1.本发明属于工业大麻差异代谢物鉴别技术领域,具体涉及一种基于代谢组学技术挖掘工业大麻激素调控应答基因的方法。
背景技术:
2.工业大麻在我国发展历史由来已久,近年年种植面积达到100万亩左右,中国已成为工业大麻种植、生产、消费和出口的大国。经系列研究证明,工业大麻其花叶中大麻酚类等医药保健成分、籽实及其内所含营养物质、纤维、杆芯等原料在军民各领域具有独特应用和功效,国内外消费者对其产品的兴趣及需求与日俱增,工业大麻产业已经成为拉动地方经济的特色经济产业之一。
3.代谢组学及其技术方法旨在通过考察生物体系(细胞、组织等)受到刺激或扰动后,其代谢产物的变化或随时间规律发生的变化,经差异代谢物筛选,研究差异代谢物参与的生物过程,揭示其参与的生命活动机制。当前,代谢组学已经成为植物生长发育调控等系列研究的强大工具之一。应用代谢组学技术分析激素调控下代谢物的调控表达机制已有报道,研究也表明,激素在调节有效成分积累代谢等方面具有特殊的作用效果,但通过非靶代谢组学方法,快速大量识别激素调节作用下工业大麻代谢作用机制目前还未见报道。
4.通过比较代谢组学技术,分析并获得激素调节作用下工业大麻有价值差异代谢物,对今后深入研究和筛选适宜其有效成分积累的生长调节剂,以及进一步通过分子手段进行生长物质调控具有重要的现实意识。
技术实现要素:
5.本发明的目的是利用外源激素作为外部重要调节物质,重点研究其对汉麻花期差异代谢物的影响作用,以期为深入挖掘花期阶段响应激素调节的代谢产物、差异性变化等提供详细信息,并进一步为生产中利用植物生长调节剂调控生长发育及分子育种等研究提供技术借鉴和理论支持,提供一种响应激素调控工业大麻差异代谢物的代谢组学分析方法。
6.为实现上述目的,本发明采取的技术方案如下:
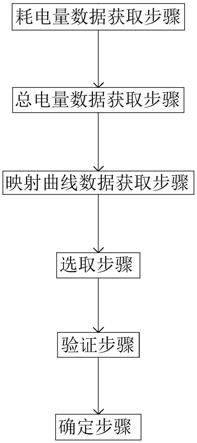
7.一种基于代谢组学技术挖掘工业大麻激素调控应答基因的方法,所述方法具体为:
8.步骤一:采用激素浸泡处理种子,以蒸馏水浸种为对照,处理时间3
‑
5h,后种子于温室中种植;
9.步骤二:选取对照及处理组雌株花期顶端叶片,液氮速冻,
‑
80℃保存,代谢组分析备用;
10.步骤三:代谢物提取及样本制备:
11.(1)分别取步骤二的对照组和处理组的组织100mg解冻后研磨,置于ep管中,加入
500μl的80%甲醇水溶液;
12.(2)涡旋震荡,冰浴静置5min,15000g、4℃离心20min;
13.(3)取一定量的上清加质谱级水稀释至甲醇含量为53%;
14.(4)15000g、4℃离心20min,收集上清,进样lc
‑
ms进行分析;
15.步骤四:进行lc
‑
ms分析,色谱条件为:色谱柱:hypesilgoldcolumn(c18),柱温:40℃,流速:0.2ml/min,正模式:流动相a:0.1%甲酸,流动相b:甲醇,负模式:流动相a:5mm醋酸铵,ph 9.0,流动相b:甲醇;
16.色谱梯度洗脱程序:0
‑
1.5min,98%a,2%b;1.5
‑
12min,100%b;12
‑
14min,100%b;14
‑
14.1min,98%a,2%b;14.1
‑
17.0min,98%a,2%b;
17.质谱条件:扫描范围m/z 100
‑
1500;esi源的设置如下,喷涂电压:3.2kv;鞘气流速:40arb;辅助气流速:10arb;毛细管温度:320℃;极性:正、负;ms/ms:二级扫描为数据依赖性全扫描;
18.步骤五:数据预处理及代谢物鉴定,将下机数据(.raw)文件导入cd搜库软件中,进行保留时间、质荷比参数的简单筛选,然后对不同样品根据保留时间偏差0.2min和质量偏差5ppm进行峰对齐,使鉴定更准确,随后根据设置的质量偏差5ppm、信号强度偏差30%、信噪比3、最小信号强度100000、加和离子等信息进行峰提取,同时对峰面积进行定量,再整合目标离子,然后通过分子离子峰和碎片离子进行分子式的预测并与mzcloud(https://www.mzcloud.org/)、mzvault和masslist数据库进行比对,用blank样本去除背景离子,并对定量结果进行归一化,最后得到数据的鉴定和定量结果;
19.步骤六:通过作图分析和数据库查询分析完成基因挖掘。
20.进一步地,步骤三和四中,所有试剂均为lc
‑
ms grade,试剂包括甲醇、水、甲酸、醋酸铵;其中,甲醇,cas67
‑
56
‑
1,甲酸,cas64
‑
18
‑
1,醋酸铵,cas631
‑
61
‑
8,品牌均thermo fisher,产地usa;水,cas7732
‑
18
‑
5,品牌merck,产地germany。
