1.本发明属于有机合成技术领域,具体地说,涉及一种普拉格雷中间体及其类似物的制备方法。
背景技术:
2.普拉格雷是一种血小板二磷酸腺苷受体抑制剂,为日本sankyo公司和礼来公司联合开发的新药,是一种前体药物,本身并不具有活性,在体内有效的转化成它的活性代谢物用于治疗血栓,具有良好的抗凝血效果,同时具有良好的生物利用度。具体结构如下:
[0003][0004]
式i所示化合物,普拉格雷,化学名为2-乙酰氧基-5-(α-环丙羰基-2-氟苄基)-4,5,6,7-四氢噻吩并[3,2-c]吡啶。
[0005]
目前公开的普拉格雷合成方法主要有:
[0006]
公开号为cn981092209的专利申请中公开了从式(ii)所示化合物开始的合成工艺,经过溴代后,与4,5,6,7a-四氢噻吩并[3,2-c]吡啶-2-酮盐酸盐反应得到5-(α-环丙羰基-2-氟苄基)-2-羰基-4,5,6,7a-四氢噻吩并[3,2-c]吡啶,然后与乙酸酐在钠氢和dmf存在下得到普拉格雷(i)。该方法的特点在于,对接时采用噻吩酮直接与卤代烃缩合,缺陷在于噻吩酮上的酮羰基存在羟酮互变,缩合时可能形成醚类化合物,选择性较差,后处理需要柱层析纯化,且乙酰化时需要使用较危险的钠氢。该方法成本太高,操作繁琐,不适宜工业化生产。
[0007][0008][0009]
公开号为us5874581的专利申请中也公开了从式(ii)所示化合物开始的合成工艺,经过氯代后,与2-叔丁基二甲硅氧基-4,5,6,7a-四氢噻吩并[3,2-c]吡啶反应得到2-叔丁基二甲硅氧基-5-(α-环丙羰基-2-氟苄基)-2-羰基-4,5,6,7a-四氢噻吩并[3,2-c]吡啶,然后与乙酸酐在4-二甲胺基吡啶和三乙胺存在下得到普拉格雷(i)。该方法能够保护噻吩
酮的酮羰基,提高缩合反应的选择性,但同时也延长了反应步骤,增加了设备成本,收率降低。
[0010][0011]
从这些工艺中可以看出,式(ii)所示化合物是合成普拉格雷的关键中间体,目前合成式(ii)所示化合物的方法主要有:
[0012]
公开号为us5288726的专利申请中公开了以邻氟苄溴为起始原料,与环丙腈发生格氏反应得到式(ii)所示化合物。该工艺需要严格的无水条件,同时使用无水乙醚作为溶剂,乙醚沸点低易挥发,危险性增大,反应条件较为苛刻,且收率不高,不利于工业化生产。
[0013][0014]
公开号为wo2011042918a2的专利申请中公开了以2-(2-氟苯基)乙酸为原料,dcc为活化剂,经酰胺化得到weinreb酰胺,与环己烷格氏试剂反应得到式(ii)所示化合物。该方法需要严格的无水条件,所得产物复杂,后处理困难,反应条件较为苛刻,且产率不高,不利于工业化生产。
[0015][0016]
公开号为cn104418718a的专利申请中公开了以2-氟苯乙酸酯为起始原料,和环丙甲酰氯发生酰化反应,水解制得式(ii)所示化合物。该方法需要使用大量二硫化碳,有强烈的恶臭气味,不利于工业化生产;需要预制备酰氯,增加了操作工序。
[0017][0018]
公开号为cn104418718a的专利申请中公开了以氟苯为起始原料,四氯化锡为路易斯酸催化剂,与1-溴-3-烯-丁-2-酮反应得到1-(2-氟苯基)-3-烯-丁-2-酮,再与硫叶立德试剂二甲基亚甲基硫或二甲基亚甲基氧硫反应制备环丙烷,得到式(ii)所示化合物。该方法需要预制备试剂,增加了操作步骤,降低了生产效率;需要使用易吸水的路易斯酸四氯化锡,具有强烈的腐蚀性,增大了工业生产的危险性。
