1.本发明涉及生物化工技术领域,尤其涉及一种透明质酸裂解酶及其基因表达和应用。
背景技术:
2.透明质酸是一种广泛存在于高等动物和低等动物细胞外间质中的高分子量聚多糖,其基本结构是由d-葡萄糖醛酸及n-乙酰氨基葡萄糖为二糖单元组成的多糖,广泛地应用在化妆品、食品和医药领域。不同分子量的的作用和功能不同。高分子量透明质酸(》160万da)在皮肤表面形成致密防护膜,起到长效保湿和良好的修复作用;中分子量透明质酸(20万da~160万da)起到保湿、润滑性、缓释和稳定乳化的作用;低分子量透明质酸(1万da~20万da)起到营养肌肤额持久保湿作用;透明质酸寡糖(《10000da)能够透皮吸收进行深层保湿,具有抗衰老、晒后修复等作用。超低分子量的透明质酸(《2000da)能够渗入真皮层,补充真皮层水分,除皱嫩肤,增加皮肤弹性;与皮肤细胞紧密结合,清除细胞内自由基,修复受损的胶原蛋白,促进伤口愈合。因此,超低分子量透明质酸在食品保健、化妆品及临床医疗领域有广阔的应用前景,近年来市场对超低分子量需求也逐年增加。
3.目前制备低分子量透明质酸主要是通过物理、化学和酶降解方法将大分子透明质酸降解为低分子量透明质酸。其中,物理法降解主要通过加热、机械剪切、紫外线、超声波、γ-射线辐射和高压均质等物理因素导致透明质酸的降解,但该方法得到的产品的稳定性差,分子量分布不均匀,降解效率较低;化学降解法主要包括碱水解、水解和氧化降解,虽然一定程度上能达到控制产物分子量目的,但由于不同化学试剂的降解条件复杂,造成产品性质易受影响和产品纯化困难,且存在废液污染的问题。酶法降解由于其反应条件温和,操作简单,且多糖的结构不发生变化,是制备低分子量透明质酸的理想方法。而超低分子量透明质酸只能通过酶解透明质酸制备。但目前透明质酸酶存在酶解产物分子量不均一、酶解转化效率低或酶的活性维持所需的温度范围和ph范围窄、且需要特定的缓冲体系,产物分离步骤繁琐等问题。因此,发掘酶解产物分子量小且均一、催化效率高、具有良好温度、ph稳定性和转化体系及回收方法简单的透明质酸酶(或裂解酶)具有重要的应用价值。
技术实现要素:
4.针对现有酶解制备超低分子量透明质酸存在的上述问题,本发明提供一种透明质酸裂解酶及其基因表达和应用,该透明质酸裂解酶具有酶活和催化效率高、稳定性好、酶解产物的分子量小且均一,在纯水中反应并回收简单,无需去除盐分的优势。
5.为达到上述发明目的,本发明实施例采用了如下的技术方案:
6.一种透明质酸裂解酶,氨基酸序列如seq id no.1。
7.相对于现有技术,本发明提供的透明质酸裂解酶的转化效率高,在适宜的酶解条件下,短时间内的酶解转化率就可达到100%;酶活稳定性好,在20℃-60℃以及ph4-10的条件下均能保持较高的酶解活性和转化效率,在水中即可进行酶解反应。酶解产物的分子量
小且均一,均为不饱和透明质酸二糖,分子量均低于《500da,产物纯度高。即本发明提供的透明质酸裂解酶的转化效率高,活性稳定性好,保持酶解活性所需的温度和ph范围宽,酶解产物的分子量小且均一,可在纯水中反应并且产物回收简单,无需除盐的优势,适用于大规模工业化生产。
8.本发明还提供所述透明质酸裂解酶的编码基因,该编码基因的核苷酸序列如seq id no.2。
9.本发明还提供了一种重组表达载体,该重组表达载体是在表达载体中插入所述透明质酸裂解酶的编码基因得到。
10.优选的,所述表达载体为质粒pet28a-sumo。
11.本发明还提供了一种表达菌体,该表达菌体是在宿主细胞中转入所述重组表达载体得到。
12.优选的,所述宿主细胞为大肠杆菌bl21(de3)。
13.本发明还提供了所述透明质酸裂解酶、所述透明质酸裂解酶的编码基因、所述重组表达载体或所述表达菌体在制备不饱和透明质酸二糖中的应用。
14.上述透明质酸裂解酶可制备得到分子量均一、纯度高和收率高的不饱和透明质酸二糖。
