1.本发明涉及医药技术领域,特别涉及一种苯并呋喃型衍生物及其制备方法和应用。
背景技术:
2.红花八角(illicium dunnianum tutcher)为木兰科(magnoliaceae)八角属(illicium)植物。八角属植物为具有芳香气味的常绿乔木或灌木,全世界有34种,我国有28种,2变种,大多数分布在亚州东部、东南部,少数分部在北美洲东南部和中南美洲。红花八角为中国特有,分布于广西、福建、贵州、湖南、广东等地,常生长于河流沿岸、山谷水旁、山地林中、湿润山坡或岩石缝中,海拔400-1000米。其果实形态似红茴香,果瘦小,通常由7-8个果组成,少数13枚,有明显钻形尖头,略弯曲;果梗纤细但其果柄较短,种子较小,易区别。味苦、辛,性温,由于其具有散瘀消肿、祛风除湿、止痛的功效,民间常用其根和树皮入药,外用治风湿骨痛、跌打损伤、挫伤骨折;有毒,其根部分离得到的莽草毒素和新莽草毒素为惊厥性成分。药理实验研究表明红花八角叶的醇提液具有中枢及外周镇痛作用,对多种疼痛及急性软组织损伤具有较好的止痛和消肿作用。
3.红花八角为我国特有植物,作为民间药常用于治疗风湿骨痛、跌打损伤、挫伤骨折,尚未被中国药典收录,但红花八角叶是中成药金红片的组方药味之一,金红片具有疏肝解郁,理气活血,和胃止痛的功效,临床上主要用于治疗慢性浅表性胃炎肝胃不和证,疗效确切,效果显著,而目前无论是对红花八角叶还是金红片的化学成分研究甚少,无法全面的阐明金红片的化学成分,限制了金红片的药效物质及其作用机制的深入研究,也无法实现其质量控制标准的提升,因此对红花八角叶中活性成分进行深入研究。
技术实现要素:
4.本发明旨在对红花八角叶中活性成分进行更深入的研究,发现其活性成分。
5.有鉴于此,本发明提出了一种苯并呋喃型衍生物或者其药学上可接受的盐、溶剂化物、互变异构体、立体异构体、前药分子、代谢物,该化合物具有如下结构,该苯并呋喃型衍生物结构如式i所示:
[0006][0007]
本发明的另一目的在于提供一种上述苯并呋喃型衍生物的制备方法,其特征在于,包括:
[0008]
a)取红花八角叶,40~60%乙醇回流提取,除去溶剂,得总浸膏;
[0009]
b)所述总浸膏溶于水,经大孔吸附树脂柱色谱分离,依次以水、25~35%乙醇、45~55%乙醇、90~100%乙醇洗脱,分别收集各洗脱液,减压浓缩,得到水洗脱部位、25~35%乙醇洗脱部位、45~55%乙醇洗脱部位和90~100%乙醇洗脱部位;每个梯度洗脱4个柱体积(下同);
[0010]
c)取所述45~55%乙醇洗脱部位,经硅胶柱色谱分离,用二氯甲烷-甲醇梯度洗脱收集得到3a-3o共15个馏分,馏分3f经ods柱色谱甲醇-水梯度洗脱得到3f1-3f9共9个馏分,馏分3f9经半制备液相分离。
[0011]
具体地,所述红花八角叶可以是红花八角的干燥叶子。
[0012]
进一步地,所述步骤a)包括:取干燥的红花八角叶,经3-5倍量的40~60%乙醇回流提取1-3次,每次1-3小时,合并提取液,减压除去溶剂,得所述总浸膏。
[0013]
优选地,所述步骤b)包括:依次水、30%乙醇、50%乙醇、95%乙醇洗脱,分别收集各洗脱液,减压浓缩至无醇味,得到水洗脱部位、30%乙醇洗脱部位、50%乙醇洗脱部位和95%乙醇洗脱部位。
[0014]
可选地,所述步骤c)的所述二氯甲烷-甲醇梯度洗脱为,以100:0到0:100体积比进行梯度洗脱;所述甲醇-水梯度洗脱为,以15:85到100:0体积比进行梯度洗脱。
[0015]
可选地,所述步骤c)的所述二氯甲烷-甲醇梯度洗脱为,以100:0到0:100体积比进行梯度洗脱;所述甲醇-水梯度洗脱为,以15~30:85~70到100:0体积比进行梯度洗脱。
[0016]
具体地,所述步骤c)的所述二氯甲烷-甲醇梯度洗脱为,以100:0;95:5;90:10;85:15;80:20;70:30to 60:40,0:100体积比进行梯度洗脱;所述甲醇-水梯度洗脱为,以30:70;40:60;50:50;70:30to 100:0体积比进行梯度洗脱。
