一种t细胞向肿瘤迁移的负调控因子的体外筛选、鉴定方法
技术领域
1.本发明属于抗肿瘤免疫治疗领域,具体涉及一种t细胞向肿瘤迁移的负调控因子的体外筛选、鉴定方法。
背景技术:
2.随着人类对免疫系统和肿瘤发生机制的深入认识,2015年,一种名叫“免疫疗法”的肿瘤治疗方式被提出,并且随着近年来的发展彻底改变了肿瘤治疗的格局,成为对抗肿瘤的有力武器。其中,免疫疗法的主力军t细胞迁移至肿瘤是抗肿瘤免疫治疗领域中改善免疫治疗疗效的主要策略之一。
3.在t细胞肿瘤免疫循环过程中,促进t细胞向肿瘤迁移在抗肿瘤免疫中发挥重要作用,然而t细胞迁移到肿瘤的能力是否受到基因调控仍缺乏报道。揭示影响t细胞向肿瘤迁移的基因调控网络对促进抗肿瘤免疫治疗领域的发展至关重要。然而一个主要的挑战是如何对驱动t细胞向肿瘤迁移的调控基因进行系统性分析。迄今为止,没有文献报道过如何对t细胞向肿瘤迁移的调控因子进行筛选,因此开发一种免疫细胞向肿瘤迁移的负调控因子的体外筛选、鉴定方法成为重中之重。
技术实现要素:
4.本发明的目的是提供一种免疫细胞向肿瘤迁移的负调控因子的体外筛选、鉴定方法。
5.为解决上述技术问题,本发明采用如下技术方案:一种免疫细胞向肿瘤迁移的负调控因子的体外筛选、鉴定方法,所述的筛选方法包括:步骤1、采用slice技术对免疫细胞进行基因组编辑;步骤2、采用嘌呤霉素puro筛选稳定表达的经基因组编辑的免疫细胞并进行扩增培养;步骤3、将扩增培养后的经基因组编辑的免疫细胞在驱动剂的作用下进行transwell迁移实验,分别收集transwell上室和下室中的细胞,比较基因未敲除和基因敲除后的迁移效率变化和/或提取基因组dna,通过pcr扩增基因组中的sgrna所对应的dna序列,并对扩增后的dna序列进行二代测序。
6.优选地,所述的免疫细胞为t细胞。
7.优选地,所述的驱动剂为趋化因子、肿瘤细胞的培养上清液或实体瘤中的一种或多种。
8.进一步优选地,所述的驱动剂为趋化因子cxcl9和/或趋化因子cxcl10,在经基因组编辑的t细胞表面的cxcr3表达水平最高的时段内进行transwell迁移实验。
9.在本研究中,发明人大胆设想了一种鉴定t细胞向肿瘤迁移的负调控因子的体外筛选平台:分别以趋化因子cxcl9/10、后期将要用到的肿瘤细胞的培养上清液、甚至是人的
肿瘤等作为“驱动剂”,利用transwell迁移实验,让已经分别被敲除不同基因的混合t细胞向加入了普通培养基的下室以及向加入了含上述“驱动剂”的培养基中迁移,最后分别收集未发生迁移和迁移到培养基中的细胞,分别提取基因组dna,通过pcr扩增基因组中的sgrna所对应的dna序列,并对扩增后的dna序列进行二代测序。pcr产物属于碱基不平衡文库,为了减少碱基不平衡对测序结果的影响,测序时混入5%的phix文库。测序采用 hiseq xten-pe150 策略,测序数据量要求不低于1000个拷贝的文库sg rna 序列数,最终数据量为2g clean data/文库。 测序过程由北京诺禾致源生物有限公司完成。在python中分析实验组较对照组sgrna富集的序列与对应的基因。
10.需要说明的是,其不仅局限于趋化因子对t细胞的驱动作用,也适用于肿瘤细胞培养上清及人的肿瘤对t细胞的驱动作用,并且其原理也适用于鉴定其他免疫细胞向肿瘤迁移的负调控因子,如dc细胞响应xcr1-xcl1,ccr5-ccl4/ccl5,ccr1-ccl4等信号轴的趋化作用发生迁移;nk细胞响应cx3cr1-cx3cl1,cxcr3-cxcl9/cxcl10信号轴的趋化作用发生迁移等,本发明的技术方案将大大推进抗肿瘤免疫治疗领域的发展。
11.根据一些具体实施方式,所述的步骤1包括:步骤a1、采用sgrna文库慢病毒感染免疫细胞;步骤a2、向步骤a1中的感染后的免疫细胞中电转cas9蛋白;步骤a3、电转结束后,加入不含il-2的培养基于30~40℃下孵育20~50 min,其中,步骤a2中使用的电转体系为100μl lonza电转buffer中溶解5
×
106~10
×
106个免疫细胞和3~5μm cas9蛋白,步骤a3中使用的培养基的配方为:rpmi-1640培养基
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
200~300 ml;x-vivo15 media无血清细胞培养基
ꢀꢀꢀꢀꢀ
200~300 ml;谷氨酰胺
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
2~8 ml;青霉素-链霉素溶液
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
2~8 ml;fbs胎牛血清
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
5 %~15 %。
12.优选地,所述的免疫细胞为cd3
