1.本发明涉及化学领域。更具体地说,本发明涉及一种新型手性硫醚-亚磺酰亚胺配体及其制备方法。
背景技术:
2.在过去的几十年中,不对称催化领域得到了广泛的关注。其中,新型手性配体的设计与合成是实现催化不对称合成的关键因素之一。甚至在很多方面,新型配体的发展成为化学反应实现工业化的决定因素。化学家们发展了成百上千种手性配体,其中一些手性配体已经被成功应用于工业,用于药物中间体的绿色合成。尽管如此,仍然还有大量的手性药物或手性中间体的合成缺少合适的催化体系,因此开发更多高效、新型的手性配体并将其应用于不对称合成中,仍然是一个现实并且具有挑战性的任务。
技术实现要素:
3.本发明的一个目的是解决至少上述问题,提供了一种新型手性硫醚-亚磺酰亚胺配体及其制备方法,并把这类配体应用于钯催化的不对称烯丙基化反应中。
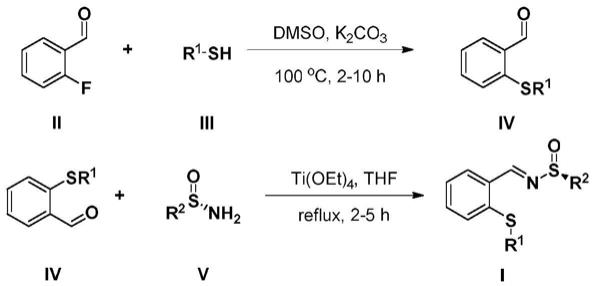
4.为了实现根据本发明的这些目的和其它优点,提供了一种新型手性硫醚-亚磺酰亚胺配体,其特征在于,该类化合物的结构式如通式i所示,
[0005][0006]
其中:r1表示4-甲基苯基、4-氯苯基、2-甲氧基苯基、2,6-二甲苯基、苄基、叔丁基中任意一种,r2表示叔丁基、4-甲基苯基中任意一种。
[0007]
本发明还提供了新型手性硫醚-亚磺酰亚胺配体的制备方法,包括以下步骤:
[0008]
将2-氟苯甲醛、硫酚或硫醇、碳酸钾混合,加入二甲亚砜,一定温度下进行反应,反应完成后抽滤脱去溶剂,滤饼用乙酸乙酯洗涤,滤液加入饱和氯化铵溶液,分液后水相用乙酸乙酯萃取,合并有机相用无水硫酸钠干燥,然后抽滤,硅胶柱层析纯化得到中间体iv;取所得中间体iv加入亚磺酰胺v、无水四氢呋喃混合反应,再逐步加入钛酸四乙脂、饱和氯化钠、乙酸乙酯,然后抽滤,滤饼用乙酸乙酯洗涤,滤液用饱和氯化铵洗涤,然后用饱和氯化钠洗涤,有机相无水硫酸钠干燥,然后抽滤,硅胶柱层析纯化得到手性硫醚-亚磺酰亚胺化合物i。
[0009]
优选的是,所述2-氟苯甲醛、硫酚或硫醇、碳酸钾的摩尔比为1:1:2。
[0010]
优选的是,所述2-氟苯甲醛、硫酚或硫醇、碳酸钾混合反应温度为100℃,反应时间为2-10小时。
[0011]
优选的是,所述中间体iv柱层析分离时洗脱液比例为,石油醚:乙酸乙酯=50:1。
[0012]
优选的是,所述中间体iv、亚磺酰胺v、钛酸四乙脂的摩尔比为1.5:1.0:1.05。
[0013]
优选的是,所述手性硫醚-亚磺酰亚胺化合物柱层析分离时洗脱液比例为,石油醚:乙酸乙酯=20:1。
[0014]
优选的是,新型手性硫醚-亚磺酰亚胺与烯丙基氯化钯二聚体反应形成的络合物能够作为不对称烯丙基烷基化反应催化剂。
[0015]
本发明至少包括以下有益效果:
[0016]
本发明的新型手性硫醚-亚磺酰亚胺配体,首先是由2-氟苯甲醛ii和硫酚iii反应得到2-硫醚苯甲醛iv,再由2-硫醚苯甲醛iv与手性亚磺酰胺v脱水缩合,得到目标产物手性硫醚-亚磺酰亚胺配体i。本发明的新型手性硫醚-亚磺酰亚胺与烯丙基氯化钯二聚体形成的络合物对不对称烯丙基烷基化反应具有良好的催化活性,可作为该类反应高选择性的手性配体。
[0017]
本发明的其它优点、目标和特征将部分通过下面的说明体现,部分还将通过对本发明的研究和实践而为本领域的技术人员所理解。