[0019][0020]
鉴于普拉格雷良好的市场价值,仍需一种方法简单、条件温和且收率高的制备普拉格雷中间体的方法。
技术实现要素:
[0021]
本发明的目的是提供一种安全、绿色、高效的普拉格雷中间体及其类似物的制备方法。
[0022]
为了实现上述目的,本发明采用的技术方案如下:
[0023]
本发明的第一方面提供了一种普拉格雷中间体及其类似物的制备方法,包括以下步骤:
[0024][0025]
将配体、过渡金属催化剂溶于溶剂中超声混匀,加入化合物ii、化合物iii、碱、光催化剂,在室温下充入氩气保护,光照反应,获得式i所述普拉格雷中间体及其类似物;
[0026]
所述配体、过渡金属催化剂、化合物ii、碱、光催化剂与化合物iii的摩尔比为(0.05~0.2):(0.05~0.2):(0.2~5.0):(1.0~5.0):(0.01~0.1):1;优选为0.067:0.067:0.67:2:0.013:1。
[0027]
所述化合物iii中,r1选自c1~c20直链烷基、c1~c20支链烷基、苯基、c3~c6环烷基;
[0028]
所述化合物ii中,x选自氯、溴;
[0029]
r2选自氟、氢、c1~c20直链烷基、c1~c20支链烷基、c1~c20直链烷氧基、c1~c20支链烷氧基;
[0030]
r3选自氟、氢、c1~c20直链烷基、c1~c20支链烷基、c1~c20直链烷氧基、c1~c20支链烷氧基、硝基;
[0031]
r4选自氟、氢、c1~c20直链烷基、c1~c20支链烷基、c1~c20直链烷氧基、c1~c20支链烷氧基、硝基;
[0032]
r5选自氟、氢、c1~c20直链烷基、c1~c20支链烷基、c1~c20直链烷氧基、c1~c20支链烷氧基、硝基;
[0033]
r6选自氟、氢、c1~c20直链烷基、c1~c20支链烷基、c1~c20直链烷氧基、c1~c20支链烷氧基、硝基。
[0034]
较优选的,所述化合物iii中,r1选自-ch(ch3)2、苯基、环丙烷基、ch3ch
2-、环戊烷基。
[0035]
较优选的,所述化合物ii中,x选自溴;
[0036]
r2选自氟、氢、甲基、甲氧基、乙基、乙氧基;
[0037]
r3选自氟、氢、甲基、甲氧基、乙基、乙氧基、硝基;
[0038]
r4选自氟、氢、甲基、甲氧基、乙基、乙氧基、硝基;
[0039]
r5选自氟、氢、甲基、甲氧基、乙基、乙氧基、硝基;
[0040]
r6选自氟、氢、甲基、甲氧基、乙基、乙氧基、硝基。
[0041]
最优选的,所述化合物iii选自以下化合物的一种:
[0042][0043]
最优选的,所述化合物ii选自以下化合物的一种:
[0044][0045]
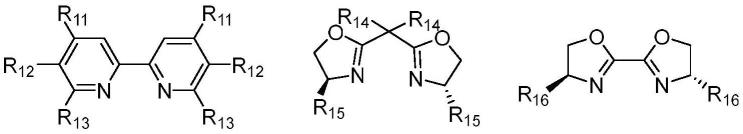
所述配体选自(1r,2r)-n,n'-二甲基-1,2-二苯基-1,2-二乙胺、2,2'-联喹啉、双((3as,8ar)-8,8a-二氢-3ah-茚并[1,2-d]恶唑-2-基)甲烷、(s)-4-(叔丁基)-2-(异喹啉-1-基)-4,5-二氢恶唑、
[0046][0047]r11
、r
12
、r
13
、r
14
、r
15
和r
16
各自独立的选自叔丁基、三氟甲基、甲氧基、甲基、羧基(cooh)、酯基(cooch3)、氰基、苄基、苯基、异丙基、cl、h;优选为4,4'-二叔丁基-2,2'-二吡啶。
