一种还原型谷胱甘肽
α
型晶体的制备方法
技术领域
1.本发明涉及结晶领域,具体涉及一种还原型谷胱甘肽α型晶体的制备方法。
背景技术:
2.谷胱甘肽(glutathione,简称gsh)是一种具有重要生理功能的活性三肽,它由谷氨酸、半胱氨酸和甘氨酸经肽键缩合而成,化学名为γ
‑
l
‑
谷氨酰
‑
l
‑
半胱氨酸
‑
甘氨酸(如
3.式1所示)。
[0004][0005]
gsh的相对分子质量为307.33,熔点189~193℃(分解),晶体呈无色透明细长柱状,等电点为5.93。它溶于水、稀醇、液氨和二甲基甲酰胺,而不溶于醇、醚和丙酮。gsh固体较为稳定,而水溶液在空气中则易被氧化。
[0006]
gsh是一种具有重要生理功能的活性三肽。它不仅能抵抗氧化剂对蛋白质硫氢基的破坏作用,维持蛋白质的正常生物活性,同时还是多种酶反应的辅酶,参与体内三羧酸循环及糖代谢,具有解毒、延缓衰老、预防糖尿病和癌症以及消除疲劳等作用。
[0007]
还原型谷胱甘肽存在两种晶型,分别为α和β。其中,α晶体在水中的溶解度较高(在10℃下,水中的溶解度约为89g/l;同等条件下,β晶型的还原型谷胱甘肽在水中溶解度仅为30g/l)。此外,α晶体为柱状,颗粒较大,便于抽滤、烘干,易于与结晶母液进行分离,因此纯度更高,杂质更少。而β晶体为针状晶体,颗粒非常细小,难以与结晶母液分离,易包裹母液中的杂质,导致成品的纯度较低。因为这些特性,大部分工业生产中需要得到的是α晶体。
[0008]
谷胱甘肽的α晶型由于溶解度高,相对于β晶型,是一种不稳定的晶型。因此结晶过程中通常得到的是β晶体,难以得到α晶体。因此迫切需要开发一种在还原性谷胱甘肽的过饱和溶液中抑制β晶体生成,并实现选择性结晶以获得α晶体的方法。
[0009]
日本专利《还原型谷胱甘肽的α型晶体的生产方法和所述晶体的保存方法》中采用氨基酸类的媒晶剂,来得到谷胱甘肽的α晶体,比如添加脂肪族氨基酸如丙氨酸、脯氨酸等,含硫氨基酸如半胱氨酸等,芳香族氨基酸如苯丙氨酸、色氨酸等。媒晶剂的添加量大约在0.01%
‑
10%。但添加这种媒晶剂,若浓度较高,在结晶过程中,随着谷胱甘肽晶体的形成,媒晶剂也会有析出,使得成品杂质的含量升高,降低谷胱甘肽的纯度。因此,需要开发一种更为有效的得到α晶体的结晶方法,以确保最终得到的谷胱甘肽纯度。
技术实现要素:
[0010]
发明目的:本发明所要解决的技术问题是针对现有技术的不足,提供一种还原型谷胱甘肽的α型晶体的制备方法。
[0011]
本发明还要解决的技术问题是提供一种抑制还原型谷胱甘肽β型晶体形成的方法。
[0012]
为了解决上述第一个技术问题,本发明公开了一种还原型谷胱甘肽的α型晶体的制备方法为以有机酸或其盐为媒晶剂。
[0013]
其中,所述有机酸包括但不限于甲酸、乙酸、丙酸、山梨酸和柠檬酸中的任意一种或几种组合。
[0014]
其中,所述盐包括但不限于钠盐、钾盐和铵盐中的任意一种或几种组合。
[0015]
其中,所述还原型谷胱甘肽的α型晶体的制备方法具体为向含还原型谷胱甘肽的溶液中加入媒晶剂,搅拌析晶,即得原型谷胱甘肽的α型晶体。
[0016]
其中,所述含还原型谷胱甘肽的溶液的溶剂为水。
[0017]
其中,所述含还原型谷胱甘肽的溶液的制备方法为将还原型谷胱甘肽加入水中,溶解,得到含还原型谷胱甘肽的溶液。