t细胞、cd4
t细胞或cd8
t细胞中的一种或多种。
13.优选地,孵育结束后,加入40~60μmβ-巯基乙醇,5~15 mm 4-羟乙基哌嗪乙磺酸,0.5~1.5 mm丙酮酸钠,80~120μm neaa细胞培养添加物, 2~8 ng/ml il-15,5~15 ng/ml il-7,400~600iu/ml il-2继续培养60~84 h。
14.进一步优选地,所述的免疫细胞的制备方法包括如下步骤:步骤s1、从外周血单核细胞中提取cd3
t细胞、cd4
t细胞或cd8
t细胞中的一种或多种;步骤s2、使用0.5~2μg/mlα-cd3抗体和0.5~2μg/mlα-cd28抗体刺激t细胞活化36~48 h;步骤s3、使用0.5~2μg/mlα-cd3抗体和0.5~2μg/mlα-cd28抗体再次刺激t细胞再次活化18~24 h。
15.再进一步优选地,步骤s2和步骤s3中所述的活化使用的培养基的配方为:rpmi-1640培养基
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
200~300 ml;
x-vivo15 media无血清细胞培养基
ꢀꢀꢀꢀꢀ
200~300 ml;谷氨酰胺
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
2~8 ml;青霉素-链霉素溶液
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
2~8 ml;fbs胎牛血清
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
5 %~15 %;人重组il-2
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
400~600iu/ml。
16.优选地,所述的cas9蛋白为nls-cas9-gfp复合蛋白,采用流式检测免疫细胞表面的cxcr3的表达水平。
17.根据一些优选地实施方式,所述的transwell小室的孔径为3~5μm。
18.根据一些优选地实施方式,选择扩增培养11~15天内的经基因组编辑的免疫细胞进行transwell迁移实验。
19.根据一些优选地实施方式,所述的sgrna文库慢病毒的moi值为0.2~0.4 。
20.通过控制sgrna文库的感染复数(moi)为0.2~0.4,从而保证一个细胞只被一条sgrna感染,故一个细胞只被敲除一个基因。在使用sgrna文库进行筛选时,也不会出现一个细胞敲除多个基因导致筛选结果不准确的问题。
21.进一步优选地,所述的transwell迁移实验为:将经基因组编辑的免疫细胞加入上室中,将驱动剂加入下室中,然后在37℃和2~8% co2条件下迁移 3~6 h,收集上室和下室细胞,计算迁移效率和/或提取基因组dna,通过pcr扩增基因组中的sgrna所对应的dna序列,并对扩增后的dna序列进行二代测序。
22.优选地,所述的筛选、鉴定方法具体包括以下步骤:(1)、制备sgrna文库慢病毒day1:将sgrna文库电转化感受态细胞,孵育后于28~35 ℃下培养10~20 h;day2:采用qiagen maxi柱富集sgrna慢病毒;(2)、使用所述的sgrna文库质粒制备sgrna慢病毒day1:培养293t细胞;day2:使用所述的sgrna文库质粒转染所述的293t细胞;day3:分离并浓缩sgrna文库慢病毒;(3)、采用slice技术对t细胞进行基因组编辑day0:0.5~2μg/mlα-cd3抗体和0.5~2μg/mlα-cd28抗体包被至细胞培养皿中;day1:从外周血单核细胞中提取cd3
t细胞、cd4
t细胞或cd8
t细胞,向包被有α-cd3抗体和α-cd28抗体的细胞培养皿中加入cd3
t细胞、cd4
t细胞或cd8
t细胞中的一种或多种和培养基进行活化;day2:按400~600iu/ml加入人重组il-2继续活化培养,将0.5~2μg/mlα-cd3抗体和0.5~2μg/mlα-cd28抗体包被至另一细胞培养皿中;day3:将原细胞培养皿中的细胞培养液转移至day2制备的包被有α-cd3抗体和α-cd28抗体的细胞培养皿中再次活化;day4:sgrna文库慢病毒感染cd3
t细胞、cd4
t细胞或cd8
t细胞;day5:电转nls-cas9-gfp蛋白,电转体系为100μl lonza电转buffer中溶解5
×
106~10
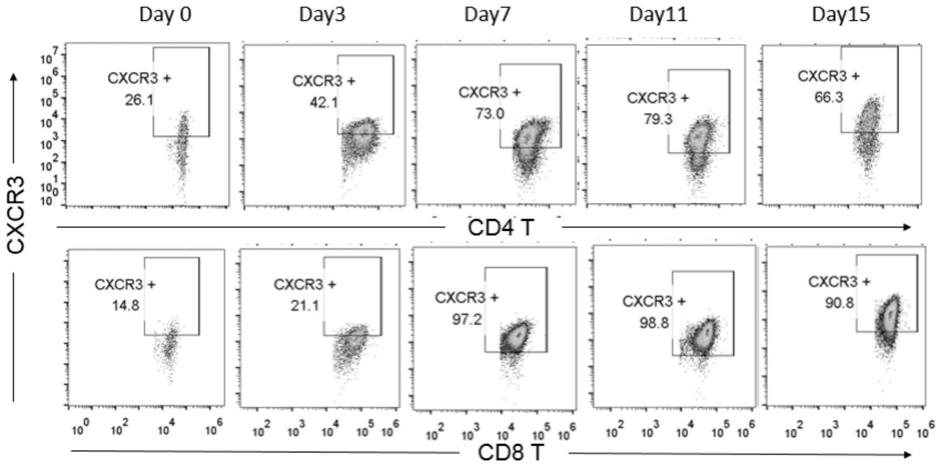
×
106个免疫细胞和3~5μm cas9蛋白,电转结束后,加入不含il-2的培养基于30~40℃下孵育20~50 min;孵育完后将细胞液转移置6孔板,向孔中加入1.5ml培养基,并加入40~60μm