15.本发明还提供了一种制备不饱和透明质酸二糖的方法,该方法为:向水相体系中加入透明质酸钠和所述透明质酸裂解酶,在温度为20℃-60℃、ph为4-10的条件下进行酶解反应,得到所述不饱和透明质酸二糖。
16.优选的,所述水相体系为纯水或氯化钙水溶液。
17.优选的,所述酶解反应的温度为40℃。
18.优选的,酶解反应的ph为7。
附图说明
19.图1是本发明实施例3中的质粒pacyc duet-1-ulp的构建和结构示意图;
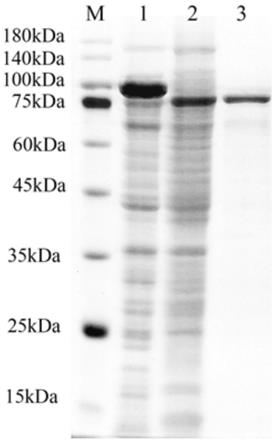
20.图2是本发明实施例4中对纯化得到的透明质酸裂解酶进行的蛋白胶检测图;其中,m:maker;1:带有sumo标签的sahyal粗酶;2:切除sumo标签的sahyal粗酶;3:纯化的重组酶;
21.图3是本发明实施例5中检测的透明质酸裂解酶在不同温度下的酶活曲线图;
22.图4是本发明实施例5中检测的透明质酸裂解酶在不同ph下的酶活曲线图;
23.图5是本发明实施例5中检测的透明质酸裂解酶的温度稳定性曲线图;
24.图6是本发明实施例5中检测的透明质酸裂解酶的ph稳定性曲线图;
25.图7是本发明实施例8中的酶解产物的tlc分析图;
26.图8是本发明实施例8中的酶解产物的hplc检测图;
27.图9是本发明实施例8中利用hplc对透明质酸裂解酶水解透明质酸钠在不同时间点的转化率监测图;
28.图10是本发明实施例8中酶解终产物的hplc检测图;
29.图11是本发明实施例8中酶解终产物的lc-ms/ms质谱分析图。
具体实施方式
30.为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
31.实施例1
32.一种透明质酸裂解酶,氨基酸序列如seq id no.1,该透明质酸裂解酶具体是从实验室筛得的一株苜蓿链霉菌(streptomyces alfalae)中得到。
33.上述透明质酸裂解酶的编码基因的获得:
34.以上述筛得的苜蓿链霉菌(streptomyces alfalae)的基因组为模板(dna模板),设计上下游引物并预测引物退火温度,在上游引物5
′
端加入bamhi限制性酶切位点和保护碱基,下游引物5
′
端引入xhoi限制性酶切位点和保护碱基,引物序列下:
35.sahyal-f:(bamhi)5
′‑
ttaagaattcgccagggccgccgagg-3
′
(seq id no.3);
36.sahyal-r:(xhoi)5
′‑
ccgctcgaggccccgcagggtcacc-3
′
(seq id no.4)。
37.利用pcr仪进行大量扩增,dna聚合酶试剂盒购自北京全式金生物技术有限公司,pcr反应体系见表1,pcr扩增程序见表2,将扩增后得到的目的基因利用0.8%的琼脂糖凝胶进行检测,然后切胶回收,进行纯化,即得到上述透明质酸裂解酶的编码基因,其核苷酸序列如seq id no.2。
38.表1 pcr体系
[0039][0040]
表2 pcr扩增程序
[0041][0042]
实施例2
[0043]
上述透明质酸裂解酶的重组表达载体的构建:
[0044]
1)双酶切实施例1中扩增、纯化得到的透明质酸裂解酶的编码基因和载体pet28a-sumo。
[0045]
利用限制性内切酶bamhi和xhoi将透明质酸裂解酶的编码基因和载体pet28a-sumo分别进行双酶切,双酶切体系如表3所示。将双酶切体系分别置于37℃水浴3h,结束后分别将上述酶切体系加样到0.8%琼脂糖凝胶中检测,并将正确大小的酶切基因片段和pet28a-sumo载体条带进行切胶回收与纯化。
[0046]
表3 酶切体系
[0047][0048]
2)酶切基因片段与线性化pet28a-sumo载体进行酶连。
[0049]
