1.本技术涉及医药中间体合成的领域,更具体地说,它涉及一种作为医药中间体制备1-萘胺的合成新方法。
背景技术:
2.1-萘胺是一种常用的化学合成中间体,广泛用于抗抑郁药等医药原料的研发,相关技术中,1-萘胺的合成主要是通过萘经硝化、还原反应得到,其化学反应如下:采用上述制备方法,制备过程中易产生大量的工业废酸以及废渣,并伴有刺激性气体产生,对环境污染较大,且制得的1-氨基萘分离颜色较黑,导致萃取分层难度较大,萃取过程需消耗大量有机溶剂,不适于大规模生产,有待改进。
技术实现要素:
3.为了改善制备1-萘胺对环境污染大的问题,本技术提供一种作为医药中间体制备1-萘胺的合成新方法。
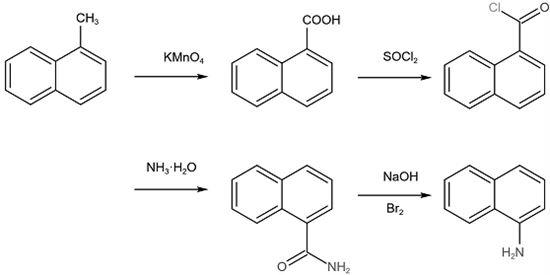
4.本技术提供的一种作为医药中间体制备1-萘胺的合成新方法采用如下的技术方案:一种作为医药中间体制备1-萘胺的合成新方法,包括以下步骤:1)1-萘甲酸的制备:将1-甲基萘投入氧化剂和乙醇的混合溶液,加热搅拌并回流,得1-萘甲酸;2)1-萘甲酰氯的制备:加入二氯亚砜和步骤1)中的1-萘甲酸加热回流至反应结束,得1-萘甲酰氯;3)萘-1-甲酰胺的制备:将步骤2)中的1-萘甲酰氯置于冰水中冷却,然后滴加冰冷的氨水并搅拌,得萘-1-甲酰胺;4) 1-萘胺的制备:将步骤3)中的萘-1-甲酰胺分多次加入次卤酸钠溶液中,保温并搅拌,随后升温回流至反应结束,得1-萘胺。
5.相关技术中,混酸硝化产生工业废酸,铁粉、硫代硫化钠还原硝基,会产生大量难以处理的铁泥废渣和硫酸盐。
6.通过采用上述技术方案,其反应方程式如下:
产品的产率相对较高,且反应产生的废渣极少,制备过程中消耗的有机溶剂和产生的废水也相对较少,减少了对环境的污染,降低了后续处理废渣和废水的难度和处理成本;另一方面,萘有毒,易升华,生产过程中,萘易升华至周遭环境中,对人体健康造成影响,本技术的制备工艺减少了萘的使用,安全性更高,适于大规模生产。
7.优选的,所述步骤1)中,所述氧化剂为硝酸或高锰酸钾,优选高锰酸钾。
8.通过采用上述技术方案,硝酸和高锰酸钾的氧化效率较高,易于氧化,有利于促进反应进行,提高产品的收率,且价格相较于其他氧化剂更为实惠,性价比更高,符合大规模生产的生产需要和经济性需求。
9.优选的,所述步骤1)中,1-甲基萘与氧化剂的投料摩尔比为1:1-2.5,优选1:1.5。
10.通过采用上述技术方案,当1-甲基萘与高锰酸钾的投料摩尔比低于1:1时,氧化效果不佳,物料不能完全氧化,当1-甲基萘与高锰酸钾的投料摩尔比高于1:2.5时,反应速率和产品的产率下降,且副产物显著增加,增大了反应后续处理难度,导致处理成本上升,采用投料摩尔比为1:1-2.5的1-甲基萘与高锰酸钾,物料能充分氧化,且产品收率较高,符合实际生产的需要。
11.优选的,所述步骤1)中,反应温度为70-85℃,优选78℃。
12.通过采用上述技术方案,在上述反应温度下,1-萘甲酸的产品收率较高。
13.优选的,所述步骤2)中,反应温度为75-90℃,优选82℃。
14.通过采用上述技术方案,在上述反应温度下,1-萘甲酰氯的产品收率较高。
15.优选的,所述步骤2)中,1-萘甲酸与二氯亚砜的投料摩尔比为1:8-14,优选1:10。
16.优选的,所述步骤3)中,反应温度为-5-5℃,优选0℃。
17.通过采用上述技术方案,抑制氨水挥发,且在上述反应温度下,萘-1-甲酰胺的产品收率较高。