β-巯基乙醇,5~15 mm 4-羟乙基哌嗪乙磺酸,0.5~1.5 mm丙酮酸钠,80~120μm neaa细胞培养添加物,2~8 ng/ml il-15, 5~15ng/ml il-7,400~600iu/ml il-2继续培养;day6:采用嘌呤霉素puro筛选稳定表达的经基因组编辑的cd3
t细胞、cd4
t细胞或cd8
t细胞并继续扩增培养至day11~15,嘌呤霉素puro的浓度为0.5~1.5μg/ml,后续每两天更换一次含有0.5~1.5μg/ml嘌呤霉素puro的培养基;(4)、transwell迁移实验 将上述扩增得到的基因组编辑的cd3
t细胞、cd4
t细胞或cd8
t细胞加入上室中,将驱动剂加入下室中,然后在37℃和2~8% co2条件下迁移 3~6 h,收集上室和下室细胞比较基因未敲除和基因敲除后的迁移效率变化和/或提取基因组dna,通过pcr扩增基因组中的sgrna所对应的dna序列,并对扩增后的dna序列进行二代测序。pcr产物属于碱基不平衡文库,为了减少碱基不平衡对测序结果的影响,测序时混入 5%的phix文库。测序采用 hiseq xten-pe150 策略,测序数据量要求不低于1000个拷贝的文库sg rna 序列数,最终数据量为2g clean data/文库。 测序过程由北京诺禾致源生物有限公司完成。在python中分析实验组较对照组sg rna富集的序列与对应的基因。
23.本发明与现有技术相比具有如下优势:本发明首次将slice技术、嘌呤霉素puro筛选以及transwell迁移实验结合作为免疫细胞向肿瘤迁移的负调控因子的体外筛选、鉴定方法,其不仅局限于趋化因子对t细胞的驱动作用,也适用于肿瘤细胞培养上清及人的肿瘤对t细胞的驱动作用,并且其原理也适用于鉴定其他免疫细胞向肿瘤迁移的负调控因子,将大大推进抗肿瘤免疫治疗领域的发展。
24.进一步地,本发明还对slice技术进行了优化,包括对t细胞的活化、增殖、慢病毒感染、cas9蛋白电转条件以及对后续编辑的t细胞的成功大量扩增的条件优化,有效地解决了至今slice技术方案不成熟,重复率不高的问题,为后续研究者进行免疫细胞研究提供便利,从而使人们能够更加快速高效地探索人类免疫细胞中的功能基因,进而能更好地改造免疫细胞,以对抗癌症和其他疾病,同时还有望加速免疫疗法中潜在的临床前候选物的发现。
附图说明
25.图1:在体外培养的day0、day3、day7、day11、day15流式检测cd4
t细胞和cd8
t细胞表面cxcr3的表达;图2:在体外培养的day0、day3、day7、day11、day15 cd4
t细胞或cd8
t细胞表面cxcr3的水平统计结果;图3:在体外培养的day0、day3、day7、day11、day15检测cd3
t细胞迁移的细胞数;图4:在体外培养的day0、day3、day7、day11、day15检测cd3
t细胞迁移率;图5:t细胞通过3-μm和5-μm孔径的小室向cxcl9/10迁移情况;图6:使用优化前的t细胞培养体系去对t细胞进行基因组crispr敲除筛选中t细胞增殖情况;图7:使用优化后的t细胞培养体系去对t细胞进行基因组crispr敲除筛选中t细胞增殖情况;图8:流式检测20
×
106或10
×
106细胞电转nls-cas9-gfp蛋白的效率;
图9:流式检测1,3,5μm nls-cas9-gfp蛋白的电转效率;图10:流式检测1,3,5μm nls-cas9-gfp蛋白的电转后的蛋白编辑效率;图11:实施例2中transwell实验检测rgs1敲除的t细胞与对照t细胞趋向cxcl9/10的迁移情况。
具体实施方式
26.下面结合实施例对本发明作进一步描述。但本发明并不限于以下实施例。实施例中采用的实施条件可以根据具体使用的不同要求做进一步调整,未注明的实施条件为本行业中的常规条件。本发明各个实施方式中所涉及到的技术特征只要彼此之间未构成冲突就可以相互组合。
27.为了能够体外筛选或鉴定免疫细胞向肿瘤迁移的负调控因子,发明人首次提出在利用crispr-screening技术对t细胞进行基因组编辑后,于体外利用“趋化剂”模拟肿瘤微环境对t细胞的招募,最终收集基因驱动的“趋化剂”募集的t细胞进行测序分析的技术方案。
28.具体地,所述的筛选方法、鉴定方法包括:步骤1、采用slice技术对免疫细胞进行基因组编辑;步骤2、采用嘌呤霉素puro筛选稳定表达的经基因组编辑的免疫细胞并进行扩增培养;步骤3、将扩增培养后的经基因组编辑的免疫细胞在驱动剂的作用下进行transwell迁移实验,分别收集transwell上室和下室中的细胞,比较基因未敲除和基因敲除后的细胞迁移效率变化和/或提取基因组dna,通过pcr扩增基因组中的sgrna所对应的dna序列,并对扩增后的dna序列进行二代测序。