[0018]
其中,所述溶解通过以下任意一种或几种组合方式促进溶解:
[0019]
(i)升温加热;
[0020]
(ii)调节ph为6
‑
7;
[0021]
(iii)搅拌。
[0022]
方式(i)中,所述升温加热为在50℃以下加热。
[0023]
方式(ii)中,所述调节ph为6
‑
7优选为采用氢氧化钠或氨水溶液调节ph为6
‑
7。
[0024]
方式(ii)中,优选地,调节ph为6
‑
7,还原型谷胱甘肽溶解后再调节ph为2.5
‑
3.0。
[0025]
其中,所述调节ph为2.5
‑
3.0优选为采用盐酸、硫酸、磷酸等调节ph为2.5
‑
3.0。
[0026]
其中,所述含还原型谷胱甘肽的溶液中还原型谷胱甘肽的浓度为100g/l以上。
[0027]
其中,当含还原型谷胱甘肽的溶液中还原型谷胱甘肽的浓度小于200g/l时,在加入媒晶剂前或加入媒晶剂后,浓缩至还原型谷胱甘肽的浓度为200g/l以上,优选为浓缩至400g/l以上。
[0028]
其中,所述媒晶剂的质量为含还原型谷胱甘肽质量的0.01%
‑
1%。
[0029]
其中,所述搅拌析晶的过程中加入晶种。
[0030]
其中,所述晶种为α型原型谷胱甘肽晶体。
[0031]
其中,所述品种为在15
‑
30℃加入;优选地,所述晶种在20
‑
25℃加入;进一步优选地,所述晶种加入的温度可以是自然降温,也可以是程序控温。
[0032]
其中,所述晶种的加入量为0.2
‑
1.3g/100g还原型谷胱甘肽;优选地,所述晶种的加入量为0.5
‑
1g/100g还原型谷胱甘肽。
[0033]
其中,所述搅拌析晶的温度为15
‑
30℃。
[0034]
其中,所述搅拌析晶的时间为1h以上;优选地,所述搅拌析晶的时间为1.5h以上;进一步优选地,所述搅拌析晶的时间为2h以上。
[0035]
其中,所述搅拌析晶后大部分还原型谷胱甘肽的α型晶体析出。
[0036]
优选地,所述还原型谷胱甘肽的α型晶体的制备方法为向含还原型谷胱甘肽的溶
液中加入媒晶剂,搅拌析晶,加入有机溶剂或有机溶剂与水的混合溶剂,搅拌降温析晶,即得原型谷胱甘肽α型晶体。
[0037]
其中,所述有机溶剂包括但不限于甲醇、乙醇、丙酮和异丙醇中的任意一种或几种组合。
[0038]
其中,所述有机溶剂或有机溶剂与水的混合溶剂可以通过流加的方式加入,如用蠕动泵加入。
[0039]
其中,所述有机溶剂或有机溶剂与水的混合溶剂加入速率为所述含还原型谷胱甘肽的溶液体积的20%/h以下。
[0040]
其中,所述有机溶剂或有机溶剂与水的混合溶剂的加入量为所述含还原型谷胱甘肽的溶液体积的30%以上。
[0041]
其中,所述降温为降温至15℃以下;优选地,所述降温为降温至10℃以下;进一步优选地,所述降温为降温至5
‑
10℃。
[0042]
其中,所述搅拌降温析晶的时间为0.5h以上;优选地,所述搅拌降温析晶的时间为1h以上;进一步优选地,所述搅拌降温析晶的时间为0.5h以上;优选地,所述搅拌降温析晶的时间为1.5h以上;更进一步优选地,所述搅拌降温析晶的时间为2h以上。
[0043]
其中,所述搅拌降温析晶结束后,停止搅拌,将所述原型谷胱甘肽的α型晶体抽滤、干燥,即得原型谷胱甘肽的α型晶体成品。
