个核苷酸的序列。引物plsc
‑
rev编码由与plsc基因的 3'
‑
末端同源的30个核苷酸组成的序列及与编码在质粒pkd13上第二个frt 位点的dna序列互补的包含20个核苷酸的序列。
124.通过电穿孔将扩增的pcr产物导入大肠杆菌菌株w3110/pafts
‑
plsc和 w3110lpp3/paf
‑
ts
‑
plsc中(参见实施例3)。使用酶flp重组酶(在质粒pcp20 上编码)再次去除染色体导入的卡那霉素抗性标记基因(kanr)。含有pcp20 的细胞的选择也在含有100mg/l氨苄青霉素(选择paf
‑
ts
‑
plsc)和34mg/l氯霉素(选择pcp20)的lb琼脂平板上进行。在含有100mg/l氨苄青霉素(选择 paf
‑
ts
‑
plsc)的lb琼脂平板上进行丧失温度敏感型
质粒pcp20及同时维持温度敏感型质粒paf
‑
ts
‑
plsc的首次选择。之后,检测预选的氨苄青霉素抗性细菌克隆的卡那霉素敏感性,即染色体导入的卡那霉素标记基因的丧失,及氯霉素敏感性,即温度敏感质粒pcp20的丧失。
125.最后使用引物plsc
‑
check
‑
for(seq id no.15)和plsc
‑
check
‑
rev(seq idno.16)检测这些克隆的plsc基因的染色体缺失。用于通过pcr检测染色体 plsc缺失的模板是来自选择的氨苄青霉素抗性、氯霉素和卡那霉素敏感性克隆的染色体dna。
126.由此产生和检测的具有染色体plsc缺失和质粒编码的plsc表达的氨苄青霉素抗性大肠杆菌菌株命名为w3110δplsc/paf
‑
ts
‑
plsc和 w3110lpp3δplsc/paf
‑
ts
‑
plsc。
127.实施例5:产生用于生产半胱氨酸的含有抗生素抗性基因的生产质粒
128.用于克隆和表达基因cysex(编码丝氨酸酰基转移酶的反馈抗性变体; cyse)和orf306(编码o
‑
乙酰丝氨酸/半胱氨酸输出蹬蛋白;eama)的起始质粒是在ep0885962b1中描述的基础质粒pmt1和生产质粒 pacyc184
‑
lh
‑
cysex
‑
orf306。
129.pmt1不仅含有四环素抗性基因(tetr),而且还含有被laciq基因产物抑制的tac启动子,所述laciq基因也存在于质粒上,所述tac启动子可以通过诱导子例如d
‑
乳糖或异丙基
‑
β
‑
d
‑
硫代半乳糖吡喃糖苷(iptg)启动。
130.质粒pmt1的限制和功能图在图4中示出。质粒pmt1的序列储存在序列表中(seq id no.17)。
131.为基于pmt1产生用于生产半胱氨酸的新生产质粒,将来自编码基因 cysex和orf306的质粒pacyc184
‑
lh
‑
cysex
‑
orf306(在ep0885962b1中描述)的ncoi
‑
bsabi片段与来自质粒pmt1的2458bp的ncoi
‑
pvuii片段(编码 cole1 ori和四环素抗性,tetr)连接。
132.将连接制备物转化进“dh5α
tm
‑
t1r大肠杆菌细胞”(life technologiesgmbh)中,在所述细胞中扩增,通过测序验证分离的质粒的dna序列。所得表达质粒命名为pcysex
‑
gapdh
‑
orf306_tetr(参见图5)。
133.实施例6:产生用于生产α
‑
cgt酶的含有抗生素抗性基因的生产质粒
134.用于克隆和表达来自肺炎克雷伯菌m5a1的环糊精糖基转移酶(cgtase) 基因(genbanknr no.m15264)的起始质粒也是us2008076158a1中描述的质粒pmt1和质粒pcgt。
135.为了基于pmt1产生用于生产cgtase的新生产质粒,将来自质粒pcgt 的maubi
‑
bsai片段(其编码来自肺炎克雷伯菌m5a1的cgtase)与来自质粒 pmt1的4004bp的maubi
‑