[0048]
所述过渡金属催化剂选自溴化镍、溴化镍六水合物、溴化镍乙烯二醇二甲基醚络合物、溴化镍二乙二醇二甲醚复合物、氯化镍、氯化镍乙二醇二甲基醚络合物、二乙酰丙酮镍、碘化镍;优选为溴化镍乙烯二醇二甲基醚络合物。
[0049]
所述溶剂选自丙酮、乙腈、二氯甲烷、水、二氯乙烷、硝基甲烷、二甲基亚砜,优选为丙酮。
[0050]
所述碱选自碳酸钠、碳酸氢钠、碳酸钾、碳酸氢钾;优选为碳酸钠。
[0051]
所述光催化剂选自四丁基铵十聚钨酸盐、十聚钨酸钠;优选为四丁基铵十聚钨酸盐。
[0052]
所述光照反应的波长范围为365~415纳米,优选为390纳米。
[0053]
所述光照反应的时间为1~24小时,优选为3小时;温度小于40℃,优选为35℃。
[0054]
由于采用上述技术方案,本发明具有以下优点和有益效果:
[0055]
本发明的普拉格雷中间体及其类似物的制备方法,同对比例1相比,收率从51%大幅提高为96%;从需要预制备weinreb酰胺、新戊基硼酸酯底物和格氏试剂的2步反应,缩短为可以使用现成的醛与卤化物通过1步反应直接得到产物;反应时间从几十个小时减少为3
小时,大幅提高了生产效率;使用的底物廉价易得(环丙基甲醛:¥5.60/g;1-溴甲基-2-氟苯:¥1.20/g;3-(甲氧基(甲基)氨基)-3-氧代丙酸:¥3635/g),减少了生产成本;合成原料主要为苄基卤化物、醛,刺激性小。本发明在常温常压下,利用光催化偶联反应合成普拉格雷中间体及其类似物,无需加热与使用格氏试剂,合成工艺简便,易于操作,显著提高合成产率,且反应后除产物外生成无机盐,对环境污染小,绿色环保。本发明能够保证整个产品的质量得到进一步的提升,保证人们的生活得到进一步的改善,具有广阔的应用前景和市场需求。
具体实施方式
[0056]
为了更清楚地说明本发明,下面结合优选实施例对本发明做进一步的说明。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
[0057]
实施例1
[0058][0059]
在干燥反应瓶中加入磁力搅拌子,充入氩气保护,依次加入4,4'-二叔丁基-2,2'-二吡啶(0.002mol,0.54g)、溴化镍乙烯二醇二甲基醚络合物(0.002mol,0.62g),加入100ml丙酮溶解,超声至溶液呈均一后,依次向反应瓶中加入碳酸钠(0.06mol,6.36g)、四丁基铵十聚钨酸盐(0.0004mol,1.328g)、1-溴甲基-2-氟苯(0.02mol,3.76g)、环丙基甲醛(0.03mol,2.10g),充入氩气保护。密封后在390纳米光源下光照,光源距离反应瓶约6厘米,风扇降温,控制温度为35℃,3小时后加入100ml丙酮稀释,减压浓缩,直接通过柱层析分离纯化(正己烷:乙酸乙酯=19:1),得到3.42g式i所述普拉格雷中间体。
[0060]
核磁图谱数据为:1h nmr(500mhz,cdcl3):δ7.11-7.32(m,4h),3.87(s,2h),1.92-2.06(m,1h),1.03-1.17(m,2h),0.82-0.98(m,2h);
13
c nmr(126mhz,cdcl3):δ206.9,161.0,131.6,128.8,124.1,121.8,115.3,43.5,20.0,11.2.hrms(m/z):[m h]
calcd for c
11h12
fo
179.0794,found 179.0796.