18.优选的,所述步骤3)中,1-萘甲酰氯与氨水的投料摩尔比为1:5-10,优选1:7。
19.通过采用上述技术方案,当1-萘甲酰氯与氨水的投料摩尔比为1:5-10时,产品的收率较高,当1-萘甲酰氯与氨水的投料摩尔比低于1:5,物料的反应不彻底,物料利用率较低,当1-萘甲酰氯与氨水的投料摩尔比高于1:10,收率降低,物料利用率较低。
20.优选的,所述步骤4)中,反应温度为90-105℃,优选100℃,萘-1-甲酰胺与次卤酸
钠的投料摩尔比为1:1.5-3,优选1:2。
21.通过采用上述技术方案,当萘-1-甲酰胺与次卤酸钠的投料摩尔比为1:1.5-3,产品的收率较高,当萘-1-甲酰胺与次卤酸钠的投料摩尔比低于1:1.5,物料的反应不彻底,物料利用率较低,当萘-1-甲酰胺与次卤酸钠的投料摩尔比高于1:3,收率降低,物料利用率较低。
22.优选的,所述步骤4)中,所述次卤酸钠为次氯酸钠或次溴酸钠,优选次溴酸钠。
23.通过采用上述技术方案,采用次溴酸钠,反应速率更快,产率更高。
24.综上所述,本技术具有以下有益效果:本技术的制备方法产品收率高,且反应产生的废渣极少,制备过程中消耗的有机溶剂和产生的废水也相对较少,减少了对环境的污染,降低了后续处理废渣和废水的难度和处理成本,另一方面,减少了萘的使用,安全性更高,适于大规模生产。
附图说明
25.图1为本技术实施例2制得的1-氨基萘的液相色谱谱图;图2为本技术实施例2制得的1-氨基萘的核磁谱图。
具体实施方式
26.以下结合附图1-2和实施例对本技术作进一步详细说明。
27.实施例中所用硝酸的浓度为68%(v/v)。
28.文中所指%(v/v)为体积百分比。
29.以下实施方式中所用原料除特殊说明外均可来源于普通市售。
实施例
30.实施例1本技术公开了一种作为医药中间体制备1-萘胺的合成新方法,包括以下步骤:1)1-萘甲酸的制备:在250ml的三口瓶中加入28.4g1-甲基萘,然后加入由27.8g氧化剂和70g乙醇混合配成的溶液,氧化剂采用硝酸,加热至78℃回流2h,回流完毕后蒸干乙醇,冷却反应液至0℃,置于0℃冰水浴中放置0.5h,抽滤,将固体置于100ml水中,加热至78℃并搅拌,搅拌同时滴加1mol/l的氢氧化钠溶液,调节ph=8,使固体基本溶解,此时混合物呈红色,静置分层,分出未溶解的油状化合物,用正丁醚萃取水相两次,水相用活性炭脱色,过滤得到透明的浅黄色液体,在液体中滴加20%(v/v)的盐酸,调节ph为4.5-5.0,析出黄棕色固体,过滤水洗,干燥得粗品,制得1-萘甲酸,产品收率80%;2)1-萘甲酰氯的制备:在安装回流冷凝装置(接干燥管)的100ml圆底烧瓶中,加入10g步骤1)制得的1-萘甲酸和69.1g二氯亚砜,在82℃下回流2h,回流完毕后,分出前馏分二氯亚砜,再继续减压蒸馏,收集馏分,得无色透明液体1-萘甲酰氯,产品收率96%;3)萘-1-甲酰胺的制备:在装有机械搅拌器、滴液漏斗的100ml三口烧瓶中加入10g步骤2)制得的1-萘甲酰氯,在0℃冷却下,逐滴加入12.85g0℃氨水并搅拌3h。反应完毕后抽滤得到固体,用氨水洗涤固体,干燥得产品萘-1-甲酰胺,产品收率95%;4) 1-萘胺的制备:在装有搅拌器的100ml三口瓶中,加入5.0g氢氧化钠和40g水,