[0017]
具体地,所述大孔吸附树脂包括d101型大孔吸附树脂、hp-20型大孔吸附树脂、hpd-100型大孔吸附树脂、hpd-100a型大孔吸附树脂或hpd-300型大孔吸附树脂的一种或几种。
[0018]
进一步地,所述半制备液相色谱的条件包括:
[0019]
规格为c
18
,5μm,10
×
250mm phenomenex gemini柱;流动相的体积比例:20~30:80~70:0.05-0.5的乙腈-水-甲酸,检测波长为240-260nm,流速2-4ml/min。
[0020]
优选地,所述步骤a)为用50%乙醇回流提取2次,每次2小时;
[0021]
苯并呋喃型衍生物流动相为体积比例分别为25:75:0.1的乙腈-水-甲酸,检测波长为254nm,流速3ml/min。
[0022]
本发明的再一目的在于提供上述苯并呋喃型衍生物或者其药学上可接受的盐、溶剂化物、互变异构体、立体异构体、前药分子、代谢物在制备抗炎药物中的应用。
[0023]
本发明还提出了一种用于治疗炎症的药物,其包括上述苯并呋喃型衍生物或者其药学上可接受的盐、溶剂化物、互变异构体、立体异构体、前药分子、代谢物。
[0024]
进一步地,所述药物含有治疗有效量的上述苯并呋喃型衍生物或者其药学上可接受的盐、溶剂化物、互变异构体、立体异构体、前药分子、代谢物,以及一种或多种药学上可接受的载体。
[0025]
具体地,所述药物可以是药剂学上所说的任何一种剂型,包括片剂、胶囊剂、软胶囊、凝胶剂、口服剂、混悬剂、冲剂、贴剂、软膏、丸剂、散剂、注射剂、输液剂、冻干注射剂、静脉乳剂、脂质体注射剂、栓剂、缓释制剂或控释制剂。
[0026]
进一步地,所述药学上可接受的载体是指药学领域常规的药物载体,例如:稀释剂、赋形剂和水等,填充剂如淀粉、蔗糖,乳糖、微晶纤维素等;粘合剂如纤维素衍生物、藻酸盐、明胶和聚乙烯吡咯烷酮;润湿剂如甘油;崩解剂如羧甲基淀粉钠,羟丙纤维素,交联羧甲基纤维素,琼脂、碳酸钙和碳酸氢钠;吸收促进剂如季铵化合物;表面活性剂如十六烷醇,十二烷基硫酸钠;吸附载体如高龄土和皂粘土;润滑剂如滑石粉、硬脂酸钙和镁、微粉硅胶和聚乙二醇等。另外还可以在组合物中加入其它辅剂如香味剂、甜味剂等。
[0027]
本发明所述的苯并呋喃型衍生物是研究人员在红花八角叶中发现的新化学成分,并且发现此化合物在各批次的红花八角叶中均稳定存在。发明人通过理化性质和现代波谱学手段(ms、1h-nmr、
13
c-nmr等等),对上述方法分离得到的化合物进行了结构鉴定,证实其为结构如式(i)所示的新化合物。本发明还运用lps诱导raw 264.7细胞炎症模型等活性筛选体系进行活性评价,发现该化合物对小鼠巨噬细胞系raw 264.7有一定的保护作用,可以显著抑制pge2的释放,显示出较强的抗炎作用。具有良好的研究开发前景。
附图说明
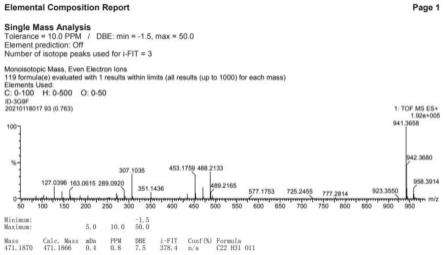
[0028]
图1为本发明化合物1的hr-esi-q-tof-ms谱图;
[0029]
图2为本发明化合物1的uv谱图;
[0030]
图3为本发明化合物1的ir谱图;
[0031]
图4为本发明化合物1的1h-nmr谱图
[0032]
图5为本发明化合物1的
13
c-nmr谱图;
[0033]
图6为本发明化合物1的dept-135谱图;
[0034]
图7为本发明化合物1的h
1-h
1 cosy谱图;
[0035]
图8为本发明化合物1的hsqc谱图;
[0036]
图9为本发明化合物1的hmbc谱图;
[0037]
图10为本发明化合物1的noesy图谱;
[0038]
图11为本发明化合物1的主要1h-1
h cosy,hmbc和noesy相关图;
[0039]
图12为本发明化合物1的实验和计算cd图谱。
具体实施方式
[0040]