将纯化的酶切基因片段和线性化载体pet28a-sumo通过t4 dna连接酶连接,酶连体系如表4所示,将酶连体系在16℃的条件下连接12h。
[0050]
表4 酶连体系
[0051][0052]
3)转化大肠杆菌感受态细胞dh5α。
[0053]
将8μl上述酶连体系和80μl大肠杆菌感受态细胞dh5α混匀后加入培养管中,在冰上静置20min,42℃加热45s后,冰上静置2min,再向管中加入无抗性的400μl液体lb,置于37℃摇床180rpm培养60min。取200μl培养的菌液均匀涂布在抗性为kan的固体lb平板上,37℃恒温倒置培养过夜。
[0054]
4)阳性克隆筛选。
[0055]
挑取上述平板上的单克隆分别于含有kan抗性的lb液体培养基中37℃摇床220rpm培养过夜,收集各菌株菌体并提取质粒。利用限制性内切酶bamhi,xhoi进行双酶切验证,双酶切验证体系如表5所示,37℃水浴锅孵育2h,挑取测序正确的质粒保存,该质粒即为插入上述透明质酸裂解酶的编码基因的重组表达载体(pet28a-sumo-sahyal)。
[0056]
表5 双酶切验证体系
[0057][0058]
实施例3
[0059]
表达菌体的获得以及透明质酸裂解酶的表达。
[0060]
利用双质粒共表达的方法将质粒pacyc duet-1-ulp(用于表达不带his标签的sumo蛋白酶ubiquitin-like protein-specific protease ulp,可特异性识别并去除sumo标签,pacyc duet-1-ulp质粒的结构图如图1所示,是在pacyc duet质粒中插入表达ulp的基因序列得到)和重组质粒pet28a-sumo-sahyal通过热激法同时转化到感受态细胞bl21(de3)中,加入400μl无抗lb液体培养基,37℃,180rpm培养60min。取80μl上述菌液均匀涂布在双抗(同时含有kan和cm抗性)性固体lb平板上,37℃倒置培养过夜。挑取单菌落于2ml的lb液体培养基中(同时含有kan和cm抗性)。37℃、200rpm培养过夜,然后按1%的体积接种量接种在200ml的lb液体培养基中(同时含有kan和cm抗性),20℃、200rpm培养到菌液od600=0.6时,按照1:1000的体积比向培养液中加入浓度为100mm的iptg诱导蛋白表达,诱导时间为24h。
[0061]
实施例4
[0062]
透明质酸裂解酶的纯化。
[0063]
利用离心机对实施例3中的iptg诱导蛋白表达体系6000rpm离心10min,收集菌体,用10ml的ni-nta平衡缓冲液对菌体进行洗涤,再用8ml的ni-nta平衡缓冲液将菌体涡旋震荡直到没有菌块,得到菌悬液。对菌悬液进行超声破碎,超声3s,间隔5s,总超声破碎时间为35min,得到细胞裂解液。将细胞裂解液12000rpm、4℃离心15min,离心处理后收集上清液放置于冰上。
[0064]
上清液(粗酶液)采用镍柱亲和层析的方法进行纯化。首先对镍柱利用5倍柱体积预冷的ni-nta平衡缓冲液进行预处理,放置于4℃的冰箱中,取上述置于冰上的上清加入到预处理的镍柱中,每次加入2ml的粗酶液,4℃孵育6min之后释放流穿液。重新加入2ml粗酶液进行纯化,重复上述步骤直到所有上清液都挂完柱子。然后利用ni-nta洗涤缓冲液对杂蛋白进行洗涤,每次加入2ml,洗涤总体积为20ml,再加入5ml 250mm的ni-nta和5ml350mm的ni-nta洗脱缓冲液对结合在镍柱上的目的蛋白进行洗脱与收集,并利用50kda的超滤柱对纯化的蛋白进行浓缩,并利用盐浓度为50mm、ph7.0的tris-hcl替换蛋白中的洗脱缓冲液,最终纯化的目的蛋白储存在1ml 50mm ph7.0的tris-hcl缓冲液中,得到纯化酶液。纯化效果利用10%的sds-page蛋白胶进行检测,检测结果如图2所示,可以看出目的蛋白(透明质酸裂解酶)成功可溶性表达,并在体内完成了sumo标签的切割。