搅拌溶解,并用冰水冷却到0℃。在不断搅拌下,加入2.81g溴素,加完后继续搅拌15分钟,使溴素完全反应,制得次溴酸钠,保持低温(0℃),加入3g步骤3)制得的萘-1-甲酰胺,保温反应30分钟,然后开始升温至100℃回流2h,改为减压蒸馏,收集1-氨基萘,产品收率91%。
31.实施例21)1-萘甲酸的制备:在250ml的三口瓶中加入28.4g1-甲基萘和80g乙醇,然后加入47.3g氧化剂,氧化剂采用高锰酸钾,将反应混合物加热至78℃回流2h,回流完毕后冷却反应液至0℃并过滤,随后蒸干溶剂,可以观察到析出黄棕色固体,干燥得粗品,制得1-萘甲酸,产品收率96%;2)1-萘甲酰氯的制备:在安装回流冷凝装置(接干燥管)的100ml圆底烧瓶中,加入10g步骤1)制得的1-萘甲酸和69.1g二氯亚砜,在82℃下回流2h,回流完毕后,分出前馏分二氯亚砜,再继续减压蒸馏,收集馏分,得无色透明液体1-萘甲酰氯,产品收率96%;3)萘-1-甲酰胺的制备:在装有机械搅拌器、滴液漏斗的100ml三口烧瓶中加入10g步骤2)制得的1-萘甲酰氯,在0℃冷却下,逐滴加入12.85g0℃氨水并搅拌3h,反应完毕后抽滤得到固体,用氨水洗涤固体,干燥得产品萘-1-甲酰胺,产品收率95%;4) 1-萘胺的制备:在装有搅拌器的100ml三口瓶中,加入5.0g氢氧化钠和40g水,搅拌溶解,并用冰水冷却到0℃。在不断搅拌下,加入2.81g溴素,加完后继续搅拌15分钟,使溴素完全反应,制得次溴酸钠,保持低温(0℃),加入3g步骤3)制得的萘-1-甲酰胺,保温反应30分钟,然后开始升温至100℃回流2h,改为减压蒸馏,收集1-氨基萘,产品收率91%。
32.最终产品经液相检测和核磁分析,液相色谱谱图见图1,液相结果见下表1,核磁谱图见图2,确认了产物结构式的正确性。
33.表1 液相结果(氨基萘氢化标 2021_10_20 14_00_23)
序号保留时间[min]峰高[mau]峰高[%]峰面积[mau.s]峰面积[%]0.05峰高处峰宽[min]13.4232439.277100.025453.843100.00.275合计 2439.277100.025453.843100.0 实施例3与实施例2的区别在于:步骤1)中,1-甲基萘与氧化剂的投料摩尔比为1:1。
[0034]
1)1-萘甲酸的制备:在250ml的三口瓶中加入28.4g1-甲基萘和80g乙醇,然后加入31.6g氧化剂,氧化剂采用高锰酸钾,将反应混合物加热至78℃回流2h,回流完毕后冷却反应液至0℃并过滤,随后蒸干溶剂,可以观察到析出黄棕色固体,干燥得粗品,制得1-萘甲酸,产品收率64%。
[0035]
实施例4与实施例2的区别在于:步骤1)中,1-甲基萘与氧化剂的投料摩尔比为1:2.5。
[0036]
1)1-萘甲酸的制备:在250ml的三口瓶中加入28.4g1-甲基萘和80g乙醇,然后加入78.9g氧化剂,氧化剂采用高锰酸钾,将反应混合物加热至78℃回流2h,回流完毕后冷却反应液至0℃并过滤,随后蒸干溶剂,可以观察到析出黄棕色固体,干燥得粗品,制得1-萘甲酸,产品收率88%,副产物增多。
[0037]
实施例5与实施例2的区别在于:步骤1)的反应温度为70℃。
[0038]
1)1-萘甲酸的制备:在250ml的三口瓶中加入28.4g1-甲基萘和80g乙醇,然后加入
47.3g氧化剂,氧化剂采用高锰酸钾,将反应混合物加热至70℃回流2h,回流完毕后冷却反应液至0℃并过滤,随后蒸干溶剂,可以观察到析出黄棕色固体,干燥得粗品,制得1-萘甲酸,产品收率90%。