以下将结合实验例的内容进行具体描述。
[0041]
特别需要指出的是,针对本发明所做出的类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明。相关人员明显能在不脱离本发明内容、精神和范围内对本文所述的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。
[0042]
本发明如未注明具体条件者,均按照常规条件或制造商建议的条件进行,所用原料药或辅料,以及所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
[0043]
实施例1本发明化合物的制备
[0044]
(1)取红花八角叶干燥叶子,用40%乙醇回流提取2次,每次2小时,合并提取液,减压除去溶剂,得总浸膏。总浸膏溶于水,经hp-20大孔吸附树脂柱色谱分离,依次以水、25%乙醇、45%乙醇、90%乙醇洗脱,每个梯度洗脱4个柱体积(下同),分别收集各洗脱液,减压
浓缩至无醇味,得到水洗脱部位、25%乙醇洗脱部位、45%乙醇洗脱部位和90%乙醇洗脱部位;
[0045]
(2)取步骤(1)的45%乙醇洗脱部位,经硅胶柱色谱分离,用二氯甲烷-甲醇梯度洗脱(95:5;90:10;85:15;80:20;70:30to 60:40,0:100,v/v),收集得到15个馏分(3a-3o),馏分3f经ods柱色谱甲醇-水(15:85;40:60;45:55to 100:0,v/v)梯度洗脱得到3f1-3f9共9个馏分,馏分3f9经半制备液相得到本发明化合物。
[0046]
其中,上述步骤(2)所述半制备液相色谱条件为,半制备型色谱柱:phenomenex gemini(c
18
,5μm,10
×
250mm),半制备型高效液相色谱仪[日本岛津,泵:lc-6ad(shimadzu,liquid chromatograph);检测器:spd-20a(prominence uv/vis detector);工作站:lc solution)]。本发明化合物的流动相为体积比例分别为:20:80:0.05的乙腈-水-甲酸,检测波长为240nm,流速3ml/min。
[0047]
实施例2本发明化合物的制备
[0048]
(1)取红花八角叶干燥叶子,用50%乙醇回流提取2次,每次2小时,合并提取液,减压除去溶剂,得总浸膏。总浸膏溶于水,经hp-20大孔吸附树脂柱色谱分离,依次以水、30%乙醇、50%乙醇、95%乙醇洗脱,分别收集各洗脱液,减压浓缩至无醇味,得到水洗脱部位、30%乙醇洗脱部位、50%乙醇洗脱部位和95%乙醇洗脱部位;
[0049]
(2)取步骤(1)的50%乙醇洗脱部位,经硅胶柱色谱分离,用二氯甲烷-甲醇梯度洗脱(100:0;95:5;90:10;85:15;80:20;70:30to 60:40,0:100,v/v),收集得到15个馏分(3a-3o),馏分3f经ods柱色谱甲醇-水(30:70;40:60;50:50;70:30to 100:0,v/v)梯度洗脱得到3f1-3f9共9个馏分,馏分3f9经经半制备液相得到本发明化合物。
[0050]
其中,上述步骤(2)所述半制备液相色谱条件为,半制备型色谱柱:phenomenex gemini(c
18
,5μm,10
×
250mm),半制备型高效液相色谱仪[日本岛津,泵:lc-6ad(shimadzu,liquid chromatograph);检测器:spd-20a(prominence uv/vis detector);工作站:lc solution)]。本发明化合物的流动相为体积比例分别为:25:75:0.1的乙腈-水-甲酸,检测波长为254nm,流速3ml/min。
[0051]
实施例3本发明化合物的制备
[0052]