29.多年来,对人原代t细胞进行基因组编辑一直是一大难点,这极大的限制了人们在系统范围内深入了解t细胞抗肿瘤免疫的机制。直到2018年,marson团队开发出了一种基于crispr的新技术—single guide rna (sgrna) lentiviral infection with cas9 protein electroporation (slice)。 slice技术是将sgrna慢病毒文库与cas9蛋白电穿孔相结合,将表达所需时间短的sgrna通过慢病毒递送,将cas9蛋白直接通过电穿孔的方式直接递送,成功解决了因人原代t细胞体外培养时间有限,慢病毒载体编码cas9蛋白时间太长而导致的crispr-cas9系统在免疫细胞上不能有效发挥作用的这一问题。故我们按照marson团队报道的技术方案进行t细胞编辑,实验结果却发现被编辑的t细胞由于遭受电刺激、抗生素筛选等压力导致细胞严重受损,无法及时恢复状态,不能继续增殖,并且在后续培养过程中增殖停滞,活率下降,所以无法进行后续实验。
30.2021年jeremy n. rich团队和西湖大学谢琦团队报道,他们在car-t细胞上使用slice技术进行t细胞编辑后,与胶质母细胞共培养,最终收集car-t细胞富集sgrna进行测序,筛选出多个基因,敲除这些基因后能够明显改进car-t细胞治疗胶质母细胞瘤的效果。我们使用了他们报道的方法,同样不能让被电转后加puro筛选的t细胞恢复状态继续增殖,t细胞受损严重,增殖停滞,无法进行后续实验。在这里,由于他们并没有写出puro的储存浓度,所以1:10000是多少的工作浓度,我们不得而知,但是我们尝试将puro用量降至0.5μg/ml进行筛选也无法解决编辑后的t细胞的扩增问题。
31.目前仅有这两篇报道使用slice技术成功编辑t细胞后进行crispr-screening的筛选,但是由于某些技术信息提供不全,某些信息差别过大,特别是培养体系无法满足我们的筛选系统等导致我们不能按照他们的系统得到大量扩增的高活性基因组编辑的t细胞,从而限制了我们的研究的推进。
32.为此我们进行了大量的研究,最终获得了能够大量扩增的高活性基因组编辑的t细胞的slice技术。
33.具体改进包括如下内容:步骤1包括:步骤a1、采用sgrna文库慢病毒感染免疫细胞;步骤a2、向步骤a1中的感染后的免疫细胞中电转cas9蛋白;步骤a3、电转结束后,加入不含il-2的培养基于30~40℃下孵育20~50 min,其中,步骤a2中使用的电转体系为100μl lonza电转buffer中溶解5
×
106~10
×
106个免疫细胞和3~5μm cas9蛋白,步骤a3中使用的培养基的配方为:rpmi-1640培养基
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
200~300 ml;x-vivo15 media无血清细胞培养基
ꢀꢀꢀꢀꢀ
200~300 ml;谷氨酰胺
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
2~8 ml;青霉素-链霉素溶液
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
2~8 ml;fbs胎牛血清
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
5 %~15 %。
34.进一步地,所述的免疫细胞为cd3
t细胞、cd4
t细胞或cd8
t细胞中的一种或多种。
35.进一步地,孵育结束后,加入40~60μmβ-巯基乙醇,5~15 mm 4-羟乙基哌嗪乙磺酸,0.5~1.5 mm丙酮酸钠,80~120μm neaa细胞培养添加物,2~8 ng/ml il-15,5~15ng/ml il-7,400~600iu/ml il-2继续培养72 h。
36.再进一步地,所述的cd3
t细胞、cd4
t细胞或cd8
t细胞的制备方法包括如下步骤:步骤s1、从外周血单核细胞中提取cd3
t细胞、cd4
t细胞或cd8
t细胞;步骤s2、使用0.5~2μg/mlα-cd3抗体和0.5~2μg/mlα-cd28抗体刺激t细胞活化36~48 h;步骤s3、使用0.5~2μg/mlα-cd3抗体和0.5~2μg/mlα-cd28抗体再次刺激t细胞再次活化18~24 h。
37.更进一步地,步骤s2和步骤s3中所述的活化使用的培养基的配方为:rpmi-1640培养基
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
200~300 ml;x-vivo15 media无血清细胞培养基
ꢀꢀꢀꢀꢀ
200~300 ml;谷氨酰胺
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
2~8 ml;青霉素-链霉素溶液
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
2~8 ml;fbs胎牛血清
