1.本发明具体涉及一种活化、再生贵金属催化剂及其制备方法和应用。
背景技术:
2.随着工业化程度的不断加重,挥发性有机化合物(voc)过度排放所引起的环境污染问题引起人们的密切关注。贵金属催化剂由于催化活性极高,被市场广泛应用于环境中voc的催化氧化。但是,实际催化条件复杂,温度不完全可控,反应气体复杂,往往出现贵金属被烧结、碳沉积或贵金属中毒等现象,导致其表面活性位点减少,催化活性降低甚至失去活性。地球上贵金属含量稀缺且价格高昂,提高贵金属催化剂的利用率对于可持续发展具有重要的意义。
3.商用贵金属催化剂的使用寿命大约为1~4年,失活之后的处理手段通常有回收贵金属或者将其再生处理,常规回收贵金属的技术主要有湿法冶金和火法冶金回收,但回收效率低下,回收成本高昂;而对于贵金属催化剂中贵金属利用率的提升、失活贵金属催化剂的再生研究却存在很大的缺口。目前,工业应用中常用的再生处理手段主要有干空气气氛中高温煅烧、湿空气气氛中高温煅烧等,然而,这些再生手段存在较多弊端,上述方法主要适用于因碳沉积所引起的贵金属催化剂的失活,高温煅烧会加剧贵金属烧结,对载体的损伤也极其严重。况且,贵金属催化剂的失活不是仅由碳沉积导致的,往往是由多种因素共同作用的结果,例如,烧结失活或中毒失活等。对因烧结或中毒导致的失活贵金属催化剂的活化鲜有报道。
4.因此,本领域亟需研发一种易于操作,成本低,可有效活化因各种原因导致失活的贵金属催化剂的活化方法。
技术实现要素:
5.本发明所要解决的技术问题在于克服现有技术中失活贵金属催化剂的活化方法主要适用于因碳沉积导致失活的贵金属催化剂,对因烧结或中毒导致失活的贵金属催化剂的活化效果较差,且现有活化方法在活化过程中温度过高,还存在破坏贵金属催化剂和载体结构等缺陷,而提供一种活化、再生贵金属催化剂及其制备方法和应用。采用本发明的活化方法处理得到的活化贵金属催化剂和/或再生贵金属催化剂均具有较理想的催化性能,且催化性能可保持稳定;本发明的活化方法适用范围广,操作简单,成本低,易实现放大应用。
6.本发明通过以下技术方案解决上述技术问题。
7.本发明提供了一种贵金属催化剂的活化方法,其包括如下步骤:在溶剂中,将贵金属催化剂与活化剂反应,即可;其中,所述活化剂为硼氢化物、水合肼、甲醛、甲酸和乙二醇中的一种或多种;所述活化剂与所述溶剂的摩尔体积比为1mol/l以上。
8.本发明中,所述活化剂较佳地为硼氢化物,更佳地为硼氢化钠和/或硼氢化钾。
9.本发明中,所述溶剂可为本领域常规使用的可使所述活化剂完全溶解,且不与所
述催化剂和所述活化剂发生化学反应的溶剂,较佳地为水、甲醇、乙醇和四氢呋喃中一种或多种,更佳地为水。
10.本发明中,所述活化剂与所述溶剂的摩尔体积比较佳地为2mol/l以上,更佳地为2~5mol/l,例如3mol/l。当所述活化剂与所述溶剂的摩尔体积比低于0.01mol/l时,活化效果不理想。
11.本发明中,所述贵金属催化剂中贵金属与所述活化剂的质量比可为本领域常规,较佳地为1:(1000~1500),更佳地为1:(1200~1500)。
12.本发明中,所述贵金属催化剂可为新鲜贵金属催化剂和/或失活贵金属催化剂。其中,按照本领域常规,所述新鲜贵金属催化剂一般指未参与过催化反应,具有催化活性的催化剂;所述失活贵金属催化剂一般指参与过催化反应,已失活的催化剂。
13.其中,所述失活贵金属催化剂的失活原因可为催化剂领域在催化反应过程中导致贵金属催化剂活性降低的常规原因,一般为烧结失活、中毒失活和碳沉积失活中的一种或多种。
