一种dpre1酶抑制剂及其用途
技术领域
1.本发明涉及一种酰肼取代的亚甲基嘧啶三酮化合物能够作为有效的dpre1酶抑制剂,具体涉及了一种dpre1酶抑制剂及其用途。本发明dpre1酶抑制剂可用于预防和/或治疗结核杆菌和/或麻风杆菌感染导致的疾病,属于药物化学和生物学技术领域,可用于制备预防和/或治疗结核杆菌和/或麻风杆菌感染的药物。
背景技术:
2.结核病作为全球排名第十三位的致死因素,最致命的单一传染病,2020年,全球新发结核病患者987万,发病率为127/10万,全球结核潜伏感染人群近20亿。结核病是由结核分枝杆菌引起的慢性感染性疾病,可侵犯全身多个脏器,其中肺结核最常见,其中多重耐药性结核分枝杆菌(mdr-tb)和广泛耐药结核分枝杆菌(xdr-tb)的出现,艾滋病(aids)的流行等因素是导致全球结核病死亡率持续上升的主要原因。麻风病是一种由麻风分枝杆菌感染引起的慢性传染病,主要侵犯皮肤和外周神经组织,本病在世界上流行甚广,主要分布在亚、非和拉丁美洲。
3.癸异戊二烯磷酰基-β-d-核糖-2
,
差向异构酶(decaprenyl-phosphoryl-β-d-ribose 2
,-epimerase 1,dpre1酶)是合成分枝杆菌细胞壁的关键酶,对分枝杆菌(包括耐药菌株)的存活至关重要。dpre1酶可以将dpr(十二烷基-磷酰基-β-d-核糖)氧化为dpx(十二烷基-磷酰基-β-d-呋喃糖),dpx再经dpre2还原生成dpa(十二烷基-磷酰基-β-d-阿拉伯呋喃糖),而dpa是细胞壁的重要组成成分,抑制dpre1酶,即可阻断dpa的合成,使分枝杆菌细胞壁无法合成,从而杀死分枝杆菌。
4.因此,目前寻求一种dpre1酶抑制剂,可以实现对分枝杆菌合成的抑制,从而避免分支杆菌引起的疾病。
技术实现要素:
5.本发明针对dpre1酶作为靶点进行研究,提供了一种dpre1酶抑制剂及其用途,该dpre1酶抑制剂是一类酰肼取代的亚甲基嘧啶三酮类化合物,为dpre1酶有效的抑制剂。为了得到抗菌活性更好,具有更好成药前景的酰肼取代的亚甲基嘧啶三酮类化合物,对现有物质进行改进,从而能够让这类分子具有更高的抗菌活性。
6.本发明提供一种dpre1酶抑制剂,其是结构式为(i)的酰肼取代的亚甲基嘧啶三酮化合物及其药学上可接受的盐、药学上可接受的酯、药学上可接受的酰胺或前体药物中的一种:
[0007][0008]
其中:
[0009]
r1、r2相同或不同,各自独立表示氢、烃基、环烷基、芳基、芳烷基中的一种或几种;
[0010]
所述的烃基,优选为c1~c8烷基、c2~c8烯烃基、c2~c8炔烃基中的一种。
[0011]
所述的环烷基为c3~c8环烷基。
[0012]
所述的芳基为单环或稠环芳烃基;
[0013]
所述的芳烷基优选为芳基c1~c8烷基;
[0014]
r1、r2还包括对烃基、环烷基、芳基、芳烷基进行取代,得到的取代基团,取代基为一个或多个,取代基选自c1~c8烷基、c3~c8环烷基、羟基、c1~c8烷氧基、c1~c8酰氧基、c1~c8酰基、氨基、c1~c8烷基胺基、c1~c8酰胺基、c2~c8酰亚胺基、硝基、巯基、c1~c8烷硫基、c1~c8酰硫基、磷酰氧基、磺酰氧基、亚磺酰氧基、脒基、胍基、羧基、膦酸基、磺酸基、亚磺酸基、氰基、卤素基团中的一种或几种。
[0015]
所述的卤素基团为氟、氯、溴和碘中的一种或几种。
[0016]
ar为取代或未取代的芳环、取代或未取代的芳杂环或取代或未取代的稠合芳环中的一种,取代基可以为一个或多个,取代基独立选择c1~c8烷基、c3~c8环烷基、单环芳烃基、稠环芳烃基、羟基、c1~c8烷氧基、c1~c8酰氧基、c1~c8酰基、氨基、c1~c8烷基胺基、c1~c8酰胺基、c2~c8烷基酰亚胺基、硝基、巯基、烃硫基、酰硫基、卤素、磷酰氧基、磺酰氧基、亚磺酰氧基、脒基、胍基、羧基、膦酸基、磺酸基、亚磺酸基、氰基的基团中的一种或几种;
[0017]
ar中,取代基中也可以进行再次取代,再次取代的取代基为一个或多个,选自羟基、c1~c8烷氧基、c1~c8酰氧基、c1~c8酰基、氨基、c1~c8烷基胺基、c1~c8酰胺基、c2~c8酰亚胺基、硝基、磷酰氧基、磺酰氧基、亚磺酰氧基、脒基、胍基、羧基、氰基、卤素的基团,以上卤素均指氟、氯、溴和碘。
[0018]
n为1或0,更优选为0。
[0019]
其中,酰肼取代的亚甲基嘧啶三酮化合物的药学上可接受的盐,包括无机盐、酸加成盐、碱加成盐或加成铵盐中的一种,其中,无机盐包括与盐酸、氢溴酸、氢碘酸、硫酸、硝酸、磷酸中的一种形成的无机盐;酸加成盐为与甲酸、乙酸、丙酸、草酸、丙二酸、琥珀酸、马来酸、酒石酸、柠檬酸、乙磺酸、对甲苯磺酸、天冬氨酸、谷氨酸中的一种形成的酸加成盐;碱加成盐为与钠、钾、钙、镁中的一种形成的碱加成盐,加成铵盐为与甲胺、乙胺、乙醇胺、赖氨酸、鸟氨酸中的一种形成的加成铵盐。