bsai片段连接。来自质粒pmt1的所述4004bp 片段编码cole1 ori、lac/tac操纵子和四环素抗性基因(tetr)。
136.将连接制备物转化进“dh5α
tm
‑
t1r大肠杆菌细胞”(life technologiesgmbh)中,在所述细胞中扩增,通过测序验证分离的质粒的dna序列。所得表达质粒命名为pcgt_tetr(参见图6)。
137.实施例7:产生用于生产fab抗溶菌酶的含有抗生素抗性基因的生产质粒
138.用于克隆和表达抗溶菌酶fab片段的基因的起始质粒也是 us20080076158a1中描述的质粒pmt1和pfab
‑
抗溶菌酶。
139.为了基于pmt1产生用于生产抗体片段fab
‑
抗溶菌酶的新生产质粒,将编码两条链即抗溶菌酶fab片段的重链(v
h
‑
c
h
1结构域)和轻链(v
l
‑
c
l
结构域)的质粒fab
‑
抗溶菌酶的maubi
‑
bsai片段与来自质粒pmt1的4004bp 的maubi
‑
bsai片段连接。来自质粒pmt1的所述
4004bp片段编码cole1 ori、lac/tac操纵子和四环素抗性基因(tetr)。
140.将连接制备物转化进“dh5α
tm
‑
t1r大肠杆菌细胞”(life technologiesgmbh)中,在所述细胞中扩增,通过测序验证分离的质粒的dna序列。所得表达质粒命名为pfab
‑
anti
‑
lysozym_tetr(参见图7)。
141.实施例8:产生含有标记基因pyrh或plsc的生产质粒,作为产生本发明的无抗生素抗性基因的生产质粒的基础
142.用于产生含有pyrh或plsc作为标记基因的生产质粒的起始质粒是如实施例5
‑
7所述的质粒pcysex
‑
gapdh
‑
orf306_tetr、pcgt_tetr和 pfab
‑
anti
‑
lysozym_tetr。所有这些质粒都具有单独的ncoi限制位点(见图 5
‑
7)。这个普遍的ncoi限制性位点用于基因pyrh或plsc的克隆或整合。
143.为此,将实施例1中描述并使用限制酶ncoi切割的pcr产物与同样使用ncoi切割后的质粒pcysex
‑
gapdh
‑
orf306_tetr、pcgt_tetr和pfab
‑ꢀ
anti
‑
lysozyme_tetr连接。将各个连接制备物转化进“dh5α
tm
‑
t1r大肠杆菌细胞”(life technologies gmbh)中,在所述细胞中扩增,通过测序验证分离的质粒的dna序列。根据方向和所使用的标记基因,所得构建体如下命名:
144.·
pcysex
‑
gapdh
‑
orf306_pyrh1_tetr(见图8)和 pcysex
‑
gapdh
‑
orf306_pyrh2_tetr
145.·
pcgt_pyrh1_tetr(见图9)和pcgt_pyrh2_tetr
146.·
pfab
‑
anti
‑
lysozyme_pyrh1_tetr(见图10)和 pfab
‑
anti
‑
lysozyme_pyrh2_tetr
147.·
pcysex
‑
gapdh
‑
orf306_plsc1_tetr(见图11)和 pcysex
‑
gapdh
‑
orf306_plsc2_tetr
148.·
pcgt_plsc1_tetr(见图12)和pcgt_plsc2_tetr
149.·
pfab
‑
anti
‑
lysozyme_plsc1_tetr(见图13)和 pfab
‑
anti
‑
lysozyme_plsc2_tetr
150.为了最终除去tetr基因,使用变体 pcysex
‑
gapdh
‑
orf306_pyrh1_tetr、pcgt_pyrh1_tetr、 pfab
‑
anti
‑
lysozyme_pyrh1_tetr、pcysex
‑
gapdh
‑
orf306_plsc1_tetr、 pcgt_plsc1_tetr和pfab
‑
anti
‑
lysozyme_plsc1_tetr继续该程序(见图8
‑