14.本发明中,所述贵金属催化剂中的贵金属可为催化剂领域常规使用的贵金属,较佳地为pt、pd、ru、rh、au、ag和ir中一种或多种,更佳地为pt、pd和ru中的一种或多种,例如,“pt和pd的混合物”或“pt和ru的混合物”。
15.其中,当所述贵金属催化剂中的贵金属为pt和ru的混合物时,pt和ru的质量比可为本领域常规,较佳地为1:(0.25~4),更佳地为1:2。
16.其中,当所述贵金属催化剂中的贵金属为pt和pd的混合物时,pt和pd的质量比可为本领域常规,较佳地为1:(0.25~4),更佳地为1:1。
17.本发明中,所述贵金属催化剂的活化方法中的“活化”可为本领域技术人员常规认为的增加失活贵金属催化剂和/或新鲜贵金属催化剂中催化活性位点的过程。
18.本发明中,所述贵金属催化剂中还可进一步包括催化剂载体和/或催化助剂。
19.其中,所述催化剂载体可为贵金属催化剂领域常规使用的载体,例如,mg2al4si5o
18
。
20.其中,所述催化剂载体的壁厚可为本领域常规,较佳地为0.1~0.2mm,更佳地为0.15~0.18mm。
21.其中,所述催化剂载体的形状可为本领域常规,较佳地为粉末、颗粒、棒状和蜂窝状中的一种或多种,更佳地为蜂窝状。
22.其中,所述催化助剂可为贵金属催化剂领域常规使用的用于提高所述贵金属催化剂的催化活性或选择性的助剂,例如,氧化铈和/氧化锆。
23.本发明中,当所述贵金属催化剂为新鲜贵金属催化剂时,所述反应后制得的物料为活化贵金属催化剂。
24.本发明中,当所述贵金属催化剂为失活贵金属催化剂时,所述反应后制得的物料为再生贵金属催化剂。
25.本发明中,所述反应的条件和方法可为本领域该类反应常规的条件和方法,一般在超声的条件下进行。当在超声条件下进行所述反应时,有利于使失活贵金属催化剂表面堵塞其孔道,覆盖活性位点的沉积碳脱落;超声也有助于所述活化剂与所述贵金属催化剂充分反应。
反应的转化率曲线;
43.图5为在不同温度下,实施例3制得的再生pt-pd贵金属催化剂的催化性能的稳定性曲线;
44.图6为在不同温度下,实施例4制得的活化pt-pd贵金属催化剂和对比例5中新鲜pt-pd贵金属催化剂分别催化甲苯氧化为co2反应的转化率曲线;
45.图7为在不同温度下,实施例3、5~6和对比例7~8制得的再生pt-pd贵金属催化剂分别催化甲苯氧化为co2反应的转化率曲线;
46.图8为实施例1中失活pt-ru贵金属催化剂和再生pt-ru贵金属催化剂的表面形貌扫描电镜图;
47.图9为实施例1制得的再生pt-ru贵金属催化剂、对比例1中失活pt-ru贵金属催化剂和对比例2中新鲜pt-ru贵金属催化剂中ru 3p的xps图。
具体实施方式
48.下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
49.下述实施例1使用的失活pt-ru贵金属催化剂为上海某环保科技有限公司在工业处理废气过程中失活的催化剂,催化过程中废气风量为1900nm3/h。由于实际处理废气过程中气体成分复杂(废气组要成分见表1),尤其三氯乙烯、四氯乙烯容易造成贵金属催化剂氯中毒,使得贵金属催化剂活性降低,甚至失去活性。为了研究催化剂的失活是否跟氯中毒有关,将上述失活pt-ru贵金属催化剂放入去离子水中超声,所得液体测试icp,发现cl元素含量达5.11mg/l,证明了失活跟氯中毒有关。