21.进一步地,步骤三中,为了对实验进行质量控制,处理样品时同时制备qc样本,具体为,从每个实验样本中取等体积样本混匀作为qc样本,用于平衡色谱
‑
质谱系统和监测仪器状态,在整个实验过程中对系统稳定性进行评价,同时设置blank样本,主要用于去除背景离子,具体为53%甲醇水溶液代替实验样本,前处理过程与实验样本相同。
22.进一步地,所述步骤六具体为:数据处理分析部分基于linux操作系统(centos版本6.6)以及软件r、python进行:
23.(1)使用kegg数据库(https://www.genome.jp/kegg/pathway.html)、hmdb数据库(https://hmdb.ca/metabolites)和lipidmaps数据库(http://www.lipidmaps.org/)对鉴定到的代谢物进行注释;
24.(2)多元统计分析部分,使用代谢组学数据处理软件metax对数据进行转换后进行主成分分析(pca)(图1)和偏最小二乘法判别分析(pls
‑
da),进而得每个代谢物的vip值,单变量分析部分,基于t检验来计算各代谢物在两组间统计学显著性(p值),并计算代谢物在两组间的差异倍数(fold change)即fc值,差异代谢物筛选的默认标准为vip>1,p值<0.05且fc≥2或fc≤0.5;
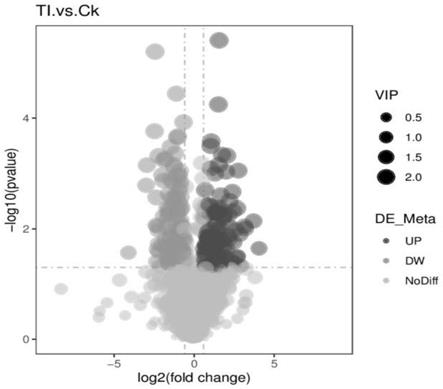
25.(3)火山图(图2)用r包ggplot2绘制,可以综合代谢物的vip值、log2(foldchange)和
‑
log10(p值)三个参数,来筛选感兴趣的代谢物;
26.(4)聚类热图,用r包pheatmap进行绘制,使用z
‑
score对代谢物数据进行归一化;
27.(5)差异代谢物之间的相关性分析(pearson相关系数)使用r语言cor()进行,统计显著性通过r语言中cor.mtest()实现,p值<0.05为在统计学上显著,并用r语言中的corrplot软件包绘制相关性图(图3);
28.(6)气泡图用r包ggplot2进行绘制,使用kegg数据库来研究代谢物的功能和代谢途径,当x/n>y/n时,认为该代谢途径富集;当代谢途径的p值<0.05时,认为该代谢途径是显著富集;
29.(7)利用kegg(https://www.kegg.jp/kegg/kegg1.html)进而分析推测与目标代谢物调控相关的重要基因。
30.本发明相对于现有技术的有益效果为:本发明基于lc
‑
ms/ms技术,对经过激素处理后种子的叶片材料进行代谢组检测,通过比较代谢组学研究方法,寻找差异代谢物,排除不同作用机制共有的代谢物,获得在不同处理条件下涉及的特异性代谢物,通过代谢物峰值的变化来实现对应答激素调节作用机制的识别和对代谢物的高通量分析。该方法的建立可在短期内实现高通量的对工业大麻代谢物的获取,同时鉴定出与激素调控紧密相关的差异代谢物。
附图说明
31.图1为激素处理组(ti)及ck主成分分析图;
32.图2为激素处理组(ti)及ck代谢物的t检验结果火山图;
33.图3为按p
‑
value值从小到大排序的top20的差异代谢物的相关性图。
具体实施方式
34.下面结合附图和实施例对本发明的技术方案作进一步的说明,但并不局限于此,凡是对本发明技术方案进行修改或者等同替换,而不脱离本发明技术方案的精神和范围,均应涵盖在本发明的保护范围中。
35.实施例1:
36.一种基于代谢组学技术挖掘工业大麻激素调控应答基因的方法,所述方法具体为:
37.步骤一:采用激素浸泡处理种子,以蒸馏水浸种为对照,处理时间3
‑
5h,后种子于温室中种植;
38.步骤二:选取对照(未处理)及处理组雌株花期顶端叶片,液氮速冻,
‑
80℃保存,代谢组分析备用;
39.步骤三:代谢物提取及样本制备:
40.(1)分别取步骤二的对照组和处理组的组织100mg解冻后研磨,置于ep管中,加入500μl的80%甲醇水溶液;
41.(2)涡旋震荡,冰浴静置5min,15000g、4℃离心20min;
42.(3)取一定量的上清加质谱级水稀释至甲醇含量为53%;
43.(4)15000g、4℃离心20min,收集上清,进样lc
‑
ms进行分析;
44.为了对实验进行质量控制,处理样品时同时制备qc样本,具体为,从每个实验样本
中取等体积样本混匀作为qc样本,用于平衡色谱
‑
质谱系统和监测仪器状态,在整个实验过程中对系统稳定性进行评价,同时设置blank样本,主要用于去除背景离子,具体为53%甲醇水溶液代替实验样本,前处理过程与实验样本相同。
45.步骤四:仪器设备涉及thermo fisher品牌的q exactivetmhf
‑
x质谱仪、vanquish uhplc色谱仪、hypesil gold column(100