[0039]
实施例6与实施例2的区别在于:步骤1)的反应温度为85℃。
[0040]
1)1-萘甲酸的制备:在250ml的三口瓶中加入28.4g1-甲基萘和80g乙醇,然后加入47.3g氧化剂,氧化剂采用高锰酸钾,将反应混合物加热至85℃回流2h,回流完毕后冷却反应液至0℃并过滤,随后蒸干溶剂,可以观察到析出黄棕色固体,干燥得粗品,制得1-萘甲酸,产品收率96%。
[0041]
实施例7与实施例2的区别在于:步骤2)的反应温度为75℃。
[0042]
2)1-萘甲酰氯的制备:在安装回流冷凝装置(接干燥管)的100ml圆底烧瓶中,加入10g步骤1)制得的1-萘甲酸和69.1g二氯亚砜,在75℃下回流2h,回流完毕后,分出前馏分二氯亚砜,再继续减压蒸馏,收集馏分,得无色透明液体1-萘甲酰氯,产品收率90%。
[0043]
实施例8与实施例2的区别在于:步骤2)的反应温度为90℃。
[0044]
2)1-萘甲酰氯的制备:在安装回流冷凝装置(接干燥管)的100ml圆底烧瓶中,加入10g步骤1)制得的1-萘甲酸和69.1g二氯亚砜,在90℃下回流2h,回流完毕后,分出前馏分二氯亚砜,再继续减压蒸馏,收集馏分,得无色透明液体1-萘甲酰氯,产品收率96%。
[0045]
实施例9与实施例2的区别在于:步骤2)中,1-萘甲酸与二氯亚砜的投料摩尔比为1:8。
[0046]
2)1-萘甲酰氯的制备:在安装回流冷凝装置(接干燥管)的100ml圆底烧瓶中,加入10g步骤1)制得的1-萘甲酸和55.3g二氯亚砜,在82℃下回流2h,回流完毕后,分出前馏分二氯亚砜,再继续减压蒸馏,收集馏分,得无色透明液体1-萘甲酰氯,产品收率80%。
[0047]
实施例10与实施例2的区别在于:步骤2)中,1-萘甲酸与二氯亚砜的投料摩尔比为1:14。
[0048]
2)1-萘甲酰氯的制备:在安装回流冷凝装置(接干燥管)的100ml圆底烧瓶中,加入10g步骤1)制得的1-萘甲酸和96.8g二氯亚砜,在82℃下回流2h,回流完毕后,分出前馏分二氯亚砜,再继续减压蒸馏,收集馏分,得无色透明液体1-萘甲酰氯,产品收率90%。
[0049]
实施例11与实施例2的区别在于:步骤3)的反应温度为-5℃。
[0050]
3)萘-1-甲酰胺的制备:在装有机械搅拌器、滴液漏斗的100ml三口烧瓶中加入10g步骤2)制得的1-萘甲酰氯,在-5℃冷却下,逐滴加入12.85g0℃氨水并搅拌3h,反应完毕后抽滤得到固体,用氨水洗涤固体,干燥得产品萘-1-甲酰胺,产品收率88%。
[0051]
实施例12与实施例2的区别在于:步骤3)的反应温度为5℃。
[0052]
3)萘-1-甲酰胺的制备:在装有机械搅拌器、滴液漏斗的100ml三口烧瓶中加入10g步骤2)制得的1-萘甲酰氯,在5℃冷却下,逐滴加入12.85g0℃氨水并搅拌3h,反应完毕后抽滤得到固体,用氨水洗涤固体,干燥得产品萘-1-甲酰胺,产品收率90%。
[0053]
实施例13与实施例2的区别在于:步骤3)中,1-萘甲酰氯与氨水的投料摩尔比为1:5。
[0054]
3)萘-1-甲酰胺的制备:在装有机械搅拌器、滴液漏斗的100ml三口烧瓶中加入10g步骤2)制得的1-萘甲酰氯,在0℃冷却下,逐滴加入9.18g0℃氨水并搅拌3h,反应完毕后抽滤得到固体,用氨水洗涤固体,干燥得产品萘-1-甲酰胺,产品收率82%。