[0020]
其中,酰肼取代的亚甲基嘧啶三酮化合物的药学上可接受的酯,包括形成的相应的碳酸酯、磷酸酯、磺酸酯,硝酸酯的一种;
[0021]
其中,酰肼取代的亚甲基嘧啶三酮化合物的药学上可接受的酰胺,包括形成的相应的甲酰胺、乙酰胺、碳酰胺、苯甲酰胺、丁二酰亚胺、磺酰胺、磷酰胺的一种;
[0022]
其中,酰肼取代的亚甲基嘧啶三酮化合物的前体药物包括载体前体药物和生物前
体药物;其中,生物前体药物为采用酯、碳酸酯、氨基甲酸酯、磷酸盐/磷酸酯、羰基、硫醚、硫酯、亚胺、酰胺、脒肟、胍肟和n-曼尼希碱其中一个或多个基团修饰得到的化合物。
[0023]
本发明的dpre1酶抑制剂中,作为优选r1、r2为选自c1~c8烷基、c2~c8烯烃基、c2~c8炔烃基、c3~c8环烷基、芳基c1~c8烷基,其中,芳基c1~c8烷基中的芳基为单环芳烃基;还包括上述基团的取代物,上述基团的取代物中,取代基为一个或多个,优选为c1~c8烷基、c3~c8环烷基、羟基、c1~c8酰氧基、氨基、c1~c8烷基胺基、c1~c8酰胺基、c2~c8酰亚胺基、胍基、羧基、膦酸基、磺酸基中的一种或几种。
[0024]
本发明的dpre1酶抑制剂中,ar优选为取代或未取代的芳环、取代或未取代的芳杂环或者取代或未取代的稠合芳环,其中取代物中,取代基可以为一个或多个,独立地选自c1~c8烷基、c3~c8环烷基、单环芳烃基、c1~c8烷氧基、c1~c8酰氧基、c1~c8酰基、氨基、c1~c8烷基胺基、c1~c8酰胺基、c2~c8烷基酰亚胺基、卤素、磷酰氧基、脒基、胍基、羧基、膦酸基、磺酸基中的一种或几种。
[0025]
本发明的酰肼取代的亚甲基嘧啶三酮化合物中,更进一步优选r1、r2独立表示为c1~c8烷基、c2~c8烯烃基、c2~c8炔烃基、芳基c1~c8烷基或上述基团的取代物,其中,芳基c1~c8烷基中的芳基为单环芳烃基,取代物中,取代基为一个或多个,取代基选自c1~c8烷基的基团。
[0026]
本发明的dpre1酶抑制剂中,更进一步优选ar选自苯基、吡啶基或喹啉基。
[0027]
本发明的dpre1酶抑制剂中,优选n为0的化合物。
[0028]
一种结构式为(i)的酰肼取代的亚甲基嘧啶三酮化合物,用于作为dpre1酶抑制剂。
[0029]
一种药物组合物,包括上述dpre1酶抑制剂,还包括药学上可接受的载剂、稀释剂或赋形剂中的一种。
[0030]
所述的药学上可接受的载剂为适用于制成口服、注射、吸入、粘膜给药剂型的载体。
[0031]
一种dpre1酶抑制剂的用途,用于制备预防和/或治疗由分支杆菌引起的慢性感染性疾病或病症的药物中的应用。
[0032]
一种药物组合物的用途,用于制备预防和/或治疗由分支杆菌引起的慢性感染性疾病或病症的药物中的应用。
[0033]
所述的分支杆菌包括结核杆菌和/或麻风杆菌。
[0034]
所述的疾病或病症为肺结核、肠结核、淋巴结核、骨结核、肾结核、结核性腹膜炎、结核性脑膜炎、麻风病中的一种。
[0035]
本发明确证了合成的酰肼取代的亚甲基嘧啶三酮化合物为dpre1酶抑制剂,具有抗结核杆菌和/或麻风杆菌感染的实用性,可用于治疗结核杆菌和/或麻风杆菌感染的疾病,包括但不限于诸如肺结核、肠结核、淋巴结核、骨结核、肾结核、结核性腹膜炎、结核性脑膜炎、麻风病等。化合物通过易合成方法获得,能用多种方法对患者给药。
[0036]
结构式(i)的酰肼取代的亚甲基嘧啶三酮化合物含有烯键,也可能含有手性中心,因此可能存在不同的顺反异构体和对映体或非对映体。本发明涉及结构式(i)的酰肼取代的亚甲基嘧啶三酮化合物的所有顺反异构和所有光学异构体,包括这类化合物的外消旋物和单一的对映体及非对映体及其混合物,以及分别含有或者采用它们的如上定义的药物组
合物和治疗方法。
[0037]
由于本发明的结构式(i)的酰肼取代的亚甲基嘧啶三酮化合物具有烯键,同时可能具有手性中心,它们能够存在各种立体异构体或构型,化合物能够存在单独的( )和(-)光学活性形式及其混合物。本发明在其范围内包括所有这类形式。单一的异构体可以借助已知方法获得,例如在最终产物或者其中间体制备中进行分离、拆分、立体选择性反应或色谱离析。
[0038]
本发明酰肼取代的亚甲基嘧啶三酮化合物能够以非溶剂化形式及溶剂化形式存在,包括水合物。一般而言,溶剂化形式包括水合形式等价于非溶剂化形式,也涵盖在本发明范围内。
[0039]