[0061]
实施例2
[0062][0063]
在干燥反应瓶中加入磁力搅拌子,充入氩气保护,依次加入4,4'-二叔丁基-2,2'-二吡啶(0.01mmol,2.7毫克)、溴化镍乙烯二醇二甲基醚络合物(0.01mmol,3.1毫克),加入2.0ml丙酮溶解,超声至溶液呈均一后,依次向反应瓶中加入碳酸钠(0.30mmol,31.8毫克)、四丁基铵十聚钨酸盐(0.002mmol,6.6毫克)、1-溴甲基-2-氟苯(0.10mmol,18.8毫克)、苯甲醛(0.15mmol,15.9毫克),充入氩气保护。密封后在390纳米光源下光照,光源距离反应瓶约6厘米,风扇降温,控制温度为35℃,3小时后加入2.0ml丙酮稀释,减压浓缩,直接通过柱层析分离纯化(正己烷:乙酸乙酯=19:1),得到19.9毫克产物。
[0064]
核磁图谱数据为:1h nmr(500mhz,cdcl3):δ8.06
–
8.03(m,2h),7.61
–
7.56(m,1h),7.50
–
7.45(m,2h),7.31
–
7.22(m,2h),7.13
–
7.07(m,2h),4.32(s,2h);
13
c nmr(126mhz,cdcl3):δ196.4,161.0,136.6,133.5,131.9,129.0,128.7,124.2,122.2,121.8,115.6,38.9.hrms(m/z):[m na]
calcd for c
14h11
fona
237.0686,found 237.0682.
[0065]
实施例3
[0066][0067]
在干燥反应瓶中加入磁力搅拌子,充入氩气保护,依次加入4,4'-二叔丁基-2,2'-二吡啶(0.01mmol,2.7毫克)、溴化镍乙烯二醇二甲基醚络合物(0.01mmol,3.1毫克),加入2.0ml丙酮溶解,超声至溶液呈均一后,依次向反应瓶中加入碳酸钠(0.30mmol,31.8毫克)、四丁基铵十聚钨酸盐(0.002mmol,6.6毫克)、1-溴甲基-2-甲基苯(0.10mmol,18.4毫克)、苯甲醛(0.15mmol,15.9毫克),充入氩气保护。密封后在390纳米光源下光照,光源距离反应瓶约6厘米,风扇降温,控制温度为35℃,3小时后加入2.0ml丙酮稀释,减压浓缩,直接通过柱层析分离纯化(正己烷:乙酸乙酯=19:1),得到20.0毫克产物。
[0068]
核磁图谱数据为:1h nmr(500mhz,cdcl3):δ8.01(d,j=8.0hz,2h),7.54(t,j=8.0hz,1h),7.45((t,j=8.0hz,2h),7.18
–
7.09(m,4h),4.26(s,2h),2.23(s,3h);
13
c nmr(126mhz,cdcl3):δ197.4,136.8,133.5,133.2,130.3,130.4,128.6,128.4,127.3,126.2,77.6,77.2,76.8,43.5,19.9.hrms(m/z):[m na]
calcd for c
15h14
ona
233.0937,found 233.0935.
[0069]
实施例4
[0070][0071]
在干燥反应瓶中加入磁力搅拌子,充入氩气保护,依次加入4,4'-二叔丁基-2,2'-二吡啶(0.01mmol,2.7毫克)、溴化镍乙烯二醇二甲基醚络合物(0.01mmol,3.1毫克),加入2.0ml丙酮溶解,超声至溶液呈均一后,依次向反应瓶中加入碳酸钠(0.30mmol,31.8毫克)、四丁基铵十聚钨酸盐(0.002mmol,6.6毫克)、2-溴甲基-1-氟-4-硝基苯(0.10mmol,23.3毫克)、异丁醛(0.15mmol,10.8毫克),充入氩气保护。密封后在390纳米光源下光照,光源距离反应瓶约6厘米,风扇降温,控制温度为35℃,3小时后加入2.0ml丙酮稀释,减压浓缩,直接通过柱层析分离纯化(正己烷:乙酸乙酯=19:1),得到19.4毫克产物。
[0072]
核磁图谱数据为:1h nmr(500mhz,cdcl3):δ8.18(ddd,j=8.8,4.5,2.9,1h),8.12(dd,j=6.2,2.9,1h),7.20(t,j=8.8,1h),3.90(s,2h),2.79(m,1h),1.20(d,j=6.8,6h);
13
c nmr(126mhz,cdcl3):δ208.8,164.5,144.3,127.8,124.8,123.8,116.1,41.0,40.1,18.2.hrms(m/z):[m h]
calcd for c
11h13
fno
3
226.0801,found 226.0804.