[0044]
其中,所述原型谷胱甘肽的α型晶体具有明显的柱状结构,在结晶罐中沉降性能好,分层明显,易于后续的抽滤和烘干,且在常温下水中的溶解速率快,溶解度比β晶体高。
[0045]
为了解决上述第二个技术问题,本发明公开了一种抑制还原型谷胱甘肽β型晶体形成的方法,以有机酸或其盐为媒晶剂。
[0046]
其中,所述有机酸包括但不限于甲酸、乙酸、丙酸、山梨酸和柠檬酸中的任意一种或几种组合。
[0047]
其中,所述盐包括但不限于钠盐、钾盐和铵盐中的任意一种或几种组合。
[0048]
本发明通有机酸的机理:由于谷胱甘肽的α晶体的溶解度高(10℃,89g/l),而β晶型的溶解度低(10℃,30g/l),因此β晶型的稳定性更高,结晶过程中易于得到的是β晶型。本发明通过加入有机酸,带来一定的阻滞作用,有机酸分子吸附在β晶体的表面,阻碍谷胱甘肽基元分子进入晶格,从而抑制结晶过程中β晶型的形成,并促进β晶型向α晶体的转变。
[0049]
有益效果:与现有技术相比,本发明具有如下优势:
[0050]
本发明所提供的方法以有机酸或其盐为媒晶剂,显著地抑制了β型晶体的形成和α型晶体向β型晶体的转变,可以高效且稳定地生产还原型谷胱甘肽的α型晶体,且最终获得的还原型谷胱甘肽晶体纯度高,质量分数可达到99%以上。
附图说明
[0051]
下面结合附图和具体实施方式对本发明做更进一步的具体说明,本发明的上述和/或其他方面的优点将会变得更加清楚。
[0052]
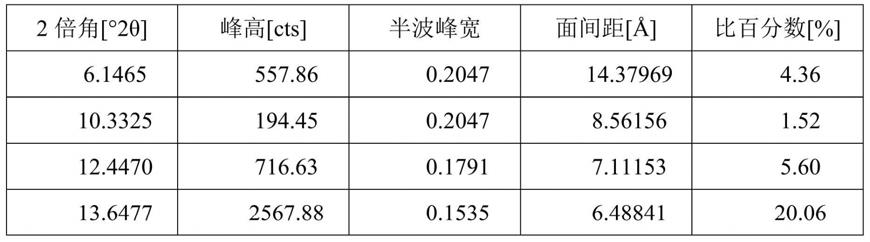
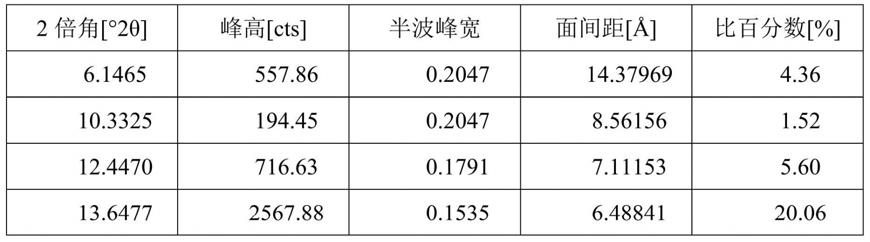
图1为实施例1所得还原型谷胱甘肽成品的α型晶体粉末x射线衍射图。
[0053]
图2为实施例1所得还原型谷胱甘肽成品α型晶体在显微镜下的形貌。
[0054]
图3为成品谷胱甘肽hplc图。
[0055]
图4为添加l
‑
半胱氨酸作为媒晶剂得到得谷胱甘肽晶体在显微镜下的形貌。
[0056]
图5为谷胱甘肽β晶体的粉末x射线衍射分析结果。
具体实施方式
[0057]
下述实施例中所述实验方法,如无特殊说明,均为常规方法;所述试剂和材料,如无特殊说明,均可从商业途径获得。
[0058]
下述实施例中所述谷胱甘肽原料中还原型谷胱甘肽含量为98%。
[0059]
下述实施例中所述搅拌,若无特殊说明,所述搅拌的转速为150rpm。
[0060]
下述实施例中所述95%乙醇为95vt%乙醇水溶液。
[0061]
实施例1:谷胱甘肽的结晶
‑
乙酸作为媒晶剂
[0062]
将150g谷胱甘肽原料溶解于1l纯水中,通过搅拌和水浴(45
‑
47℃)加速溶解,然后加入0.5ml乙酸,再通过真空浓缩的方式(50℃)将谷胱甘肽的浓度提高至450g/l。将浓缩好的溶液转移到1l结晶罐中,在不断搅拌下,自然降温至25℃,加入0.75g α型的谷胱甘肽晶体作为晶种,继续搅拌2小时,使得其中大部分α型谷胱甘肽晶体析出。随后用蠕动泵向结晶罐中流加95%的乙醇,流速为:40ml/h,总添加量为120ml。待酒精添加结束后,通过结晶罐的夹套进行降温至5℃,在此温度下维持2小时,然后停止搅拌,将结晶罐中的谷胱甘肽晶浆放出,晶体抽滤,并放入45℃下真空干燥箱中进行烘干,即得到α型谷胱甘肽晶体成品126g,收率为84%,纯度为99.5%;所得成品的x