本发明还包括同位素标记的酰肼取代的亚甲基嘧啶三酮化合物,它们等同于结构式(i)所述酰肼取代的亚甲基嘧啶三酮化合物,但结构中有一个或多个原子被相应的自然界中常见的同位素原子所替代。可以引入本发明中的同位素实例包括氢、碳、氮、氧、磷、硫、氟和氯,分别对应于例如:2h、3h、
11
c、
13
c、
14
c、
15
n、
17
o、
18
o、
31
p、
32
p、
35
s、
18
f和
36
cl。含有上述同位素和/或其他原子的其他同位素的本发明化合物、其前体药物和所述化合物或所述前体药物的药学上可接受的盐都属于本发明的范围。
[0040]
结构式(i)的酰肼取代的亚甲基嘧啶三酮化合物能够进一步生成药学上可接受的形式,包核、肾结核、结核性腹膜炎、结核性脑膜炎、麻风病等。
[0041]
结构式(i)的酰肼取代的亚甲基嘧啶三酮化合物的盐、溶剂化物和n-氧化物,盐包括但不限于酸加成盐和/或碱加成盐。
[0042]
本发明还提供药物制剂,包含治疗有效量的结构式(i)的酰肼取代的亚甲基嘧啶三酮化合物或其治疗学上可接受的盐和其药学上可接受的载体、稀释剂或赋型剂。所有这些形式都属于本发明。
[0043]
本发明提供治疗哺乳动物、包括人类由结核杆菌和/或麻风杆菌感染引发病症的方法,包括但不限于肺结核、肠结核、淋巴结核、骨结核、肾结核、结核性腹膜炎、结核性脑膜炎。该方法包含给予所述哺乳动物治疗此类疾病有效量的结构式(i)的酰肼取代的亚甲基嘧啶三酮化合物或其药学上可接受的盐。
[0044]
由于本发明化合物对dpre1酶的抑制活性,它们也是有用的研究工具,用于体外和体内研究dpre1酶的作用机理。
[0045]
优选地,上述治疗方法给予需要治疗的患者治疗有效量的结构式(i)的酰肼取代的亚甲基嘧啶三酮化合物。本发明化合物是酰肼取代的亚甲基嘧啶三酮,它们是有效的dpre1酶抑制剂。这些化合物容易合成,能够借助多种途径给药,包括口服和胃肠外。
[0046]
因此,本发明也提供了结构式(i)的酰肼取代的亚甲基嘧啶三酮化合物用于制备治疗哺乳动物、包括人类由结核杆菌和/或麻风杆菌感染引发病症的药物的用途。本发明提供了用作为dpre1酶抑制剂的结构式(i)的酰肼取代的亚甲基嘧啶三酮化合物或其药物组合物,以及用作治疗结核杆菌和/或麻风杆菌感染引发病症治疗药物的结构式(i)的酰肼取代的亚甲基嘧啶三酮化合物或其药物组合物。
[0047]
本发明结构式(i)的酰肼取代的亚甲基嘧啶三酮化合物的制药用途或者治疗用途所述的因结核杆菌和/或麻风杆菌感染引发的病症。
附图说明
[0048]
图1是实施例9中的dpre1基因比对结果;
[0049]
图2是实施例11中的dmso对结核杆菌生长的影响;
[0050]
图3是实施例11中的25号化合物浓度对结核杆菌生长的影响;
[0051]
图4是实施例11中的异烟肼(inh)浓度对结核杆菌生长的影响;
[0052]
图5是实施例11中的利福平(rfp)浓度对结核杆菌生长的影响;
[0053]
图6是实施例12中的小鼠肺脏菌落计数结果;
[0054]
图7是实施例12中的小鼠脾脏菌落计数结果;
[0055]
图8是实施例12中的小鼠肺部切片抗酸染色结果对比图,从下到上依次为(a)pbs组、(b)低剂量组、(c)高剂量组、(d)inh组,图中,黑色部分表示抗酸染色后显蓝色,灰色部分表示抗酸染色后显红色。
具体实施方式
[0056]
下面结合实施例对本发明作进一步的详细说明。
[0057]
本发明酰肼取代的亚甲基嘧啶三酮化合物可通过不同的制备方法获得。
[0058]
方法a:
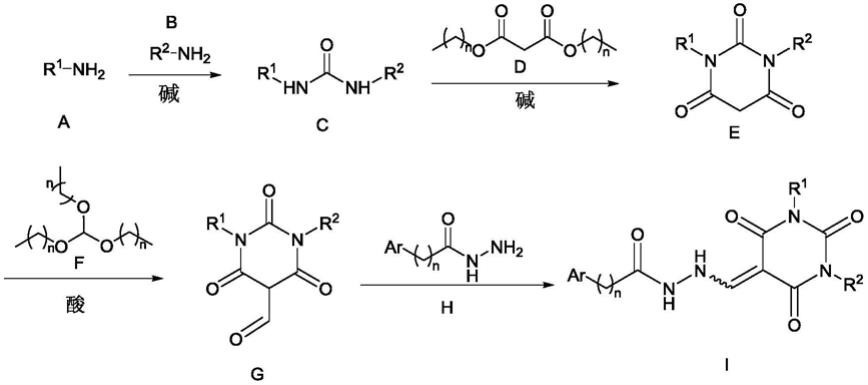
[0059][0060]
胺(a)与另一胺(b)在三光气和碱的条件下反应得到双取代脲(c),碱通常选择dipea或三乙胺,双取代脲(c)与丙二酸二酯(d)在碱性条件下反应得到结构通式e化合物,通常丙二酸酯(d)可选择甲酸低级烷基酯,如丙二酸二甲酯或者丙二酸二乙酯,碱性条件优选醇钠或者醇钾,例如甲醇钠、乙醇钠、叔丁醇钠、叔丁醇钾。结构通式e化合物与原酸酯(f)在酸性条件下反应得到结构通式g化合物,通常原酸酯(f)选择低级烷基酯,如原甲酸三甲酯或原甲酸三乙酯,催化剂酸性条件优选质子酸,如浓硫酸、对甲苯磺酸、磷酸、干燥氯化氢气体和高氯酸。化合物g与酰肼化合物(h)缩合生成终产物(i)。
[0061]
酰肼化合物(h)可方便地通过相应的羧酸酯化后再发生肼解进行制备,反应通式如下:
[0062][0063]
方法b:
[0064][0065]
嘧啶三酮(a
′
)与甲酸酯(b
′
)在碱性条件下进行缩合,得到烯醇(c
′
),通常甲酸酯(b
′
)可选择甲酸低级烷基酯,如甲酸甲酯或者甲酸乙酯,碱性条件优选醇钠或者醇钾,例如甲醇钠、乙醇钠、叔丁醇钠、叔丁醇钾。烯醇(c