13)。
151.实施例9:除去抗生素抗性基因tetr并将含有作为剩余标记基因的pyrh 或plsc的生产质粒转化进具有染色体pyrh或plsc缺失的大肠杆菌菌株中
152.用于产生没有抗生素抗性基因tetr及含有pyrh或plsc作为标记基因的生产质粒的起始质粒是如实施例8所述的质粒pcysex
‑
gapdh
‑
orf306_ pyrh1_tetr、pcgt_pyrh1_tetr、pfab
‑
anti
‑
lysozyme_pyrh1_tetr、 pcysex
‑
gapdh
‑
orf306_plsc1_tetr、pcgt_plsc1_tetr和 pfab
‑
anti
‑
lysozyme_plsc1_tetr。从如实施例8所述质粒 pfab
‑
anti
‑
lysozyme_pyrh1_tetr和pfab
‑
anti
‑
lysozym_plsc1_tetr中去除抗生素抗性基因tetr是通过使用限制酶clai消化及随后重新连接而实现。
153.对于质粒pcysex
‑
gapdh
‑
orf306_pyrh1_tetr和 pcysex
‑
gapdh
‑
orf306_plsc1_tetr,进行相似程序,不同之处是基因tetr 是通过使用clai部分消化而去除,因为两个进一步的clai限制性位点位于结构基因cysex和orf306中(见图8和11)。
154.在pcgt_plsc1_tetr情况中,tetr基因是通过使用酶stui(切割以余下钝端)和fspi(切割以余下钝端)消化而从质粒中去除。
155.在pcgt_pyrh1_tetr的情况中,tetr基因同样通过使用酶stui(切割以余下钝端)和fspi(切割以余下钝端)部分消化而除去,因为进一步的fspi限制性位点位于pyrh基因内(见图9)。
156.在限制性消化之后,通过琼脂糖凝胶电泳纯化不含tetr的各个线性载体片段,并使用“qiaquick gel extraction kit”(qiagen gmbh,hilden, germany)根据厂商指导从琼脂糖凝胶中分离。之后,将所述无tetr的载体片段重新连接。
157.使用改进的cacl2方法,将相应的连接制备物转化进在实施例4中描述的菌株w3110δpyrh/paf
‑
ts
‑
pyrh和w3110lpp3δpyrh/paf
‑
ts
‑
pyrh或者w3110δpyrh/paf
‑
ts
‑
plsc和w31101pp3δpyrh/paf
‑
ts
‑
plsc。为了转化,即将含有pyrh或plsc作为标记基因的不含抗生素的生产质粒导入具有相应染色体缺失(pyrh或plsc)的大肠杆菌菌株中,进行以下程序:
158.在将5
‑
20μl所述连接制备物加入100μl的菌株 w3110 pyrh/paf
‑
ts
‑
pyrh和w31101pp3 pyrh/paf
‑
ts
‑
pyrh或者 w3110plsc/paf
‑
ts
‑
plsc和w31101pp3 plsc/paf
‑
ts
‑
plsc的cacl2感受态细胞中之后,将细胞在冰上再温育30分钟。在42℃短暂热休克45秒后,将细胞在冰上冷却2分钟。之后,将900μl的lb培养基加入转化制备物中,将细胞不是按照惯例在37℃而是在47℃
‑
55℃下温育/再生30
‑
90分钟。然后在lb琼脂平板上或在没有抗生素的液体lb培养基(例如不含四环素)中在升高的非许可温度于40
‑
45℃进一步温育15
‑
24小时。
159.在47℃
‑
55℃的30
‑
90分钟再生阶段和在升高的非许可温度下培养15 至24小时便于温度敏感型质粒paf
‑
ts
‑
pyrh或paf
‑
ts
‑
plsc换成含有pyrh 或plsc作为新选择标记基因的最终无抗生素抗性的生产质粒。
160.当转化的细胞在52℃再生60分钟然后在lb琼脂平板上在42℃温育 20小时时实现最佳转化结果。
161.首先在不具有抗生素的lb琼脂平板上初步选择温度敏感型质粒 paf