具体实施方式
[0018]
下面通过实施例来具体说明本发明的新型手性硫醚-亚磺酰亚胺配体i类化合物的制备方法。这些实例仅对本发明进行说明,而不对本发明进行限制。
[0019]
本发明提供了一种新型手性硫醚-亚磺酰亚胺配体的制备方法,该类化合物的制备方法包括以下步骤:
[0020]
用1.0当量的2-氟苯甲醛与1.0当量的硫酚(醇)和2.0当量的碳酸钾,以二甲亚砜为溶剂,在100℃下反应2-10小时,反应完成后抽滤脱去溶剂,固体用乙酸乙酯洗涤,滤液加入饱和氯化铵溶液,分液后水相用乙酸乙酯萃取,合并有机相用无水硫酸钠干燥,然后抽滤,滤液拌硅胶粉在减压下脱去溶剂,过硅胶柱,以石油醚/乙酸乙酯(50:1)进行柱层析分离,脱去层析液后得到中间体iv;
[0021]
取1.5当量所得硫醚中间体iv再与1.0当量的亚磺酰胺v溶于无水四氢呋喃中,加热至回流后逐滴加入1.05当量钛酸四乙脂,回流2-5个小时,反应完成后,降至室温,逐滴加入饱和氯化钠至产生大量沉淀,加入乙酸乙酯后搅拌10分钟,体系过硅藻土抽滤,滤饼用乙酸乙酯洗涤,滤液用饱和氯化铵洗涤,然后用饱和氯化钠洗涤,有机相无水硫酸钠干燥,然后抽滤,滤液拌硅胶粉在减压下脱去溶剂,以石油醚/乙酸乙酯(20:1)进行柱层析分离,减压脱去层析液后得到手性硫醚-亚磺酰亚胺化合物i。
[0022]
化学反应方程式如下所示:
[0023]
[0024]
其中:r1表示4-甲基苯基、4-氯苯基、2-甲氧基苯基、2,6-二甲苯基、苄基、叔丁基中任意一种,r2表示叔丁基、4-甲基苯基中任意一种。
[0025]
实施例1:i-1的制备
[0026][0027]
在室温下,将2-氟苯甲醛(50mmol)、碳酸钾(100mmol)与4-甲基苯硫酚(50mmol)溶解于50ml二甲亚砜溶液中,加热至100℃,反应完成后抽滤,滤饼用乙酸乙酯(3x20ml)洗涤,滤液加入100ml饱和氯化铵溶液后转入分液漏斗分液,收集有机相,水相用乙酸乙酯洗涤(3x20ml),合并有机相后无水硫酸钠干燥,然后抽滤,滤液拌硅胶粉在减压下脱去溶剂,过硅胶柱,以石油醚/乙酸乙酯(50:1)进行柱层析分离,减压脱去层析液后得到中间体iv-1,该化合物为淡黄色固体,收率为85%。
[0028]
将硫醚中间体iv-1(7.5mmol)溶解在50ml无水四氢呋喃中,然后加入s-叔丁基亚磺酰胺(5mmol),升温至回流,然后逐滴加入钛酸四乙脂(5.25mmol),利用薄层色谱法监测反应,反应完成后,降至室温,逐滴加入饱和氯化钠至产生大量沉淀,加入乙酸乙酯(20ml)后搅拌10分钟,体系在减压下过1厘米厚硅藻土抽滤,滤饼用乙酸乙酯洗涤,滤液用饱和氯化铵(3x20ml)洗涤,然后用饱和氯化钠洗涤,有机相无水硫酸钠干燥,然后抽滤,滤液拌硅胶粉在减压下脱去溶剂,快速过硅胶柱,以石油醚/乙酸乙酯(20:1)进行柱层析分离,减压脱去层析液后得到中间体i-1,该化合物为白色固体,收率为53%。
[0029]
核磁数据:1h nmr(400mhz,cdcl3)δ9.27(s,1h),7.97-7.86(m,1h),7.64-7.57(m,2h),7.36-7.20(m,8h),7.20-7.06(m,3h),2.40(s,3h),2.36(s,3h).