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
5 %~15 %;人重组il-2
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
400~600iu/ml。
38.早在10年前,有多篇文献报道cxcr3 在静止的t细胞上几乎不表达,但是它在激活后的t细胞上表达显著增加,表明cxcr3对激活的t细胞功能至关重要。多年来随着研究的深
入,研究者们发现cxcr3在t细胞上的表达与t细胞在肿瘤中的浸润密切相关。更重要的是研究表明t细胞通过其表面的cxcr3的表达响应肿瘤微环境中分泌的其配体cxcl9/10的趋化作用从而向黑色素瘤、卵巢癌等实体瘤发生迁移,从而发挥抗肿瘤免疫的功能。但是多篇文献报道原代t细胞响应cxcl9/10的迁移效率低于10%,使得肿瘤中极少t细胞浸润从而导致实体瘤的免疫抑制。故找到促进t细胞向肿瘤迁移的新靶点是免疫治疗中的重要一环。目前对影响肿瘤基质分泌的cxcl9/10与t细胞表面的cxcr3受体结合而介导的信号转导的调控基因鲜有报道。
39.因此,作为一种实施方式,以cxcl9/10作为驱动剂,在免疫细胞表面的cxcr3的表达水平达到最高的时段内时进行transwell实验,利用t细胞响应趋化因子cxcl9/10的趋化作用而发生迁移这一原理,对驱动t细胞向趋化因子cxcl9/10迁移的调控基因进行系统性分析从而筛选出负调控t细胞向肿瘤迁移的基因。
40.优选地,所述的cas9蛋白为nls-cas9-gfp复合蛋白,采用流式检测免疫细胞表面的cxcr3的表达水平。
41.具体地,将扩增得到的基因组编辑的cd3t细胞加入上室中,将驱动剂加入下室中,然后在37℃和2~8%co2条件下迁移3~6h,收集上室和下室细胞,比较基因未敲除和基因敲除后的迁移效率变化和/或提取基因组dna,通过pcr扩增基因组中的sgrna所对应的dna序列,并对扩增后的dna序列进行二代测序。
42.下面结合具体实施例和对比例进一步阐述本发明的技术方案。
43.本发明中,所示的细胞迁移效率的计算方法为:迁移率(%)=(实验组下室中细胞数-对照组下室中细胞数)/(上室铺孔细胞总数-对照组下室细胞数)
×
100。铺孔细胞总数为1
×
105/孔。
44.以下具体实施例和对比例,除有特殊说明的,所使用的原料、试剂等均可通过市售获得。
45.实施例1:以敲除t细胞中的cd8a基因作为实施例,证实我们优化的slice技术相比优化前的技术方案能够促进编辑的t细胞状态恢复且大量增殖。
46.一、设计并合成四条sgcd8a,序列如下:cd8asgrna-1fcaccgggagcagcaaggccagcggc(seqidno.1)cd8asgrna-1raaacgccgctggccttgctgctccc(seqidno.2)cd8asgrna-2fcaccggaccgccttgctcctgccgc(seqidno.3)cd8asgrna-2raaacgcggcaggagcaaggcggtcc(seqidno.4)cd8asgrna-3fcaccgaaacaagcccaaggcggccg(seqidno.5)cd8asgrna-3raaaccggccgccttgggcttgtttc(seqidno.6)cd8asgrna-4fcaccgggtgtcgtcagtgcacacga(seqidno.7)cd8asgrna-4raaactcgtgtgcactgacgacaccc(seqidno.8)二、制备sgcd8a文库质粒day1:1.电转sgcd8a文库质粒;(1)室温下解冻recoverymedium;(2)将电穿孔比色杯(bio-rad,165-2089)和离心管置于冰上预冷;
(3)从-80度冰箱取出endura
tm
感受态细胞(lucigen,60242-1),置于冰上完全解冻20min;(4)将50
µ
l解冻的感受态细胞转入冷却的电穿孔比色杯中,加入2
µ
l浓度为5ng/μl的sgcd8a混合液;(5)用枪头混匀,避免气泡,使细胞沉在比色杯底部后进行电转,设置如下:10uf,600ohms,1800volts,时间约5.1~5.5ms;电转4个平行(共计40ng);(6)在脉冲的10s内,2mlrecoverymedium重悬混匀细胞后取出于15ml离心管中置于摇床,250rpm37度下培养1h。
47.2、将上述4管菌液混在一起,共8ml,涂几个稀释液的平板,计算转化效率。
48.3、随后将所有菌液平均涂于10块24cm直径菌板置于30℃过夜。
49.day2:1、16h后,向每个菌板加入4mllb后用玻璃棒刮下菌收集于50ml离心管,再加4mllb冲洗一次板子,故最终10块24cm皿板子收集到约120ml菌液于4个50ml离心管2、4000g4度离心15min后弃上清,称重菌的重量3、根据qiagen的1个maxi柱子(最大承重500ug)能够富集0.45g菌,故取相应数目的柱子提取sgcd8a质粒。