[0065]
实施例5
[0066]
对实施例4中纯化得到的透明质酸裂解酶的酶学性质进行检测。
[0067]
1)最适酶解温度的测定
[0068]
反应体系:50mm tris-hcl 7.0的缓冲液200μl,0.5wt.%透明质酸钠(80-150万da)200μl,实施例4中得到的纯化酶液100μl,混匀后分别在20℃,25℃,30℃,35℃,40℃,45℃,50℃,55℃,60℃,65℃,70℃,75℃和80℃下进行反应,反应时间为15min,利用500μl浓度为20mm的hcl终止反应并进行酶活测定,并将最适温度下的蛋白活性定义为100%,其他温度的蛋白活性计算为相对活性,测得当反应温度为50℃时,催化活性最高,如图3所示。
[0069]
2)最适酶解ph的测定
[0070]
反应体系:0.5wt.%的透明质酸钠(80-150万da)200μl,纯化酶液100μl,其中反应体系中200μl的缓冲液为不同ph的缓冲液。利用的缓冲液为:柠檬酸-na2hpo4缓冲液(ph4.0-6.0),nah2po
4-na2hpo4缓冲液(ph6.0
–
8.0),tris-hcl缓冲液(ph8.0-9.0)和gly-naoh缓冲液(ph9.0-10.0),上述缓冲液浓度均为50mm。将各个不同ph的反应体系,在最适温度下反应15min,利用500μl浓度为20mm的hcl终止反应并进行酶活测定,将最适ph条件下的酶活力定义为100%,其他ph条件下的酶活力计算为相对酶活,测得当ph为7.0时,催化活性最高,如图4所示。
[0071]
3)温度稳定性的测定
[0072]
反应体系:最适ph的缓冲液200μl,0.5wt.%透明质酸钠(80-150万da)200μl,纯化酶液100μl,其中酶液要先分别在20℃,25℃,30℃,35℃,40℃,45℃,50℃,55℃,60℃,65℃,70℃,75℃和80℃条件下孵育60min,然后再加入上述反应体系中,在最适条件下反应15min,利用500μl浓度为20mm的hcl终止反应并进行酶活测定,将未进行温度处理的蛋白酶活性定义为100%,将进行温度处理的酶活性计算为相对活性,测得反应温度在20℃-45℃
时,酶孵育60min后,相对活性仍然具有90%以上的活性,在45-50℃,孵育60min后仍然具有80%以上的活性,如图5所示,说明该透明质酸裂解酶表现出好的温度稳定性。
[0073]
4)ph稳定性的测定
[0074]
反应体系:最适ph缓冲液200μl,0.5wt.%透明质酸钠(80-150万da)200μl,纯化酶100μl,2.5μl终浓度为1m的cacl2。为了测定蛋白的ph稳定性,需要将酶与上述不同ph的缓冲液按1:1进行混合,然后在37℃孵育1h。将上述处理的酶分别加入到上述反应体系中,在最适反应条件下反应15min,利用500μl浓度为20mm的hcl终止反应并进行酶活测定,将未用ph处理的蛋白酶的活性定义为100%,其他不同ph条件下处理的酶的活性计算为相对活性。测得在ph5.0-10.0孵育60min仍然保持高于90%以上的活性,在ph4.0-5.0范围内孵育60min仍然有60%以上活性,如图6所示,说明该透明质酸裂解酶具有好的ph稳定性。
[0075]
实施例6
[0076]
实施例4中纯化得到的透明质酸裂解酶的底物谱。
[0077]
反应体系:0.2ml的透明质酸钠(5mg/ml,分子量80-150万da)和0.1ml的纯化酶液,加入0.2mm的tris-hcl缓冲液中(ph7.0)。在50℃孵育15min后,加入0.5ml的20mm的hcl终止反应,以含有0.1ml热灭活酶溶液的相同混合物作为空白对照。在相同反应条件和体系下,将纯化的透明质酸裂解酶与不同底物在最适反应条件下(50℃、ph7.0)进行反应,然后分别在232nm波长下测定酶活,结果如表6所示。该酶对透明质酸钠表现出最高活性,对硫酸软骨素a,硫酸软骨素b,硫酸软骨素c均表现出较低的蛋白活性,而对于肝素钠,海藻酸钠,羧甲基纤维素则没有酶活,说明该透明质酸裂解酶的底物较为特异,对透明质酸钠降解活性达到最高(152.12