(1)取红花八角干燥叶子,用60%乙醇回流提取2次,每次2小时,合并提取液,减压除去溶剂,得总浸膏。总浸膏溶于水,经hp-20大孔吸附树脂柱色谱分离,依次以水、35%乙醇、55%乙醇、100%乙醇洗脱,分别收集各洗脱液,减压浓缩至无醇味,得到水洗脱部位、35%乙醇洗脱部位、55%乙醇洗脱部位和100%乙醇洗脱部位;
[0053]
(2)取步骤(1)的55%乙醇洗脱部位,经硅胶柱色谱分离,用二氯甲烷-甲醇梯度洗脱(90:10;80:20;75:25;70:30to 60:40,v/v),收集得到15个馏分(3a-3o),馏分3f经ods柱色谱甲醇-水(20:80;50:50;55:45to 100:0,v/v)梯度洗脱得到3f1-3f9共9个馏分,馏分3f9经半制备液相得到本发明化合物。
[0054]
其中,上述步骤(2)所述半制备液相色谱条件为,半制备型色谱柱:phenomenex gemini(c
18
,5μm,10
×
250mm),半制备型高效液相色谱仪[日本岛津,泵:lc-6ad(shimadzu,liquid chromatograph);检测器:spd-20a(prominence uv/vis detector);工作站:lc solution)]。本发明化合物的流动相为体积比例分别为:30:70:0.5的乙腈-水-甲酸,检测波长为260nm,流速3ml/min。
[0055]
实施例4本发明化合物的结构鉴定
[0056]
棕色无定形粉末,[α]25d 74.8(c 0.65,meoh),hr-esi-ms给出m/z 399.1411[m na]
(计算值:399.1420),确定分子式为c
20h24
o7,不饱和度为9。
[0057]
如图1-12所示,化合物的1h-nmr(600mhz,in cd3od)图谱低场区氢信号[δ
h 6.74(1h,brs,h-2
′
),6.73(1h,d,j=8.4hz,h-5
′
),6.62(1h,dd,j=8.4,2.7hz,h-6
′
);6.76(1h,brs,h-4),6.74(1h,brs,h-6)]提示存在一组1,2,4-三取代和一组1,2,3,5-四取代的苯环,2个甲氧基氢信号[δh3.84,(6h,s,3
′
,7-och3)]。
13
c-nmr(150mhz,in cd3od)共给出20个碳信号,包括12个sp2杂化碳信号(δ
c 151.7,149.4,146.1,145.5,143.1,137.7,128.8,117.9,116.1,114.6,110.1,104.0),2个次甲基碳信号(δ
c 109.4,53.9),4个亚甲基碳信号(δ
c 63.7,62.2,35.8,32.8)以及2个甲氧基碳信号(δ
c 56.9,56.4)。
[0058]1h-1
h cosy谱中,观察到h-2/h-3/h
2-13;h
2-10/h
2-11/h
2-12分别存在明显相关,结合hsqc谱,推测结构中含有两个c3结构片段c-2-c-3-c-13和c-10-c-11-c-12。结合hmbc相关h-4/c-10;h-6/c-10;h-10/c-4,5,6,11,12推断出结构中存在一个c
6-c3结构片段,此外还观察到h-4/c-3;h-2/c-8,9,13的远程相关,推断结构中存在苯并呋喃环结构。最后,h-2与c-1
′
的远程相关确定3
′‑
甲氧基-4
′‑
羟基苯环结构片段与c-2位相连,因此确定了化合物的平面结构。noesy图谱显示相关峰h-2/h-13,结合2,3位的耦合常数j
2,3
=1.3hz,确定2,3位的相对构型为反式,结合cd测试结果与ecd结果确定2,3位的绝对构型为2s,3r。
[0059]
综合一维二维核磁信息,归属了化合物的全部碳氢信号(表1)。将化合物鉴定为新的苯并呋喃类化合物,命名为(2s,3r)-2-(3-甲氧基-4-羟基苯氧基)-2,3-二氢-3-羟甲基-7-甲氧基-5-苯并呋喃丙醇。结构如下:
[0060][0061]
表1.化合物的1h and 13
c nmr数据
[0062][0063]