50.三、sgrna文库慢病毒的制备与浓缩day0:铺4e6293t细胞于10cm皿,培养24h;day1:细胞转染1、取500μl的opti-mem培养基放入无菌ep中,在opti-mem中加入30μlpei后斡旋混匀。
51.2、在另一个ep管中加入500μl的opti-mem后,加入三种质粒(sgcd8a文库质粒3.8μg、pspax24μg和pmd2.g2.2μg),完全混合;3、将530μlopti-mem/pei混合加入质粒管中,轻轻混合后在室温下静置15分钟;4、弃10cm皿细胞的上清,加入5mlopti-mem,将步骤2中孵育好的转染物逐滴加入;5、6~8h后换20�s和viralboostreagent(500x,alstemcat#vb100)的dmem继续培养48h。
52.day3:浓缩病毒1、收集转染48h的293t细胞的慢病毒上清,放入10ml注射器,通过0.45μm低吸附性滤膜的过滤器过滤去除细胞碎片。
53.2、将慢病毒上清与慢病毒浓缩液(alstemcat#vc100)按4:1比例混合,4度孵育过夜。
54.3、于4度,1500g离心30min后弃上清4、用原上清液体积的1/100的pbs重悬病毒颗粒后50ul/管分装,冻存于-80保存。
55.四、优化后的slice技术对t细胞进行基因组编辑day0(包被抗体):1、将1μg/mlanti-humancd3(tonbo,#40-0038)和1μg/mlanti-humancd28(tonbo,#40-0289)加入1
×
pbs中混匀,按10ml/10cmdish加到10cmdish中,4℃静置过夜
day1(分离cd3
t细胞):1、使用ficoll-paqueplus密度梯度分离液(cytiva,17144002)从健康人外周血中分离pbmc;2、弃掉10cmdish中的pbs,加入9ml/孔培养基(250mlofrpmi-1640 250mlofx-vivo15media 5mlglutamine 10�s 5mlp/s),放入37℃预热;3、使用humancd3
t细胞分离试剂盒(biolegend,480131)从pbmc中分离cd3
t细胞,取少许细胞与台盼蓝混匀后计数;4、按细胞每孔(1~5)
×
107细胞铺入10cmdish。
56.day2(培养cd3
t细胞):1、向10cmdish中加入il-2(500iu/ml)后用移液器轻柔的吹匀细胞;2、将1μg/mlanti-humancd3(tonbo,#40-0038)和1μg/mlanti-humancd28;(tonbo,#40-0289)加入1
×
pbs中混匀,按10ml/10cmdish加到10cmdish中,4℃静置过夜;day3(再次活化cd3
t细胞):弃掉day2包被抗体的10cmdish中的pbs,将原10cmdish中的细胞液转移到day2包被抗体的10cmdish中,轻柔的吹匀细胞;day4(文库慢病毒感染cd3
t细胞);1、若cd3
t细胞较day3发生了明显的增殖,则收集细胞于15ml离心管中,1200rpm离心3min;2、用0.45μm的滤器过滤细胞上清液于新15ml离心管中待用;3、取适量培养基重悬细胞后,取少许细胞与台盼蓝混匀后计数;4、将(5-8)
×
106细胞/孔的细胞数,步骤2过滤的培养基,按体积比1:50添加文库病毒,8μg/ml的polybere这些成分按2ml/孔的总体积按需配置mix后混匀平均加入六孔板,常温2000g离心1.5h后放置37℃培养箱培养3.5h后,向每孔添加含500iu/mlil-2的培养基1ml。
57.day5(电转nls-cas9-gfp蛋白):1、慢病毒感染24h后收集所有孔的细胞,1200rpm离心3min,弃上清;2、取适量培养基重悬细胞后,取少许细胞与台盼蓝混匀后计数;3、取出p3primarycell4d-nucleofectortmxkitl(lonza,v4xp-3024)中的电转杯,电转液和吸管,按说明书按需配置电转液置于冰上;4、打开电转仪,找到程序eh-115待用;5、向电转液中加入4μmnls-cas9-gfp蛋白(金斯瑞,z03467-1)混匀后,按1
×
107细胞/100μl电转液重悬细胞并加入电转杯;6、进行电转后,向电转杯中加入500μl不含il-2的培养基,轻柔混匀后置于37℃培养箱静置30min;7、向t75瓶中加入不含il-2的18ml培养基置于37℃培养箱预热;8、用吸管一次性吸出电转杯中的细胞液,每2个电转杯中的细胞液转入1个上述t75瓶,培养;day6(检测cas9蛋白电转效率):1、电转24h后,用移液器轻柔吹匀细胞,取10μl与等量台盼蓝混匀计数;