‑
射线衍射图如图1所示,衍射数据如表1所示,在显微镜下的形貌如图2所示,样品的hplc图如图3所示。
[0063]
表1α型谷胱甘肽晶体成品的衍射数据
[0064]
[0065]
[0066][0067]
实施例2:谷胱甘肽的结晶
‑
乙酸钠作为媒晶剂
[0068]
将100g谷胱甘肽原料溶解于500ml纯水中,然后加入0.6g乙酸钠,接着采用真空浓缩的方式,将谷胱甘肽溶液浓缩至250ml,并将浓缩好的溶液转移至1l的结晶罐中,搅拌器的转速为150rpm,自然冷却至20℃,并加入0.5gα型原型谷胱甘肽晶体,然后以40ml/h的流速滴加95%的乙醇,共添加250ml。然后将结晶罐的温度在2小时内降温至10℃,并在此温度下维持2小时,随后停止搅拌,将结晶罐内的谷胱甘肽晶浆放出,晶体抽滤、烘干,即得到α型谷胱甘肽晶体成品91g,收率为:91%,纯度为:99%。
[0069]
实施例3:谷胱甘肽的结晶
‑
柠檬酸纳为媒晶剂
[0070]
通过在45℃下加热搅拌,将200g谷胱甘肽原料溶解于1l纯水中,接着加入2g柠檬酸钠。搅拌溶解后,将配好的溶液转移至2l的结晶罐中,并在不断搅拌下,将溶液的温度在2小时内程序降温至25℃,接着向结晶罐中加入2gα型原型谷胱甘肽晶体作为晶种,搅拌1小时后,通过蠕动泵以150ml/h的速度,向结晶罐中流加95%的乙醇,共加入1.5l。接着通过夹套降温的方式,将溶液的温度程序梯度降温至5℃,并在此温度下维持2小时以上,然后停止搅拌,下罐,收集晶浆,晶体抽滤、烘干,即得到α型谷胱甘肽晶体成品160g,收率为80%,纯度为99%。
[0071]
对比实施例1:谷胱甘肽的结晶
‑
l
‑
半胱氨酸作为媒晶剂
[0072]
将150g谷胱甘肽原料溶解于1l纯水中,通过搅拌和水浴(45
‑
47℃)加速溶解,然后再加入0.5g l
‑
半胱氨酸,再通过真空浓缩的方式(50℃)对谷胱甘肽溶液进行浓缩。发现当浓缩至谷胱甘肽浓度达到约260g/l时,溶液中开始有白色沉淀析出。继续浓缩后,析出大量的沉淀。析出的沉淀经过抽滤、烘干后,发现主要为非常细小的谷胱甘肽β晶体,其在显微镜下得照片如图4所示,晶体粉末x
‑
射线衍射分析结果如图5所示。因此,采用添加l
‑
半胱苷酸的方法(添加量为谷胱甘肽的0.33%)难以实现制备得到α
‑
谷胱甘肽晶体。
[0073]
对比实施例2:无媒晶剂的谷胱甘肽的结晶
[0074]
将20g谷胱甘肽原料(外购自金城医药,α晶体,谷胱甘肽的含量为98%),溶解于200ml纯水中,通过加热和搅拌的方式使之全部溶解,然后在不断搅拌下,冷却至10℃,在此过程中析出大量的沉淀,经在显微镜下观察,为细碎的β晶体。因此若不加入媒晶剂,得到的是β晶体。
[0075]
本发明提供了一种还原型谷胱甘肽α型晶体的制备方法的思路及方法,具体实现该技术方案的方法和途径很多,以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。本实施例中未明确的各组成部分均可用现有技术加以实现。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。