同时,对比失活前后催化剂表面形貌(具体见附图8),扫描电镜图如图8所示,图8中a(30倍放大图)和c(2000倍放大图)对应新鲜pt-ru贵金属催化剂表面形貌,b(30倍放大图)和d(2000倍放大图)对应失活pt-ru贵金属催化剂表面形貌,相对于新鲜pt-ru贵金属催化剂,失活pt-ru贵金属催化剂在放大30倍时可清晰看到表面出现很多裂缝,放大2000倍时可清晰看到氧化铝颗粒变大,有明显烧结现象,氧化铝颗粒的生长是通过颗粒间相互接触部分羟基基团脱水实现的,随着小分子的不断脱除,al-o-al键生成,从而导致氧化铝的表面积衰减。该贵金属催化剂使用过程中不完全可控的高温是造成烧结的主要原因。同时,xrf测试结果显示,催化前后贵金属催化剂中c含量由3.10%增至4.73%,证明了失活与贵金属催化剂炭沉积有关。综上分析,实施例1使用的失活pt-ru贵金属催化剂失活是由烧结、贵金属氯中毒、炭沉积共同引起的。
50.采用上述方法对实施例3中失活pt-pd贵金属催化剂的失活原因进行分析,结果与实施例1相同,也是由烧结、贵金属氯中毒、炭沉积共同引起的。
51.表1
52.废气种类最高浓度(mg/m3)甲基异丁基酮1635.74二甲苯1813.68乙苯1507.78甲基丙烯酸甲酯367.36
甲醇230.03甲苯168.69四氯乙烯71.64三氯乙烯71.56异丙醇54.52苯酚40.89甲基丙烯酸38.17丁酮16.36丁腈12.95环己酮1.23合计6030.60
53.实施例1
54.采用上述购买的工业上用于处理vocs的失活pt-ru贵金属催化剂,催化剂载体为200目的堇青石(mg2al4si5o
18
),催化助剂包括氧化铈(ceo2)和氧化锆(zro2),贵金属包括100g/m3的pt和ru,其中,pt和ru的质量比为1:2,蜂窝载体壁厚0.15mm;利用吹风机吹去失活pt-ru贵金属催化剂孔隙中的灰尘,准备待用;
55.配制2.0mol/l的硼氢化钠水溶液,恒温至50℃,将上述失活pt-ru贵金属催化剂放入其中,pt和ru的总质量与硼氢化钠的质量比为1:1200;在超声的条件下,进行反应,反应的时间为1h;之后利用去离子水反复清洗3次以上,以保证制得物料表面不再残留硼氢化钠;置于60℃烘箱中干燥8h,烘箱的升温速度为2℃/min,制得再生pt-ru贵金属催化剂。
56.实施例2
57.采用新鲜pt-ru贵金属催化剂,其与实施例1中失活pt-ru贵金属催化剂失活前的新鲜pt-ru贵金属催化剂为同批生产得到;
58.配制2.0mol/l的硼氢化钠水溶液,恒温至50℃,将上述新鲜pt-ru贵金属催化剂放入其中,pt和ru的总质量与硼氢化钠的质量比为1:1200;在超声的条件下,进行反应,反应的时间为1h;之后利用去离子水反复清洗3次以上,以保证制得物料表面不再残留硼氢化钠;置于60℃烘箱中干燥8h,烘箱的升温速度为2℃/min,制得活化pt-ru贵金属催化剂。
59.实施例3
60.采用上述购买工业上用于处理vocs的失活pt-pd贵金属催化剂,催化剂载体为200目的堇青石(mg2al4si5o
18
),催化助剂包括氧化铈(ceo2)、氧化锆(zro2),贵金属包括120g/m3的pt和pd,其中,pt和pd的质量比为1:1,蜂窝载体壁厚0.18mm;利用吹风机吹去失活pt-pd贵金属催化剂孔隙中的灰尘,准备待用;
61.配制2.0mol/l的硼氢化钾水溶液,恒温至25℃,将上述失活pt-pd贵金属催化剂放入其中,pt和pd的总质量与硼氢化钾的质量比为1:1200;在超声的条件下,进行反应,反应的时间为4h;之后利用去离子水反复清洗3次以上,以保证制得物料表面不再残留硼氢化钾;置于烘箱中120℃干燥8h,烘箱的升温速度为2℃/min,制得再生pt-pd贵金属催化剂。
62.实施例4