measured at 600mhz for 1
h and 150mhz for 13
c in cd3od
[0064]
multiplets and or overlapped signals are reported without designating multiplicity
[0065]
实施例5本发明化合物体外抗pge2实验
[0066]
1.材料
[0067]
1.1药物 本发明化合物;
[0068]
1.2细胞模型 小鼠巨噬细胞系raw 264.7,来源于中医科学院;培养条件:dmem 10%胎牛血清(fbs),37℃,5%co2。
[0069]
2.原理和方法
[0070]
2.1实验原理
[0071]
革兰阴性菌外膜的脂多糖(lps)(美国sigma公司,批号:114m4009)是介导感染性炎症损伤的最主要的病原分子之一,许多疾病与lps诱导的持续亚临床炎症密切相关。在动物和细胞实验中,lps被广泛用来诱导炎症的发生。
[0072]
巨噬细胞在炎症反应中起着至关重要的作用,被刺激后,巨噬细胞产生大量的炎症因子和炎症介质,如:tnf-α,il-1β,il-6,no和pge2等。这些炎症因子和介质激活是炎症的关键过程,对它们的抑制作用常常作为评价药物抗炎活性的重要指标。
[0073]
2.2药物对分泌pge2的抑制试验
[0074]
方法步骤:
[0075]
(1)药液的配制:将本发明的化合物以10%fbs的dmem培养基溶解,制得2mg/ml的储备液。
[0076]
(2)实验方法:将细胞以0.25%胰酶(含0.02%edta)消化,含10%fbs的dmem培养基调整细胞密度为1
×
105个/ml,均匀接种至24孔板,每孔400μl,种板后放入培养箱培养24小时。
[0077]
空白对照组(n组):每孔加入495μl无血清的dmem培养基;
[0078]
溶媒组/溶剂对照组(rm组):每孔加入495μl含千分之一dmso的无血清dmem培养基;
[0079]
模型组(m组):每孔加入495μl 100μg/ml的lps;
[0080]
给药样品组:每孔加495μl含不同浓度含药的培养基;
[0081]
同时设6个复孔,加药完毕后将24孔板放入co2细胞培养箱培养1小时。1小时后,除空白对照和溶剂对照组外,其余每孔加入5μl的100μg/ml的lps(终浓度为1μg/ml),溶剂对照组和空白对照组每孔加入5μl的无血清的dmem培养基,加药完毕后将24孔板放入co2细胞培养箱继续培养18小时。
[0082]
18小时后收集细胞培养液,按试剂盒说明,用elisa法检测细胞上清中pge2的含量。
[0083]
pge2抑制率(%)=(模型组pge2的平均含量-样品组pge2的平均含量)/(模型组pge2的平均含量-溶剂组pge2的平均含量)
×
100%。
[0084]
3.实验结果
[0085]
3.1药物样品对小鼠巨噬细胞系raw 264.7细胞上清pge2的影响
[0086]
结果表明,药物样品可以显著抑制lps诱导小鼠巨噬细胞raw 264.7pge2的分泌,显示出较强的抗炎作用。数据结果如表2。
[0087]
表2化合物各浓度对小鼠巨噬细胞系raw 264.7细胞上清pge2的影响(n=3)
[0088][0089]
本发明采用graphadprism 7.00分析软件,通过线性回归分析方法测的本发明化合物体外抑制lps诱导小鼠巨噬细胞raw 264.7分泌炎性介质pge2的平均ic
50
为18.13μm。
[0090]
4.结论
[0091]
本发明化合物对lps诱导小鼠巨噬细胞raw 264.7分泌炎性介质pge2有显著的抑制作用,显示出较强的抗炎作用,且随着药物浓度的上升,对pge2分泌的抑制作用也增加,ic
50
为18.13μm。
[0092]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。