‑
ts
‑
pyrh或paf
‑
ts
‑
plsc的丢失及同时换成各个不具有四环素抗性基因 tetr的含有pyrh或plsc的生产质粒。
162.之后,检测预选的细菌克隆的氨苄青霉素敏感性,即丢失温度敏感型质粒paf
‑
ts
‑
pyrh或paf
‑
ts
‑
plsc
163.最终通过限制性消化检测来自氨苄青霉素敏感性克隆的编码pyrh或plsc的生产质粒。各自均没有抗生素抗性基因tetr的编码pyrh的生产质粒 pcysex
‑
gapdh
‑
orf306_pyrh、pcgt_pyrh和pfab
‑
anti
‑
lysozyme_pyrh 的限制图示于图14
‑
16。各自均没有抗生素抗性基因tetr的编码plsc的生产质粒pcysex
‑
gapdh
‑
orf306_plsc、pcgt_plsc和pfab
‑
anti
‑
lysozyme _plsc的限制图示于图17
‑
19。
164.如此产生和经检测的含有编码pyrh或plsc的生产质粒且具有染色体 pyrh或plsc缺失的无抗生素抗性的大肠杆菌菌株具有以下名称:
165.·
w3110δpyrh/pcysex
‑
gapdh
‑
orf306_pyrh
166.·
w31101pp3δpyrh/pcgt_pyrh
167.·
w3110lpp3δpyrh/pfab
‑
anti
‑
lysozym_pyrh
168.·
w3110δplsc/pcysex
‑
gapdh
‑
orf306_plsc
169.·
w3110lpp3δplsc/pcgt_plsc
170.·
w3110lpp3δplsc/pfab
‑
anti
‑
lysozym_plsc
171.实施例10:半胱氨酸发酵
172.预培养物1:
173.在erlenmeyer瓶(100ml)中,用特定的大肠杆菌菌株 w3110/pcysex
‑
gapdh
‑
orf306_tetr、 w3110δpyrh/pcysex
‑
gapdh
‑
orf306_pyrh或者 w3110δplsc/pcysex
‑
gapdh
‑
orf306_plsc接种20ml的lb培养基,并在摇床上温育7小时(150rpm,30℃)。对于现有技术的构建体 w3110/pcysex
‑
gapdh
‑
orf306_tetr的培养,即用抗生素作为选择剂,培养基补加15mg/l四环素。
174.预培养物2:
175.之后,将预培养物1全部移至100ml的sm1培养基(12g/l k2hpo4、3 g/l kh2po4、5g/l(nh4)2so4、0.3g/l mgso4×
7h2o、0.015g/l cacl2×
2h2o、 0.002g/l feso4×
7h2o、1g/l柠檬酸钠
×
2h2o、0.1g/l nacl、1ml/l微量元素溶液,其包含0.15g/l na2moo4×
2h2o、2.5g/l h3bo3、0.7g/l cocl
2 x 6h2o、 0.25g/l cuso4×
5h2o、1.6g/l mncl2×
4h2o、0.3g/l znso4×
7h2o,补加5g/l 葡萄糖和5mg/l维生素b1。将培养物在erlenmeyer瓶(1l)中在30℃以 150rpm摇动17小时。在此温育后,600nm处的光密度(od600)在3
‑
5之间。对于现有技术的w3110/pcysex
‑
gapdh
‑
orf306_tetr的培养,即用抗生素作为选择剂,培养基补加15mg/l四环素。
176.主培养物:
177.在来自sartorius stedim的biostat b型发酵罐中进行发酵。使用总体积为2l的培养容器。发酵培养基(900ml)含有15g/l葡萄糖、10g/l胰蛋白胨(difco)、5g/l酵母提取物(difco)、5g/l(nh4)2so4、1.5g/l kh2po4、0.5g/l nacl、0.3g/l mgso4×