′
)与酰肼(d
′
)反应得到结构通式e
′
化合物,再进一步依次与烃基化试剂r1x(f
′
)和r2x(h
′
)在碱性条件下发生烃基化反应,最终得到结构通式i
′
化合物,其中x为离去基团,优选卤素或磺酸酯,碱性条件优选氢化钠,显而易见的是,当r1与r2为相同的基团时,获得结构通式g
′
和结构通式i
′
的转化是合并在一步内完成的。
[0066]
本发明酰肼取代的亚甲基嘧啶三酮化合物能够被制成多种口服与胃肠外剂型给药,包括透皮、直肠或者吸入给药。剂型中包含结构式(i)酰肼取代的亚甲基嘧啶三酮化合物或对应的结构式(i)酰肼取代的亚甲基嘧啶三酮化合物的药学上可接受的盐或者溶剂化物。
[0067]
本发明的药物制剂中包含治疗有效量的结构式(i)酰肼取代的亚甲基嘧啶三酮化合物以及药学上可接受的载体、稀释剂或赋型剂。就制备含有本发明结构式(i)酰肼取代的亚甲基嘧啶三酮化合物的药物组合物而言,药学上可接受的载体可以是固体或液体。固体形式制备物包括粉剂、片剂、丸剂、胶囊剂、扁囊剂、栓剂、可分散的颗粒剂、吸入剂。固体载体可以是一种或多种物质,它们也可以充当稀释剂、矫味剂、粘合剂、防腐剂、崩解剂或者包封材料。
[0068]
本发明药物组合物优选地含有质量百分比为1%以上的结构式(i)酰肼取代的亚甲基嘧啶三酮化合物。药物组合物还包括药学上可接受的载体,适合的药学上可接受的载体包括碳酸镁、硬脂酸镁、滑石、蔗糖、乳糖、甘露醇、果胶、糊精、淀粉、明胶、黄芪胶、甲基纤维素、羧甲基纤维素钠、硅胶、微晶纤维素、聚维酮、低熔点蜡、可可脂中的一种。可以使用片剂、粉剂、胶囊剂、丸剂、扁囊剂、锭剂和颗粒剂作为适合于口服给药的固体剂型,优选的口服剂型是片剂和胶囊剂。
[0069]
片剂的制备是通过将结构式(i)酰肼取代的亚甲基嘧啶三酮化合物与适当的辅料混合通过造粒工艺结合,包括湿法、干法、挤出、喷雾等方法,也可以是粉末不经造粒工艺直接混合,然后压制成片。胶囊的制备是将结构式(i)酰肼取代的亚甲基嘧啶三酮化合物与适当的辅料通过造粒工艺结合,包括湿法、干法、挤出、喷雾等方法,也可以是粉末不经造粒工艺直接混合,然后经过填充胶囊而成。粉剂的制备是通过将细粉化的结构式(i)酰肼取代的亚甲基嘧啶三酮化合物和稀释剂、着色剂、矫味剂、稳定剂、增稠剂、分散剂和芳香剂等混
合,经过分装而成。颗粒剂的制备是通过将结构式(i)酰肼取代的亚甲基嘧啶三酮化合物和稀释剂、着色剂、矫味剂、稳定剂、增稠剂、分散剂和芳香剂等通过造粒工艺结合,包括湿法、干法、挤出、喷雾等方法,制成具有一定粒度的颗粒,再将颗粒分装而成。栓剂的制备是通过将结构式(i)酰肼取代的亚甲基嘧啶三酮化合物与适宜的基质粉末混合均匀,然后分装于模具内,通过模具挤压而成;也可以是将结构式(i)酰肼取代的亚甲基嘧啶三酮化合物溶解或分散于已经熔融的基质内,然后分装至模具内,冷却凝固而成。吸入剂的制备是通过将结构式(i)酰肼取代的亚甲基嘧啶三酮化合物微粉化,然后附着在适宜的载体上而成,如乳糖、糖醇类等,再借助吸入器装置给药。
[0070]
液体形式制备物包括溶液、混悬液和乳液,例如水、水/乙醇、水/丙二醇和水/聚乙二醇等溶液。就注射而言,可以将液体制备物配制成在乙醇、丙二醇或者聚乙二醇水溶液、等渗盐水、5%含水葡萄糖等中的溶液。适合于口服的水溶液可以这样制备,将结构式(i)酰肼取代的亚甲基嘧啶三酮化合物溶于水,根据需要加入适合的着色剂、矫味剂、稳定剂和增稠剂。适合于口服的水混悬液可以这样制备,将微细粉碎的活性组分分散在水中,再与粘性材料混合,例如天然或合成树脂、树胶、甲基纤维素、羧甲基纤维素钠或其他熟知的悬浮剂。乳剂可以通过将结构式(i)酰肼取代的亚甲基嘧啶三酮化合物分散于或溶解于油相或水相中,例如豆油、蒸馏水,再加入另一相液体和乳化剂,例如吐温80,经过搅拌、超声或其它乳化装置乳化而成。
[0071]
本发明药物制备物优选地为单位剂型。在此类剂型中,制备物被细分为含有适量结构式(i)酰肼取代的亚甲基嘧啶三酮化合物的单位剂量。单位剂型可以是带包装的制备物,该包装含有离散量的制备物,例如小包装片剂、胶囊剂和小瓶或安瓿装粉剂。单位剂型可以是片剂、胶囊剂、扁囊剂或锭剂本身,或者是适当数量的任意这些的带包装形式。
[0072]
结构式(i)酰肼取代的亚甲基嘧啶三酮化合物的治疗有效剂量从0.01mg/kg~100mg/kg体重每天不等。典型的剂量是0.1mg/每天至5000mg/每天。根据特定的应用和活性组分的效力,活性组分在单位剂量制备物中的量可以从0.1mg~1000mg不等,优选1mg~500mg。如果需要,组合物还可以含有其它可相容的治疗剂。给予需要用结构式(i)酰肼取代的亚甲基嘧啶三酮化合物治疗的受治疗者1mg/每天~3000mg/每天的剂量,在24小时内单次或多次给药,治疗可以按连续间隔反复达必要的时间长度。