×
2.1mm,1.9μm)色谱柱、scilogex品牌的d3024r低温离心机。进行lc
‑
ms分析,色谱条件为:色谱柱:hypesilgoldcolumn(c18),柱温:40℃,流速:0.2ml/min,正模式:流动相a:0.1%甲酸,流动相b:甲醇,负模式:流动相a:5mm醋酸铵,ph 9.0,流动相b:甲醇;
46.色谱梯度洗脱程序:0
‑
1.5min,98%a,2%b;1.5
‑
12min,100%b;12
‑
14min,100%b;14
‑
14.1min,98%a,2%b;14.1
‑
17.0min,98%a,2%b;
47.质谱条件:扫描范围m/z 100
‑
1500;esi源的设置如下,喷涂电压:3.2kv;鞘气流速:40arb;辅助气流速:10arb;毛细管温度:320℃;极性:正、负;ms/ms:二级扫描为数据依赖性全扫描;
48.上述步骤三和四中,所有试剂均为lc
‑
ms grade,试剂包括甲醇、水、甲酸、醋酸铵;其中,甲醇,cas67
‑
56
‑
1,甲酸,cas64
‑
18
‑
1,醋酸铵,cas631
‑
61
‑
8,品牌均thermo fisher,产地usa;水,cas7732
‑
18
‑
5,品牌merck,产地germany。
49.步骤五:数据预处理及代谢物鉴定,将下机数据(.raw)文件导入cd搜库软件中,进行保留时间、质荷比参数的简单筛选,然后对不同样品根据保留时间偏差0.2min和质量偏差5ppm进行峰对齐,使鉴定更准确,随后根据设置的质量偏差5ppm、信号强度偏差30%、信噪比3、最小信号强度100000、加和离子等信息进行峰提取,同时对峰面积进行定量,再整合目标离子,然后通过分子离子峰和碎片离子进行分子式的预测并与mzcloud(https://www.mzcloud.org/)、mzvault和masslist数据库进行比对,用blank样本去除背景离子,并对定量结果进行归一化,最后得到数据的鉴定和定量结果;
50.步骤六:通过作图分析和数据库查询分析完成基因挖掘。数据处理分析部分基于linux操作系统(centos版本6.6)以及软件r、python进行:
51.(1)使用kegg数据库(https://www.genome.jp/kegg/pathway.html)、hmdb数据库(https://hmdb.ca/metabolites)和lipidmaps数据库(http://www.lipidmaps.org/)对鉴定到的代谢物进行注释;
52.(2)多元统计分析部分,使用代谢组学数据处理软件metax对数据进行转换后进行主成分分析(pca)(图1)和偏最小二乘法判别分析(pls
‑
da),进而得每个代谢物的vip值,单变量分析部分,基于t检验来计算各代谢物在两组间统计学显著性(p值),并计算代谢物在两组间的差异倍数(fold change)即fc值,差异代谢物筛选的默认标准为vip>1,p值<0.05且fc≥2或fc≤0.5;
53.(3)火山图(图2)用r包ggplot2绘制,可以综合代谢物的vip值、log2(foldchange)和
‑
log10(p值)三个参数,来筛选感兴趣的代谢物;
54.(4)聚类热图,用r包pheatmap进行绘制,使用z
‑
score对代谢物数据进行归一化;
55.(5)差异代谢物之间的相关性分析(pearson相关系数)使用r语言cor()进行,统计显著性通过r语言中cor.mtest()实现,p值<0.05为在统计学上显著,并用r语言中的corrplot软件包绘制相关性图(图3);
56.(6)气泡图用r包ggplot2进行绘制,使用kegg数据库来研究代谢物的功能和代谢途径,当x/n>y/n时,认为该代谢途径富集;当代谢途径的p值<0.05时,认为该代谢途径是显著富集;
57.(7)利用kegg(https://www.kegg.jp/kegg/kegg1.html)进而分析推测与目标代谢物调控相关的重要基因。
58.表1对差异代谢物kegg富集结果表(部分)
[0059][0060]
注:mapid:富集的kegg pathway的id;maptitle:富集的kegg pathway名称;pvalue:富集分析的pvalue;x:与该通路相关的差异代谢物的数目;y:与该通路相关的背景(所有)代谢物的数目;n:kegg注释的差异代谢物数目;n:kegg注释的背景(所有)代谢物的数目;metaids:富集到的代谢物list;kegg_cpd_id:kegg数据库代谢物的id。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。