[0055]
实施例14与实施例2的区别在于:步骤3)中,1-萘甲酰氯与氨水的投料摩尔比为1:10。
[0056]
3)萘-1-甲酰胺的制备:在装有机械搅拌器、滴液漏斗的100ml三口烧瓶中加入10g步骤2)制得的1-萘甲酰氯,在0℃冷却下,逐滴加入18.4g0℃氨水并搅拌3h,反应完毕后抽滤得到固体,用氨水洗涤固体,干燥得产品萘-1-甲酰胺,产品收率91%。
[0057]
实施例15与实施例2的区别在于:步骤4)的反应温度为90℃。
[0058]
4) 1-萘胺的制备:在装有搅拌器的100ml三口瓶中,加入5.0g氢氧化钠和40g水,搅拌溶解,并用冰水冷却到0℃。在不断搅拌下,加入2.81g溴素,加完后继续搅拌15分钟,使溴素完全反应,制得次溴酸钠,保持低温(0℃),加入3g步骤3)制得的萘-1-甲酰胺,保温反应30分钟,然后开始升温至90℃回流2h,改为减压蒸馏,收集1-氨基萘,产品收率80%。
[0059]
实施例16与实施例2的区别在于:步骤4)的反应温度为105℃。
[0060]
4) 1-萘胺的制备:在装有搅拌器的100ml三口瓶中,加入5.0g氢氧化钠和40g水,搅拌溶解,并用冰水冷却到0℃。在不断搅拌下,加入2.81g溴素,加完后继续搅拌15分钟,使溴素完全反应,制得次溴酸钠,保持低温(0℃),加入3g步骤3)制得的萘-1-甲酰胺,保温反应30分钟,然后开始升温至105℃回流2h,改为减压蒸馏,收集1-氨基萘,产品收率91%。
[0061]
实施例17与实施例2的区别在于:步骤4)中,萘-1-甲酰胺与次溴酸钠的投料摩尔比为1:1.5。
[0062]
4) 1-萘胺的制备:在装有搅拌器的100ml三口瓶中,加入5.0g氢氧化钠和40g水,搅拌溶解,并用冰水冷却到0℃。在不断搅拌下,加入2.1g溴素,加完后继续搅拌15分钟,使溴素完全反应,制得次溴酸钠,保持低温(0℃),加入3g步骤3)制得的萘-1-甲酰胺,保温反应30分钟,然后开始升温至100℃回流2h,改为减压蒸馏,收集1-氨基萘,产品收率78%。
[0063]
实施例18与实施例2的区别在于:步骤4)中,萘-1-甲酰胺与次溴酸钠的投料摩尔比为1:3。
[0064]
4) 1-萘胺的制备:在装有搅拌器的100ml三口瓶中,加入5.0g氢氧化钠和40g水,搅拌溶解,并用冰水冷却到0℃。在不断搅拌下,加入4.2g溴素,加完后继续搅拌15分钟,使溴素完全反应,制得次溴酸钠,保持低温(0℃),加入3g步骤3)制得的萘-1-甲酰胺,保温反应30分钟,然后开始升温至100℃回流2h,改为减压蒸馏,收集1-氨基萘,产品收率91%。
[0065]
实施例19与实施例2的区别在于:步骤4)中,将制得的次溴酸钠替换为次氯酸钠。
[0066]
4) 1-萘胺的制备:在装有搅拌器的100ml三口瓶中,加入26.1g 10%次氯酸钠溶液,并用冰水冷却到0℃,加入3g步骤3)制得的萘-1-甲酰胺,保温反应30分钟,然后开始升
温至100℃回流2h,改为减压蒸馏,收集1-氨基萘,产品收率83%。
[0067]
本具体实施例仅仅是对本技术的解释,其并不是对本技术的限制,本领域技术人员在阅读完本说明书后可以根据需要对本实施例做出没有创造性贡献的修改,但只要在本技术的权利要求范围内都受到专利法的保护。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。