[0073]
部分中间体可通过直接购买或通过以下实施例合成。
[0074]
实施例1
[0075]
本实施例为中间体酰肼化合物(h)(喹啉-4-甲酰肼)的合成,也可以直接购买。
[0076]
15g 4-喹啉羧酸甲酯中加入100ml甲醇和20g edci,840mg dmap以及15ml的dipea,常温搅拌过夜,减压浓缩得油状物,向此油状物中加入200ml二氯甲烷,用100ml饱和食盐水洗涤一次,再用200ml饱和食盐水洗涤一次,用无水硫酸钠干燥,有机层减压浓缩至干。浓缩物用硅胶色谱纯化,石油醚-乙酸乙酯5:1至2:1梯度洗脱,得白色固体8g。用50ml无水乙醇将白色固体溶解,加入20ml的水合肼加热至回流反应4小时,减压浓缩得油状物,向此油状物中加入100ml乙酸乙酯,用100ml饱和食盐水洗涤一次,用200ml饱和食盐水洗涤一次,用无水硫酸钠干燥,有机层减压浓缩至干。浓缩物用硅胶色谱纯化,二氯甲烷-甲醇50:1至20:1梯度洗脱,得白色固体5g。1h-nmr(600mhz,dmso)δ8.94(t,j=4.2hz,1h),8.75(d,j=4.6hz,1h),8.25(dd,j=8.6,1.3hz,1h),8.16(dd,j=8.1,1.5hz,1h),7.81(d,j=
4.7hz,1h),7.70(ddd,j=8.1,7.0,1.5hz,1h),7.65(ddd,j=8.3,7.0,1.5hz,1h),4.38(dd,j=7.1,4.0hz,1h),4.26(dd,j=7.2,4.1hz,1h)。
[0077]
本实施例得到的中间体酰肼化合物(h)(喹啉-4-甲酰肼)的结构式为:
[0078][0079]
实施例2
[0080]
本实施例为中间体双取代脲(c)(1,3-二烯丙基脲)的合成
[0081]
冰浴下,将1g烯丙基胺加入10ml干燥的二氯甲烷中,加入1.2g三光气和3ml dipea,0℃下反应3小时,再加入1g烯丙基胺,升至室温,反应过夜,减压浓缩得白色固体,乙酸乙酯溶解,饱和食盐水洗涤,收集有机相,无水硫酸钠干燥,有机层减压浓缩至干。浓缩物用硅胶色谱纯化,石油醚-乙酸乙酯3:1至1:1梯度洗脱,得白色固体600mg。1h-nmr(600mhz,dmso)δ5.99(t,j=5.6hz,2h),5.81(ddt,j=17.2,10.3,5.1hz,2h),5.10(dq,j=17.2,1.8hz,2h),5.01(dq,j=10.3,1.7hz,2h),3.63(ddt,j=5.8,5.1,1.7hz,4h)。
[0082]
本实施例合成的中间体双取代脲(c)(1,3-二烯丙基脲)结构式为:
[0083][0084]
实施例3
[0085]
本实施例为中间体双取代脲(c)(1-环丙基脲)的合成
[0086]
冰浴下,将1g环丙胺加入15ml h2o中,逐滴加入浓盐酸至ph至1~2,再加入2g氰酸钾,加热至50℃反应过夜,冷却,减压浓缩得白色固体,乙酸乙酯溶解,滤去不溶物,浓缩成白色固体粉末,干燥,得2.2g产物。1h-nmr(600mhz,dmso)δ6.38(d,j=7.3hz,1h),5.22(d,j=7.9hz,1h),5.11(d,j=7.9hz,1h),2.75(dp,j=7.3,5.0hz,1h),0.84
–
0.78(m,2h),0.78
–
0.71(m,2h)。
[0087]
中间体双取代脲(c)(1-环丙基脲)结构式为:
[0088][0089]
实施例4
[0090]
本实施例为中间体结构通式e化合物(1-环丙基嘧啶-2,4,6(1h,3h,5h)-三酮)的合成
[0091]
氮气保护下,将2g 1-环丙基脲加入14ml干燥的甲醇中,加入2g的甲醇钠和2.4ml的丙二酸二甲酯,加热至65℃回流反应6小时,减压浓缩得淡黄色固体,加入冰水使其溶解至澄清透明液体,逐滴加入1mol/l盐酸溶液后有大量白色固体沉淀,抽滤并用大量清水洗涤白色固体,烘干得到1.2g产物。1h-nmr(600mhz,dmso)δ9.29(s,1h),3.94(p,j=6.2hz,1h),3.33(s,2h),1.25
–
1.19(m,2h),1.18
–
1.11(m,2h)。
[0092]
中间体结构通式e化合物(1-环丙基嘧啶-2,4,6(1h,3h,5h)-三酮)结构式为:
[0093][0094]
实施例5
[0095]
本实施例为中间体结构通式g化合物(1-环丙基嘧啶-2,4,6(1h,3h,5h)-三酮-5-碳醛)的合成
[0096]