2、取适量细胞,加入cd4、cd8a、dapi抗体常温染色15min后流式检测cas9蛋白电转效率;3、向培养基中再补加如下成分后继续培养细胞2天(后续全用该培养基):50um2-me10mmhepes1mmsodiumpyruvate100umneaa10ng/mlil-155ng/mlil-7day8(puro筛选):1、收集细胞于15ml离心管,1200rpm离心3min,用0.45μm的滤器过滤细胞上清液于新50ml离心管中待用;2、将一半上述过滤的培养基与一半含上述添加物的新培养基混匀(conditionmedium)后重悬细胞,加入1μg/mlpuro,细胞密度维持在106cells/ml。
58.day9(检测cas9蛋白编辑效率):1、用移液器轻柔吹匀细胞,取10μl与等量台盼蓝混匀计数;2、取适量细胞加入cd8a、dapi抗体常温染色15min后流式检测cas9蛋白编辑效率。
59.day10(换新鲜的含puro的培养基):收集细胞于15ml离心管,1200rpm离心3min,弃上清换新鲜含上述添加物的培养基培养细胞,每日观察细胞增殖情况,后续每2天换一次含1μg/mlpuro的培养基。
60.本实施例中,还研究了sgcd8a的慢病毒感染和不同浓度的cas9蛋白电转,通过检测cas9蛋白电转效率与编辑效率(见图8和9)以确定cas9蛋白的使用浓度,随后进行puro筛选,只让sgcd8a感染的细胞进行大量增殖。
61.图8显示:20e6或10e6细胞分别与4μmnls-cas9-gfp蛋白溶于100μl电转buffer后进行电转,电转24h后流式检测gfp的阳性率,结果表明1e7数量的细胞电转效率81.3%,远高于2e7数量的细胞电转效率64.3%,(图8最左侧为空白对照,对细胞进行了电转操作所有流程,但没有加入cas9蛋白)。
62.图9显示:10e6细胞分别与1,3,5μmnls-cas9-gfp蛋白溶于100μl电转buffer后进行电转,电转24h后流式检测gfp的阳性率,结果显示nls-cas9-gfp蛋白用量越多,电转效率越高,(图9上面左侧图为空白对照,对细胞进行了电转操作所有流程,但没有加入cas9蛋白,图9上面左侧图为阳性质粒对照,阳性质粒为电转试剂盒里面自带的gfp阳性质粒)。
63.图10显示:10e6细胞分别与1,3,5μmnls-cas9-gfp蛋白溶于100μl电转buffer后进行电转,电转72h后加puro筛选24h后流式检测gfp蛋白的编辑效率。结果表明5μmcas9蛋白的编辑效率达到80%以上。
64.对比例1:按照marson团队公开的方法(shifrute,carnevalej,tobinv,rothtl,woojm,buict,lipj,etal.genome-widecrisprscreensinprimaryhumantcellsrevealkeyregulatorsofimmunefunction.cell2018;175:1958-1971e1915.)进行实验,结果显示t细胞被编辑后无法继续增殖,细胞停滞,我们通过对细胞的动
态计数体现这一差异(见图6和图7)。
65.图6显示,使用优化前的t细胞培养体系去对t细胞进行基因组crispr敲除筛选,在day4电转cas9蛋白后至day7的时间中t细胞接近死亡30%以上,在day7加入puro筛选后细胞数目进一步下降,至day9保持稳定,但是一直到day13都没有发生增殖,于是我们延长了培养时间,并增加血清浓度至15�s,一直培养到day20,细胞无法增殖。
66.图7显示,使用本发明的优化后的t细胞培养体系去对t细胞进行基因组crispr敲除筛选,在day4电转cas9蛋白24h后,我们换用15%血清的以及添加了多种营养成分的培养基,如图可见一直到day7的时间中虽然电刺激让t细胞能够发生40%以上的死亡,但细胞数目明显增多,说明细胞仍在增殖。接着我们在day7加入puro筛选,到day9时细胞受到压力,只发生了微弱增殖,后续我们每两天换一次加了puro的新培养基,至13天细胞数目大大增加,说明细胞状态完全恢复能够大量扩增。
67.实施例2:以敲除t细胞中的rgs1基因文献中报道的负调控t细胞向肿瘤迁移的因子作为实施例,证实我们优化的slice技术能够用于构建我们的调控t细胞向肿瘤迁移的基因筛选模型。
68.一、设计并合成四条sgrgs1,序列如下:rgs1sgrna-1fcaccggttcttctctgctaacccaa(seqidno.9)rgs1sgrna-1raaacttgggttagcagagaagaacc(seqidno.10)rgs1sgrna-2fcaccggatgtgggatcatagatctc(seqidno.11)rgs1sgrna-2raaacgagatctatgatcccacatcc(seqidno.12)rgs1sgrna-3fcaccgctgctgctgaagtaatgcaa(seqidno.13)rgs1sgrna-3raaacttgcattacttcagcagcagc(seqidno.14)rgs1sgrna-4fcaccgggagaatattgagttctggc(seqidno.15)rgs1sgrna-4raaacgccagaactcaatattctccc(seqidno.16)二、制备sgrgs1质粒方法同实施例1。