7h2o、0.015g/l cacl2×
2h2o、0.075g/l feso4×
7h2o、 1g/l柠檬酸钠
×
2h2o和1ml微量元素溶液(见上述)及0.005g/l维生素b1。通过泵入25%nh4oh溶液将发酵罐中的ph初始调节至6.5。在发酵过程中,使用25%nh4oh自动校正为6.5水平维持ph。对于接种,将100ml 的预培养物2泵入发酵罐容器中。起始量因此为大约1l。最初在400rpm 下搅拌培养物并用2vvm通过无菌过滤器灭菌的压缩空气充气。在这些起始条件下,氧探针在接种前已校准至100%饱和。发酵过程中氧饱和度的标称值设定为50%。在o2饱和度低于标称值后,开始调节级联以使o2饱和度回到标称值。就此而言,首先持续增加气体供应(最大5vvm),然后持续提高搅拌速率(最高1500rpm)。
178.发酵在30℃的温度进行。在2小时的发酵时间后,以1.5ml/小时速率加入无菌的60%硫代硫酸钠
×
5h2o储存液形式的硫源。一旦发酵罐中起始 15g/l的葡萄糖含量已降至大约2g/l,则连续计量加入56%的葡萄糖溶液。设定进料速率以使发酵罐中的葡萄糖浓度从此开始不再超过2g/l。
179.使用来自ysi(yellow springs,ohio,usa)的葡萄糖分析仪测定葡萄糖。对于现有技术中的构建体w3110/pcysex
‑
gapdh
‑
orf306_tetr的发酵,即用抗生素作为选择剂,培养基补加15mg/l的四环素。
180.发酵时间为48小时。此后,收集样品并分别测定培养上清和沉淀物中的l
‑
半胱氨酸及其衍生物的含量,尤其是上清中的l半胱氨酸和四氢噻唑以及沉淀物中的l
‑
胱氨酸。为此,在每种情况中使用gaitonde(gaitonde,m. k.(1967),biochem.j.104,627
–
633)的比色测定法。存在于沉淀中的l
‑
胱氨酸首先必须溶解在8%盐酸中,然后才能以相同的方式定量。表1中列出的半胱氨酸总量相当于培养上清中l
‑
半胱氨酸和沉淀中l
‑
胱氨酸的总和。就此而言,每个l
‑
胱氨酸分子对应于两个l
‑
半胱氨酸分子。
181.表1:48小时后培养液中总半胱氨酸(l
‑
半胱氨酸
培养物上清
l
‑
胱氨酸
沉淀
)的含量以及生产质粒的稳定性
[0182][0183]
*现有技术环境中的构建体(对比实施例)
[0184]
**本发明的构建体
[0185]
实施例11:在10l规模(发酵)分泌生产环糊精糖基转移酶
[0186]
借助大肠杆菌的lpp突变体,可以产生生物技术相关酶如cgtases,并将其分泌至培养基中(us2008076158a1)。
[0187]
使用菌株w3110lpp3/pcgt_tetr(对照)、w3110lpp3δpyrh/pcgt_pyrh 和w3110lpp3δpyrh/pcgt_plsc,在10l搅拌发酵罐中进行cgtase的分泌产生。
[0188]
以1:10比率给填充有6l发酵培养基fm4(1.5g/l kh2po4,5g/l (nh4)2so4,0.5g/l mgso4×
7h2o,0.15g/l cacl2×
2h2o,0.075g/lfeso4×
7h2o,1g/l柠檬酸钠
×
2h2o,0.5g/l nacl,1ml/l微量元素溶液(0.15 g/l na2moo4×
2h2o,2.5g/l na3bo3,0.7g/l cocl2×
6h2o,0.25g/lcuso4×
5h2o,1.6g/l mncl2×
4h2o,0.3g/l znso4×
7h2o),5mg/l维生素 b1,3g/l phytone,1.5g/l酵母提取物,10g/l葡萄糖)的发酵罐接种在相同培养基中培养过夜的预培养物。在发酵期间,设定温度为30℃,通过计量加入nh4oh或h3po4使ph保持恒定在7.0的水平。在整个发酵过程中计量加入葡萄糖,争取在发酵培养基中的最大葡萄糖浓度<10g/l。通过在对数生长期结束时加入异丙基
‑
β
‑
d
‑