将1g 1-环丙基嘧啶-2,4,6(1h,3h,5h)-三酮加入7ml的原甲酸三甲酯中,加入120mg对甲苯磺酸,加热至80度反应6小时,减压浓缩得黄色固体粉末,甲醇中重结晶得到产品,干燥得到820mg产物。1h-nmr(600mhz,dmso)δ9.77(d,j=9.3hz,1h),9.13(s,1h),4.44(d,j=9.3hz,1h),3.89(p,j=6.0hz,1h),1.25
–
1.19(m,2h),1.18
–
1.12(m,2h)。
[0097]
中间体结构通式g化合物(1-环丙基嘧啶-2,4,6(1h,3h,5h)-三酮-5-碳醛)结构式为:
[0098][0099]
实施例6
[0100]
本实施例为结构式(i)的酰肼取代的亚甲基嘧啶三酮化合物—5-(喹啉-4-甲酰肼基)亚甲基-1-环丙基嘧啶-2,4,6(1h,3h,5h)-三酮的合成
[0101]
将500mg 1-环丙基嘧啶-2,4,6(1h,3h,5h)-三酮-5-碳醛加入5ml干燥的甲醇中,加入470mg喹啉-4-甲酰肼,常温搅拌30分钟,有白色固体析出,抽滤并用大量甲醇洗涤,干燥得到720mg终产物。对得到的产物进行核磁测试,其核磁氢谱的测试结果如下:
[0102]1h-nmr(600mhz,dmso)δ11.92(s,1h),11.62(d,j=12.6hz,1h),10.95(d,j=50.5hz,1h),9.06(d,j=4.2hz,1h),8.34(d,j=8.4hz,1h),8.24(d,j=12.4hz,1h),8.13(d,j=8.4hz,1h),7.86(ddd,j=8.4,6.7,1.4hz,1h),7.79(dd,j=4.3,1.5hz,1h),7.72(ddd,j=8.2,6.8,1.2hz,1h),2.59
–
2.51(m,1h),1.03
–
0.90(m,2h),0.71(dq,j=22.6,4.2,3.8hz,2h)。
[0103]
其结构式为:
[0104][0105]
实施例7
[0106]
本实施例为结构式(i)的酰肼取代的亚甲基嘧啶三酮化合物—5-[(喹啉-4-甲酰肼基)亚甲基]嘧啶-2,4,6(1h,3h,5h)-三酮的合成
[0107]
步骤a:向500ml dmf中加入250ml甲酸乙酯,搅拌下加入6g嘧啶三酮和2.5g甲醇钠
反应过夜,加入稀盐酸淬灭,加入1000ml乙酸乙酯和500ml饱和食盐水,搅拌分液,水层用500ml乙酸乙酯提取两次,合并有机层,用饱和食盐水洗涤,无水硫酸钠干燥,过滤。
[0108]
步骤b:将步骤a所得滤液浓缩,加入500ml四氢呋喃和2.5g喹啉-4-甲酰肼,反应3小时,浓缩除去溶剂,加入250ml乙腈和250ml甲醇,搅拌下加热至回流后冷却至室温,过滤得固体产物3.2g。1h-nmr(600mhz,dmso)δ11.97(s,1h),11.64(s,1h),11.14(d,j=50.6hz,1h),10.89(s,1h),9.06(d,j=4.3hz,1h),8.38
–
8.29(m,2h),8.13(d,j=8.4hz,1h),7.86(ddd,j=8.4,6.8,1.4hz,1h),7.79(d,j=4.3hz,1h),7.72(ddd,j=8.3,6.8,1.3hz,1h)。
[0109]
其结构式为:
[0110][0111]
实施例8
[0112]
本实施例为结构式(i)的酰肼取代的亚甲基嘧啶三酮化合物—5-(喹啉-4-甲酰肼基)亚甲基-1-苯甲基嘧啶-2,4,6(1h,3h,5h)-三酮的合成
[0113]
氮气保护下,将5-[(喹啉-4-甲酰肼基)亚甲基]嘧啶-2,4,6(1h,3h,5h)-三酮500mg加入50ml dmf中,降温至-5℃,加入六甲基二硅基氮烷钠380mg,搅拌1小时,缓慢滴加400mg溴化苄,升至室温搅拌过夜。加入冰醋酸淬灭,搅拌下加入200ml乙酸乙酯和200ml饱和食盐水,分液,水层用200ml乙酸乙酯提取两次,合并有机层,用饱和食盐水洗涤,无水硫酸钠干燥,浓缩。浓缩物用硅胶色谱纯化,二氯甲烷-甲醇50:1至10:1梯度洗脱,得白色固体580mg
[0114]1h-nmr(600mhz,dmso)δ11.97(s,1h),11.64(s,1h),11.20(d,j=47.7hz,1h),9.06(dd,j=4.4,1.7hz,1h),8.34(t,j=6.7hz,2h),8.12(d,j=8.4hz,1h),7.86(t,j=7.7hz,1h),7.78(d,j=4.3hz,1h),7.71(t,j=7.8hz,1h),7.37
–
7.21(m,5h),4.97(d,j=7.2hz,2h).