[0073]
实施例5
[0074][0075]
在干燥反应瓶中加入磁力搅拌子,充入氩气保护,依次加入4,4'-二叔丁基-2,2'-二吡啶(0.01mmol,2.7毫克)、溴化镍乙烯二醇二甲基醚络合物(0.01mmol,3.1毫克),加入2.0ml丙酮溶解,超声至溶液呈均一后,依次向反应瓶中加入碳酸钠(0.30mmol,31.8毫克)、四丁基铵十聚钨酸盐(0.002mmol,6.6毫克)、1-溴甲基-2-甲氧基苯(0.10mmol,20.0毫克)、环丙基甲醛(0.15mmol,10.5毫克),充入氩气保护。密封后在390纳米光源下光照,光源距离反应瓶约6厘米,风扇降温,控制温度为35℃,3小时后加入2.0ml丙酮稀释,减压浓缩,直接通过柱层析分离纯化(正己烷:乙酸乙酯=9:1),得到15.6毫克产物。
[0076]
核磁图谱数据为:1h nmr(500mhz,cdcl3):δ7.29-7.21(m,1h),7.18-7.13(m,1h),6.96-6.85(m,2h),3.80(s,3h),3.79(s,2h),2.00-1.90(m,1h),1.06-0.99(m,2h),0.84-0.76(m,2h);
13
c nmr(126mhz,cdcl3):δ208.7,157.5,131.1,128.3,123.7,120.6,110.5,55.3,45.0,19.6,10.8.hrms(m/z):[m na]
calcd for c
12h15o2
191.0994,found 191.0991.
[0077]
对比例1
[0078][0079]
在反应瓶中加入三氟甲烷磺酸铜(54.3毫克,0.150mmol,0.300当量),2-氟苯基新戊基硼酸酯(312毫克,1.50mmol,3.00当量),1.25ml n,n-二甲基苯胺,3-(甲氧基(甲基)氨基)-3-氧代丙酸(73.6毫克,0.500mmol,1.00当量),混合搅拌10分钟后加入0.42ml三乙胺,80小时后加入氯化铵水溶液和乙酸乙酯稀释,合并有机相,用氢氧化钾水溶液和饱和食盐水洗涤,无水硫酸钠干燥,真空浓缩后,柱层析纯化(正己烷/乙酸乙酯=3:2),得到weinreb酰胺(64.1毫克,65%)。
[0080]
在氮气保护下,向密封反应瓶中加入环丙基溴化镁的四氢呋喃溶液(1.37ml,0.365m,2.2当量),将weinreb酰胺(46.0毫克,0.23mmol,1.0当量)溶解于四氢呋喃(0.17ml),加入反应瓶,45℃加热1小时。冷却至0℃后使用1m hcl溶液(1.0ml)淬灭,加水(5ml),然后用乙酸乙酯(3
×
10ml)萃取。有机层用盐水(5ml)和水(5ml)洗涤,然后用无水硫酸钠干燥,真空浓缩。混合物通过柱色谱纯化(己烷/乙酸乙酯=10:1),得到式ii所示化合物(31.9毫克,78%),为淡黄色油状物。
[0081]
表1
[0082] 步骤收率%实施例1196实施例2193实施例3195实施例4186
实施例5182对比例1251
[0083]
本发明同对比例1相比,收率从51%大幅提高为96%;从需要预制备weinreb酰胺、新戊基硼酸酯底物和格氏试剂的2步反应,缩短为可以使用现成的醛与卤化物通过1步反应直接得到产物;反应时间从几十个小时减少为3小时,大幅提高了生产效率;使用的底物廉价易得(环丙基甲醛:¥5.60/g;1-溴甲基-2-氟苯:¥1.20/g;3-(甲氧基(甲基)氨基)-3-氧代丙酸:¥3635/g),减少了生产成本。
[0084]
实施例6
[0085]
光催化剂的筛选:
[0086]
在4毫升干燥反应瓶中,依次加入4,4'-二叔丁基-2,2'-二吡啶(10μmol,2.7毫克,0.1当量)、溴化镍乙烯二醇二甲基醚络合物(10μmol,3.1毫克,0.1当量),加入2.0毫升丙酮(0.05m),超声溶解至溶液为均一,随后依次加入碳酸钠(0.30mmol,31.8毫克,3.0当量)、光催化剂(1.0~5.0μmol,0.01~0.05当量)、1-溴甲基-2-氟苯(0.10mmol,18.8毫克,1.0当量)、环丙基甲醛(0.15mmol,10.5毫克,1.5当量)。在室温下,氩气保护,390纳米光源光照,风扇降温(控制温度低于40℃,约为35℃),反应3小时。
[0087]
光催化剂的筛选如表2所示:
[0088]
表2
[0089][0090][0091]
使用四丁基铵十聚钨酸盐和十聚钨酸钠分别作为光催化剂,在相同用量(2.0μmol,0.02当量)下催化同一反应,使用四丁基铵十聚钨酸盐的产率最佳(96%)。降低和增加四丁基铵十聚钨酸盐的用量,都会导致产率降低(1.0μmol,0.01当量,73%;5.0μmol,0.05当量,83%)。最佳条件为四丁基铵十聚钨酸盐(2.0μmol,0.02当量)。
[0092]
实施例7
[0093]
过渡金属镍催化剂的筛选:
[0094]
在4毫升干燥反应瓶中,依次加入4,4'-二叔丁基-2,2'-二吡啶(10μmol,2.7毫克)、过渡金属镍催化剂(10μmol),加入2.0毫升丙酮(0.05m),超声溶解至溶液为均一,随后依次加入碳酸钠(0.30mmol,31.8毫克)、四丁基铵十聚钨酸盐(2.0μmol,6.6毫克)、1-溴甲基-2-氟苯(0.10mmol,18.8毫克,1.0当量)、环丙基甲醛(0.15mmol,10.5毫克,1.5当量)。在室温下,氩气保护,390纳米光源光照,风扇降温(控制温度低于40℃,约为35℃),反应3小时。
[0095]
过渡金属镍催化剂的筛选如表3所示:
[0096]
表3
[0097]
过渡金属镍催化剂用量收率%溴化镍10μmol84溴化镍六水合物10μmol91溴化镍乙烯二醇二甲基醚络合物10μmol96溴化镍二乙二醇二甲醚复合物10μmol92氯化镍10μmol84氯化镍乙二醇二甲基醚络合物10μmol91二乙酰丙酮镍10μmol32碘化镍10μmol27
[0098]
分别使用各种过渡金属镍催化剂,在相同用量(10μmol)下催化同一反应,使用溴化镍乙烯二醇二甲基醚络合物的产率最佳(96%),其他溴化、氯化镍类催化剂也可以使用,碘化镍的产率不佳。最佳条件为溴化镍乙烯二醇二甲基醚络合物(10μmol)。
[0099]
实施例8
[0100]
配体的筛选:
[0101]
在4毫升干燥反应瓶中,依次加入配体(10μmol)、溴化镍乙烯二醇二甲基醚络合物(10μmol,3.1毫克),加入2.0毫升丙酮(0.05m),超声溶解至溶液为均一,随后依次加入碳酸钠(0.30mmol,31.8毫克)、四丁基铵十聚钨酸盐(2.0μmol,6.6毫克)、1-溴甲基-2-氟苯(0.10mmol,18.8毫克,1.0当量)、环丙基甲醛(0.15mmol,10.5毫克,1.5当量)。在室温下,氩气保护,390纳米光源光照,风扇降温(控制温度低于40℃,约为35℃),反应3小时。
[0102]
配体的筛选如表4所示:
[0103]
表4
[0104]