63.采用新鲜pt-pd贵金属催化剂,其与实施例3中失活pt-pd贵金属催化剂失活前的新鲜pt-pd贵金属催化剂为同批生产得到;
64.配制2.0mol/l的硼氢化钾水溶液,恒温至25℃,将上述新鲜pt-pd贵金属催化剂放入其中,pt和pd的总质量与硼氢化钾的质量比为1:1200;在超声的条件下,进行反应,反应的时间为4h;之后利用去离子水反复清洗3次以上,以保证制得物料表面不再残留有硼氢化钾;置于120℃烘箱中干燥8h,烘箱的升温速度为2℃/min,制得活化pt-pd贵金属催化剂。
65.实施例5
66.与实施例3相比,区别仅在于硼氢化钾水溶液的浓度为1mol/l,其他参数条件均相同。
67.实施例6
68.与实施例3相比,区别仅在于硼氢化钾水溶液的浓度为3mol/l,其他参数条件均相同。
69.对比例1
70.同实施例1中失活pt-ru贵金属催化剂。
71.对比例2
72.新鲜pt-ru贵金属催化剂,其与实施例1中失活pt-ru贵金属催化剂失活前的新鲜pt-ru贵金属催化剂为同批生产得到。
73.对比例3
74.选取实施例1中失活pt-ru贵金属催化剂,将其放置于卧式管式炉中,升温至220℃,通入氢气,氢气气相空速保持在600小时-1
(体积),处理失活催化剂48h得到再生pt-ru贵金属催化剂。
75.对比例4
76.同实施例3中失活pt-pd贵金属催化剂。
77.对比例5
78.新鲜pt-pd贵金属催化剂,其与实施例3中失活pt-pd贵金属催化剂失活前的新鲜pt-pd贵金属催化剂为同批生产得到。
79.对比例6
80.选取实施例3中失活pt-pd贵金属催化剂,将其放置于卧式管式炉中,升温至220℃,通入氢气,氢气气相空速保持在600小时-1
(体积),处理失活催化剂48h得到再生pt-pd贵金属催化剂。
81.对比例7
82.与实施例3相比,区别仅在于不添加硼氢化钾水溶液,其他参数条件均相同。
83.对比例8
84.与实施例3相比,区别仅在于硼氢化钾水溶液的浓度为0.1mol/l,其他参数条件均相同。
85.效果实施例1
86.采用上述实施例1和对比例3制得的再生pt-ru贵金属催化剂、对比例1中失活pt-ru贵金属催化剂和对比例2中新鲜pt-ru贵金属催化剂,分别在固定床反应器中催化甲苯氧化反应,并评估各自的催化性能,图1是在不同温度下,催化床层中,催化甲苯氧化为co2反应的转化率曲线。图1可以发现,失活的pt-ru贵金属催化剂经过实施例1方法再生后,性能提升至新鲜pt-ru贵金属催化剂99%以上,与对比例3制得的再生pt-ru贵金属催化剂相比,
催化性能显著提高。采用上述测试方法重复测试实施例1制得的再生pt-ru贵金属催化剂的催化性能,重复次数为三次,结果见图2的稳定性曲线图谱;从图2可以发现,再生pt-ru贵金属催化剂在三次重复测试中均能保持稳定。
87.效果实施例2
88.采用上述实施例2制得的活化pt-ru贵金属催化剂和对比例2中新鲜pt-ru贵金属催化剂,分别在固定床反应器中催化甲苯氧化反应,并评估其催化性能,图3是在不同温度下,催化床层中,催化甲苯氧化为co2反应的转化率曲线。图3可以发现,新鲜pt-ru贵金属催化剂经过实施例2活化后制得的活化pt-ru贵金属催化剂在催化性能上有明显提升。
89.效果实施例3