[0115]
结构式(i)的酰肼取代的亚甲基嘧啶三酮化合物结构式为:
[0116][0117]
依据上述方法制得了下述终化合物:
[0118]
[0119]
[0120]
[0121]
[0122]
[0123]
[0124]
[0125][0126]
实施例9
[0127]
dpre1酶结合实验
[0128]
(1)dpre1基因的获得
[0129]
从耻垢分枝杆菌(mycobacterium smegmatis strain atcc607购买自中国微生物菌种保藏管理委员会普通微生物中心)菌种中获得全基因组,运用pcr的方法,以提取的全基因组为模板,加入前后引物,进行基因序列扩增,获得全长目的基因片段,对该基因片段进行dna测序,对测序结果运用ncbi的基因组数据库中进行基因序列匹配比对,比对结果见图1所示,其与耻垢分枝杆菌全基因中dpre1基因序列一致。将比对正确的基因与pet28a质粒同时双酶切并连接后,用dh5α进行扩增,提取扩增后的质粒,用双酶切方法对提取质粒dpre1-pet28a进行酶切,用琼脂糖凝胶进行验证,在dpre1基因序列大小处有对应的条带,连接成功。(参考文献:li,h;jogl,g.proteins.2013,81:538-543.)
[0130]
(2)dpre1蛋白表达和纯化
[0131]
测序检测dpre1基因正确且连接成功后,将构建好的质粒dpre1-pet28a,用热激法转入表达宿主感受态细胞rossetta(购买自上海迈其生物科技有限公司),挑取单克隆子至5ml含有相应抗性的常用细菌培养液体培养基即lb液体培养基(胰蛋白胨10g/l,酵母提取物10g/l,氯化钠10g/l)中,37℃,200rpm,摇床过夜培养。次日按1:100(体积比)接入含有相应抗性的1l lb液体培养基中,继续摇床培养至对数期,使od600=0.8,加入异丙基-β-d-硫代半乳糖苷(iptg)终浓度0.4mm,转移至18℃,220rpm继续诱导培养过夜。离心收集细胞,加入适量体积5的裂解液,超声破碎细胞,将破碎液体4℃,20000rpm离心30min,分离上清和沉淀,利用akta pure蛋白纯化系统(自ge医疗集团),分别采用亲和层析ni柱(型号:hitrapimac ff 5ml)、离子交换层析柱(型号:qff-16-10-hiprep)和分子筛层析(型号:superdextm200 10/300gl)纯化得到dpre1蛋白,供结合实验使用。
[0132]
(3)mst(微量热泳动仪)测定结构式(i)的酰肼取代的亚甲基嘧啶三酮化合物与dpre1酶的结合能力
[0133]
将待测化合物用dmso溶解,配制成100mm母液,随后用缓冲液(20mm hepes)稀释成4mm的初始工作液1,测定缓冲液为含4%dmso的20mm hepes。初始工作液1作为结合力测定的最高浓度,然后用测定缓冲液倍比稀释工作液1,共稀释23次,得到24个工作液,工作液24的浓度为0.95nm。各工作液加入50nm dpre1酶蛋白,在微量热泳动仪上测定平衡解离常数kd。待测化合物和dpre1酶蛋白结合的平衡解离常数为kd见下表:
[0134]
化合物编号kd化合物编号kd14.73μm180.22mm223.78μm192.7μm39.98μm2042.7nm4103nm214.9nm544.4nm221.16mm66.53nm231.08μm767.76μm240.434mm857.65μm252.28nm911.7nm261.88μm101.9mm2752.6μm111.77mm2819.3μm120.67μm29156nm1318.7nm300.508μm1467.7nm310.887μm1521.5nm32no binding162.23μm33no binding170.22mm348.26μm
[0135]
实施例10
[0136]
体外抑菌试验
[0137]
采用耻垢分枝杆菌(mycobacterium smegmatis strain atcc 607购买自中国微生物菌种保藏管理委员会普通微生物中心)进行抑菌体外活性检测。结构式(i)的酰肼取代的亚甲基嘧啶三酮化合物溶解于dmso中。用小纸片浸润化合物溶液,置于培养有mycobacterium smegmatis strain(atcc607)的middlebrook 7h10培养皿中,化合物终浓度是25mm,抑菌圈直径大于1.1cm,视为抑菌活性为阳性用“ ”标记,抑菌圈直径小于1.1cm视为抑菌活性为阴性用
“‑”
标记。
[0138]
化合物抑菌活性化合物抑菌活性1 20 3 21 4 22 6 23 9 24