配体用量收率%2,2'-联喹啉10μmol714,4'-二叔丁基-2,2'-二吡啶10μmol964,4'-双(三氟甲基)-2,2'-联吡啶10μmol724,4'-二甲氧基-2,2'-联吡啶10μmol532,2'-联吡啶10μmol735,5'-二甲基-2,2'-联吡啶10μmol696,6'-二氰基-2,2'-联吡啶10μmol582,2-双(2-恶唑啉)10μmol62
[0105]
分别使用各种配体,在相同用量(10μmol)下催化同一反应,使用4,4'-二叔丁基-2,2'-二吡啶的产率最佳(96%),其他联喹啉、联吡啶、双恶唑类等催化剂也可以使用,产率相对较低。最佳条件为4,4'-二叔丁基-2,2'-二吡啶(10μmol)。
[0106]
实施例9
[0107]
碱的筛选:
[0108]
在4毫升干燥反应瓶中,依次加入4,4'-二叔丁基-2,2'-二吡啶(10μmol,2.7毫克)、溴化镍乙烯二醇二甲基醚络合物(10μmol,3.1毫克),加入2.0毫升丙酮(0.05m),超声溶解至溶液为均一,随后依次加入碱(0.30mmol)、四丁基铵十聚钨酸盐(2.0μmol,6.6毫克)、1-溴甲基-2-氟苯(0.10mmol,18.8毫克,1.0当量)、环丙基甲醛(0.15mmol,10.5毫克,
1.5当量)。在室温下,氩气保护,390纳米光源光照,风扇降温(控制温度低于40℃,约为35℃),反应3小时。
[0109]
碱的筛选如表5所示:
[0110]
表5
[0111]
碱用量收率%碳酸钠0.30mmol96碳酸氢钠0.30mmol91碳酸钾0.30mmol88碳酸氢钾0.30mmol78碳酸钠0.11mmol41碳酸钠0.20mmol82
[0112]
分别使用各种碱,在相同用量(0.30mmol)下催化同一反应,使用碳酸钠的产率最佳(96%),碳酸氢钠、碳酸钾、碳酸氢钾也可以使用。降低碳酸钠的用量,会导致产率降低(0.11mmol,41%;0.20mmol,82%)。最佳条件为碳酸钠(0.30mmol)。
[0113]
实施例10
[0114]
溶剂的筛选:
[0115]
在4毫升干燥反应瓶中,依次加入4,4'-二叔丁基-2,2'-二吡啶(10μmol,2.7毫克)、溴化镍乙烯二醇二甲基醚络合物(10μmol,3.1毫克),加入溶剂,超声溶解至溶液为均一,随后依次加入碳酸钠(0.30mmol,31.8毫克)、四丁基铵十聚钨酸盐(2.0μmol,6.6毫克)、1-溴甲基-2-氟苯(0.10mmol,18.8毫克,1.0当量)、环丙基甲醛(0.15mmol,10.5毫克,1.5当量)。在室温下,氩气保护,390纳米光源光照,风扇降温(控制温度低于40℃,约为35℃),反应3小时。
[0116]
溶剂的筛选如表6所示:
[0117]
表6
[0118][0119][0120]
分别使用各种溶剂,在相同浓度下(0.05m)下催化同一反应,使用丙酮的产率最佳
(96%),二氯甲烷也可以使用,乙腈、水、二氯乙烷、硝基甲烷、二甲基亚砜的产率不佳,不适合使用。增加反应体系的浓度,会导致产率降低(0.09m,86%)。最佳条件为丙酮(0.05m)。
[0121]
以上所述仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,虽然本发明已以较佳实施例揭露如上,然而并非用以限定本发明,任何熟悉本专利的技术人员在不脱离本发明技术方案范围内,当可利用上述提示的技术内容作出些许更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明方案的范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