±
5.17u/mg)。
[0078]
表6 透明质酸裂解酶的底物谱
[0079][0080][0081]
注:“/”代表没有酶解活性。
[0082]
实施例7
[0083]
实施例4中纯化得到的透明质酸裂解酶的动力学参数测定。
[0084]
反应体系:0.2ml的透明质酸钠(5mg/ml,80-150万da)和0.1ml的纯化酶液,加入
0.2mm的tris-hcl缓冲液中(ph7.0)。在50℃孵育15min后,加入0.5ml的20mm的hcl终止反应,以含有0.1ml热灭活酶溶液的相同混合物作为空白对照。将纯化的透明质酸裂解酶和透明质酸钠在最适反应环境下孵育不同时间后,测定生成的产物量,确定了透明质酸裂解酶降解透明质酸钠反应时间为15min时,其反应速度达到最大。并根据linewaver-burk作图法,计算出透明质酸裂解酶对透明质酸钠的动力学参数,如表7所示。分析结果可知,透明质酸裂解酶降解透明质酸钠的米氏常数km为0.31mg/ml,最大反应速度vmax为135.14μmol/min/mg,转化率kcat为201.36s-1
,催化效率kcat/km为647.87mg/ml/s。
[0085]
表7 透明质酸裂解酶的动力学参数
[0086][0087]
实施例8
[0088]
实施例4中纯化得到的透明质酸裂解酶的产物分析。
[0089]
将0.1ml的透明质酸裂解酶(纯化酶液)和透明质酸钠(0.2ml,5mg/ml,80-150万da)同时加入反应体系中,37℃、200rpm摇床中进行水解,分别在反应0min、1min、2min、10min、30min和60min时在总反应体系中取出500μl,在100℃的环境下加热处理5min,使反应终止。利用薄层层析法(tlc)对产物进行检测分析,结果如图7所示,同时将上述不同时间点的产物利用hplc进行检测,结果如图8所示,由tlc和hplc检测结果可知透明质酸裂解酶水解透明质酸钠的终产物始终仅有一种不饱和透明质酸二糖,且该透明质酸裂解酶是一种外切酶,从底物末端开始逐渐水解产生不饱和二糖产物,直到多聚透明质酸钠完全被降解。
[0090]
将45g的透明质酸钠(80-150万da)加入到900ml水中,置于37℃、200rpm的摇床,加入100mg透明质酸裂解酶(纯化酶液),利用hplc对透明质酸裂解酶水解透明质酸钠24h的产物进行检测与分析,如图9所示,发现反应初期透明质酸钠迅速降解,6h时转化率达到最大。当转化率达到最大时对产物进行收集与纯化。利用3kda超滤膜去除体系中的蛋白物质,之后100℃加热10分钟灭活处理,利用喷雾干燥去除产物水分,最终得到的不饱和透明质酸二糖的收率为97.4%、纯度≥98.5%,hplc检测只有单一的产物峰,且出峰时间与不饱和二糖标品出峰时间一致,如图10所示,说明该透明质酸裂解酶降解透明质酸终产物只有透明质酸二糖一种产物。
[0091]
同时,利用lc-ms/ms对透明质酸裂解酶降解透明质酸钠产物分析,结果如图11所示,在产物检测峰中只有一个主峰,其质荷比m/z为378.10,与不饱和透明质酸二糖大小一致,且通过质谱图发现并没有不饱和四糖和不饱和六糖等产物峰出现,说明该透明质酸裂解酶降解透明质酸钠的终产物只有单一产物,为不饱和透明质酸二糖。
[0092]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换或改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。