10 25 11 26 12 27-13 28 14 29 15 30-16 31 17 32-18 33 19 34 异烟肼 链霉素 dmso
‑ꢀꢀ
[0139]
实施例11
[0140]
体外结核杆菌抑制实验
[0141]
将m.tuberculosis h37rv atcc 27294培养于middle brook 7h11琼脂(干粉购买自碧迪医疗器械有限公司,直接配制),将菌斑重悬于含有0.04%(v/v)tween 80、0.2%牛血清白蛋白的悬浮溶液中,培养至其浑浊度与mcfarland no.1的浑浊度一致,将上述符合浊度的菌液用7h9gc broth middle brook 7h9(干粉购买自碧迪医疗器械有限公司,直接配制4.7g middle brook 7h9肉汤培养基[difco,detroit,mich.],20ml 10%[vol/vol]甘油,1g bacto casitone[difco],880ml蒸馏水,100ml油酸,白蛋白,葡萄糖,过氧化氢酶)1:25稀释,得到适宜浊度菌液。取96孔细胞培养板,将其外侧36孔加入200μl无菌水,在第2列b-e排孔分别加入不同浓度的06号化合物dmso溶液200μl,设置f行作为空白对照,加入200μl 7h9液体培养。设置异烟肼、利福平为阳性对照,将96孔板盖好盖子,用封口膜封口,放入37℃培养箱中,培养5天后,向f2中加入显色液almar blue阿尔玛兰(购买自sigma)10μl,37℃培养24h,颜色变为粉色,向其他孔中加入显色液110μl,观察其由粉变蓝的浓度,确定其mic为3.1μg/ml。将上述显颜色变化的96孔板,用酶标仪在激发波长为520nm,发射波长为590nm的条件下得到相应读数,拟合曲线作变化趋势图,结核杆菌生长随dmso、待测化合物、inh(异烟肼)、rfp(利福平)浓度变化结果如图2、图3、图4和图5所示。
[0142]
通过以上图的对比,说明dmso对照组对结核杆菌的生长没有影响,加入06号化合物对结核杆菌的生长有抑制作用,且与阳性对照异烟肼和利福平作用相当。
[0143]
实施例12
[0144]
结核杆菌模型动物实验
[0145]
1.实验动物:
[0146]
spf级c57bl/6小鼠,购自北京华阜康公司。6-8周龄,雌性。动物购入后,随机预分笼饲养。生物安全实验室饲养1周,适应生物安全实验室环境。
[0147]
2.感染模型的建立:
[0148]
接种结核分枝杆菌h37rv(菌种由武汉大学病毒所提供)于middle brook 7h11(干粉购买自碧迪医疗器械有限公司,可直接配置)固体平板培养基中37℃培养2-3周至对数生长期。按照菌体1mg干重相当于1*107cfu(cfu:单位体积的活菌个数)进行定量。称取h37rv
后,依据菌量加入无菌磷酸缓冲盐溶液,用匀浆器磨成终浓度为5mg/ml的均匀菌悬液。
[0149]
采用腹腔注射0.8%(8mg/ml)戊巴比妥钠120μl,麻醉小鼠。按照结核杆菌h37rv毒株10μl/只,经滴鼻方式感染小鼠,获得小鼠tb感染模型。感染后第2天,处死2只小鼠,取肺脏,菌落计数进行菌落计数。明确实际感染剂量为100cfu/只。于感染后第18天处死3只小鼠,取小鼠肺脏及脾脏菌落计数。
[0150]
3.动物分组:
[0151]
感染后的小鼠随机分组,每组9只。组别分为:pbs对照组、异烟肼组(10mg/kg)、低剂量组dose 1(10mg/kg)、高剂量组dose 2(30mg/kg),每天灌胃给药一次,200μl/只/次,每周五天。
[0152]
治疗周期和处理:
[0153]
连续治疗3周,分别处死各组小鼠。6只无菌分离肺脏、脾脏。研磨均匀,倍比稀释后,涂布含有100mg/l氨苄青霉素、50mg/l羧苄青霉素、2.5mg/l两性霉素b、25mg/l多黏菌素b、tch 2mg/l的7h11平板,37℃培养3周后,细菌菌落计数。3只无菌分离肺脏放入4%多聚甲醛固定,石蜡包埋,切片,分别抗酸染色。结果如图6、图7和图8所示,低剂量组也具有一定的抗结核杆菌感染的治疗作用,且dose 2高剂量组显示出了和阳性对照药相当的明显的抗结核杆菌感染的治疗作用。图8中黑色部分表示抗酸染色后显蓝色,灰色部分表示抗酸染色后显红色。
[0154]
对比例1
[0155]
同化合物1,不同之处在于,n=5。
[0156]
对比例2
[0157]
同化合物2,不同之处在于,n=10。
[0158]
通过研究,对比例1和对比例2中,链长过长的化合物不利于与酶的结合。
[0159]
以上具体实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。