90.采用上述实施例3和对比例6制得的再生pt-pd贵金属催化剂、对比例4中失活pt-pd贵金属催化剂、对比例5新鲜pt-pd贵金属催化剂,分别在固定床反应器中催化甲苯氧化反应,并评估各自的催化性能,图4是在不同温度下,催化床层中,催化甲苯氧化为co2反应的转化率曲线。图4可以发现,失活的pt-pd贵金属催化剂经过实施例3的方法再生后,性能提升至新鲜pt-pd贵金属催化剂99%以上,与对比例6制得的再生pt-pd贵金属催化剂相比,催化性能显著提高。采用上述测试方法重复测试实施例3制得的再生pt-pd贵金属催化剂的催化性能,重复次数为三次,结果见图5的稳定性曲线图谱;从图5可以发现,再生pt-pd贵金属催化剂在三次重复测试中均能保持稳定。
91.效果实施例4
92.采用上述实施例4制得的活化pt-pd贵金属催化剂和对比例5中新鲜pt-pd贵金属催化剂,分别在固定床反应器中催化甲苯氧化反应,并评估其催化性能,图6是在不同温度下,催化床层中,催化甲苯氧化成co2的转化率的曲线。图6可以发现,新鲜pt-pd贵金属催化剂经过实施例4活化后制得的活化pt-pd贵金属催化剂在催化性能上有明显提升。
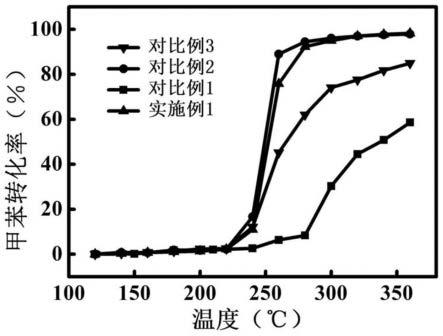
93.效果实施例5
94.采用上述实施例3、5~6和对比例7~8制得的再生pt-pd贵金属催化剂分别在固定床反应器中催化甲苯氧化反应,并评估其催化性能,图7是在不同温度下,催化床层中,催化甲苯氧化成co2的转化率的曲线。图7可以发现,当不采用硼氢化钾水溶液处理失活pt-pd贵金属催化剂,或是当硼氢化钾水溶液浓度较低时,对失活pt-pd贵金属催化剂的活化效果均较差。
95.效果实施例6
96.分别采用上述实施例或对比例处理后得到的催化剂,在固定床反应器中催化甲苯氧化反应,甲苯转化率达到10%、50%和90%时的温度分别见表2所示。其中,t
10
代表甲苯转化率达到10%时的温度,同理,t50和t90分别代表甲苯转化率达到50%和90%时的温度。
97.表2
[0098][0099][0100]
效果实施例7
[0101]
测试实施例1、对比例1和对比例2中样品ru 3p的xps图,见图9,由于pt元素出峰位置与al2o3有重合,不好判断,故主要研究贵金属ru元素在失活、再生前后化合态的变化。ru作为该催化剂的主要贵金属成分,研究失活前后表面价态对于研究其失活机理以及后续寻找再生策略具有重要的作用,对ru 3p轨道分峰拟合发现,对于对比例2中新鲜催化剂,位于462.1ev,484.3ev的子峰归属于ru0,位于465.1ev,487ev的子峰归属于ru
δ
,对于对比例1中失活催化剂,位于461.7ev,483.9ev的子峰归属于ru0,位于464.1ev,486.1ev的子峰归属于ru
δ
,与对比例2中新鲜催化剂相比,失活催化剂上ru的3p轨道结合能出现负移的现象,负移值大约为0.4~1ev,这表明了催化剂失活之后,ru与载体铈锆氧化物的相互作用减弱,这种变化表现为ru向铈锆氧化物转移电子的能力减弱,导致失活催化剂上ru原子轨道的电子云密度降低。从表3xps结果中也能看出催化剂失活之后,ru0被部分氧化,这跟失活前催化剂所处复杂的高温催化环境相关。对于再生催化剂,位于463.4ev,485.4ev的子峰归属于ru0,位于465.1ev,487ev的子峰归属于ru
δ
,对于失活催化剂,位于461.7ev,483.9ev的子峰归属于ru0,位于465.7ev,487.6ev的子峰归属于ru
δ
。与失活催化剂相比,其ru的3p轨道结合能偏移的减弱,这表明了催化剂再生之后,ru与载体铈锆氧化物的相互作用又有增强。同样从表3xps结果中也能看出催化剂再生之后,ru
δ
被部分还原。
[0102]
表3
[0103]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。