硫代半乳糖吡喃糖苷(iptg)至0.1mm诱导表达。
[0189]
对于现有技术中的构建体w3110lpp3/pcgt_tetr的发酵,即用抗生素作为选择剂,培养基补加15mg/l四环素。
[0190]
发酵72小时后,收集样品,通过离心从发酵培养基中除去细胞,并如 us2008076158a1的实施例4中所述测定发酵上清中的cgtase含量。表2 列出了在发酵上清中的功能性cgtase的产量以及活性。
[0191]
表2:发酵72小时后在发酵上清中环糊精糖基转移酶产量
[0192][0193]
*现有领域环境中的构建体(对比实施例)
[0194]
**本发明的构建体
[0195]
实施例12:在10l规模分泌性发酵生产fab抗体片段抗溶菌酶
[0196]
借助大肠杆菌的lpp突变体,功能性fab抗体片段也可以在细胞外产生 (us2008076158a1)。在这种情况中,细胞必须同时合成包含结构域vl和 cl的轻链的相应片段以及包含结构域vh和ch1的重链的相应片段,然后将其分泌至周质中并最终分泌至发酵培养基中。在细胞质外,两条链装配形成功能性fab片段。
[0197]
这个实施例描述了充分表征的抗
‑
溶菌酶抗体d1.3的fab片段的产生。
[0198]
质粒pfab
‑
anti
‑
lysozym_tetr、pfab
‑
anti
‑
lysozym_pyrh和 pfab
‑
anti
‑
lysozym_plsc不仅分别含有标记基因tetr、pyrh和plsc,而且还含有操纵子形式的fab片段的hc和lc的结构基因。在这种情况中,hc 符合读框地与ompa信号序列(ompa
ss
)的3'末端融合,lc符合读框地与 cgtase信号序列(cgt
ss
)的3'末端融合。ompa
ss
‑
hc
‑
cgt
ss
‑
lc操纵子的表达处于tac启动子的控制下。
[0199]
基于cgtase,与实施例11所述方法相似地进行抗溶菌酶fab片段的生产,使用菌株w3110lpp3/pfab
‑
anti
‑
lysozym_tetr、 w3110lpp3δpyrh/pfab
‑
anti
‑
lysozym_pyrh和 w3110lpp3δplsc/pfab
‑
anti
‑
lysozym_plsc。对于现有技术中大肠杆菌 w3110lpp3/pfab
‑
anti
‑
lysozym_tetr的发酵,即使用抗生素作为选择剂,所述培养基补加15mg/l四环素。
[0200]
发酵72小时后,收集样品,然后通过离心从发酵培养基中除去细胞。
[0201]
通过亲和性层析从发酵上清中纯化抗溶菌酶fab片段,如skerra(1994, gene 141,79
–
84)所述。
[0202]
定量所纯化的抗
‑
溶菌酶fab片段并通过用溶菌酶作为抗原的elisa测定确定其活性(skerra,1994,gene 141,79
–
84)。
[0203]
表2列出了发酵上清中功能性抗溶菌酶fab片段的预计(projected)产量,基于每种情况中发酵72小时后20ml发酵上清的分离的量。
[0204]
表3:发酵72小时后发酵上清中抗溶菌酶fab片段产量。
[0205][0206][0207]
*现有技术环境中的构建体(对比实施例)
[0208]
**本发明的构建体
[0209]
实施例13:质粒稳定性的确定
[0210]
通过质粒制备及随后的限制性消化检测质粒稳定性。为此目的,完成生产菌株的培养(例如发酵72小时后),随后在lb琼脂平板上铺板各种稀释度的培养物。为了随后确定质粒稳定性,即鉴定携带质粒的细胞(集落),仅具有单独集落的lb平板用于评估。
[0211]
总共50个单独菌落在液体lb培养基中培养15
‑
20小时,然后从所述培养物中分离质粒dna。单个生产质粒的特征性限制模式用作验证分离的质粒正确性的基础。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。