69.三、sgrna文库慢病毒的制备与浓缩方法同实施例1。
70.四、优化后的slice技术对t细胞进行基因组编辑方法同实施例1,分别使用cd3
t细胞、cd4
t细胞和cd8
t细胞进行实验验证。
71.五、transwell实验1、确定体外培养14天中t细胞表面cxcr3动态表达水平及t细胞响应cxcl9/10的迁移情况为了方便实验条件摸索,我们选择用外周血pbmc按步骤二的细胞培养条件,一直用培养基(250mlofrpmi-1640 250mlofx-vivo15media 5mlglutamine 10�s 5mlp/s 500iu/mlil-2)在体外培养的多个时间点(0-20天)分别取细胞检测cxcr3的表达水平;2、在不同时间点取培养的细胞进行transwell迁移实验,取1e6细胞加入transwell小室中,将600μl培养基(对照)或600μl补充有100ng/mlcxcl9或cxcl10(r&d,392-mg-010或266-ip-010)的迁移培养基加入下室。使细胞在37
°
c和5%co2中迁移4小时。收集下室的细胞,加入适量的countingbeads(biolegend#424902)后进行流式分析
细胞数。从而观察cxcr3表达水平与t细胞迁移效率的关系(图1-4),结果显示在体外培养过程中cxcr3的表达水平先升高后下降。
72.3、确定适合大规模的t细胞迁移的小室规格和孔径实验结果进行说明t细胞通过3μm和5μm孔径的小室向cxcl9/10迁移没有差异(图5)。因此我们决定使用3μm孔径的75mm直径的小室(corning,3420),对于大批量细胞的transwell实验操作更为方便,减少误差。
73.4、依据上述条件,在t细胞体外培养及基因编辑过程中,选择11-15天之内对细胞进行transwell迁移实验:取上述细胞2
×
107于10ml培养基中混匀后加入transwell小室,将600 μl培养基或600μl补充有 100 ng/ml cxcl9、cxcl10(r&d,392-mg-010和 266-ip-010)的培养基(与重悬细胞的培养基相同)加入下室。使细胞在 37
°
c 和 5% co
2 中迁移 4小时,同时以未被编辑的t细胞作为对照。结果显示,与未被编辑的t细胞相比,rgs1基因敲除的t细胞向趋化因子cxcl9或cxcl10迁移的细胞数目明显增多(图11,左图);与未被编辑的t细胞相比,rgs1基因敲除的t细胞向趋化因子cxcl9或cxcl10迁移的率明显提高(图11,右图)。
74.对比例2:按照marson团队公开的方法(shifrut e, carnevale j, tobin v, roth tl, woo jm, bui ct, li pj, et al. genome-wide crispr screens in primary human t cells reveal key regulators of immune function. cell 2018;175:1958-1971 e1915.)进行实验,结果显示t细胞被编辑后无法继续增殖,细胞停滞,不能够有足够的细胞进行transwell实验。
75.以上结果显示,通过本发明的方法可以体外鉴定免疫细胞向肿瘤迁移的负调控因子,直接通过比较基因未敲除时的细胞迁移效率和基因敲除后的细胞迁移效率来确定目的基因是否为免疫细胞向肿瘤迁移的负调控因子,若基因敲除后,细胞迁移效率提高,则该目的基因鉴定为免疫细胞向肿瘤迁移的负调控因子。
76.随后又使用liu human crispr knockout library替换实施例2中的sgrgs1,在transwell实验后分别收集上室和下室中的细胞,提取rna后,通过pcr扩增基因组中的sgrna序列,最后对sgrna产物纯化后进行二代测序。pcr产物属于碱基不平衡文库,为了减少碱基不平衡对测序结果的影响,测序时混入5%的phix文库。测序采用 hiseq xten-pe150 策略,测序数据量要求不低于1000个拷贝的文库sg rna 序列数,最终数据量为2g clean data/文库。测序过程由北京诺禾致源生物有限公司完成。在python中分析实验组较对照组sgrna富集的序列与对应的基因,从而实现体外筛选免疫细胞向肿瘤迁移的负调控因子。
77.以上对本发明做了详尽的描述,其目的在于让熟悉此领域技术的人士能够了解本发明的内容并加以实施,并不能以此限制本发明的保护范围,凡根据本发明的精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。