13
c nmr(100mhz,cdcl3)δ159.12,141.76,141.59,140.66,138.18,132.84,132.59,132.33,131.11,130.46,130.32,130.26,129.78,126.59,124.86,21.44,21.19.[α]
d30
=111.867(c=0.1,ch2cl2)。比旋光为[α]
d30
=111.867(c=0.1,ch2cl2)。
[0030]
实施例2:i-2的制备
[0031][0032]
在室温下,将2-氟苯甲醛(50mmol)、碳酸钾(100mmol)与4-甲基苯硫酚(50mmol)溶
解于50ml二甲亚砜溶液中,加热至100℃,反应完成后抽滤,滤饼用乙酸乙酯(3x20ml)洗涤,滤液加入100ml饱和氯化铵溶液后转入分液漏斗分液,收集有机相,水相用乙酸乙酯洗涤(3x20ml),合并有机相后无水硫酸钠干燥,然后抽滤,滤液拌硅胶粉在减压下脱去溶剂,过硅胶柱,以石油醚/乙酸乙酯(50:1)进行柱层析分离,减压脱去层析液后得到中间体iv-1,该化合物为淡黄色固体,收率为85%。
[0033]
将硫醚中间体iv-1(7.5mmol)溶解在50ml无水四氢呋喃中,然后加入s-对甲苯基亚磺酰胺(5mmol),升温至回流,然后逐滴加入钛酸四乙脂(5.25mmol),利用薄层色谱法监测反应,反应完成后,降至室温,逐滴加入饱和氯化钠至产生大量沉淀,加入乙酸乙酯(20ml)后搅拌10分钟,体系在减压下过1厘米厚硅藻土抽滤,滤饼用乙酸乙酯洗涤,滤液用饱和氯化铵(3x20ml)洗涤,然后用饱和氯化钠洗涤,有机相无水硫酸钠干燥,然后抽滤,滤液拌硅胶粉在减压下脱去溶剂,快速过硅胶柱,以石油醚/乙酸乙酯(20:1)进行柱层析分离,减压脱去层析液后得到中间体i-2,该化合物为白色固体,收率为79%。
[0034]
核磁数据:1h nmr(400mhz,cdcl3)δ9.02(s,1h),7.88(dd,j=7.4,1.9hz,1h),7.35
–
7.23(m,4h),7.20-7.09(m,3h),2.36(s,3h),1.27(s,9h).
13
c nmr(100mhz,cdcl3)δ161.52,141.07,138.54,133.56,132.10,131.12,130.43,130.33,130.17,130.04,126.13,57.67,22.53,21.24.。比旋光为[α]
d30
=223.7(c=0.1,ch2cl2)。
[0035]
实施例3:i-3的制备
[0036][0037]
在室温下,将2-氟苯甲醛(50mmol)、碳酸钾(100mmol)与4-氯苯硫酚(50mmol)溶解于50ml二甲亚砜溶液中,加热至100℃,反应完成后抽滤,滤饼用乙酸乙酯(3x20ml)洗涤,滤液加入100ml饱和氯化铵溶液后转入分液漏斗分液,收集有机相,水相用乙酸乙酯洗涤(3x20ml),合并有机相后无水硫酸钠干燥,然后抽滤,滤液拌硅胶粉在减压下脱去溶剂,过硅胶柱,以石油醚/乙酸乙酯(50:1)进行柱层析分离,减压脱去层析液后得到中间体iv-2,该化合物为淡黄色固体,收率为90%。
[0038]
将硫醚类中间体iv-2(7.5mmol)溶解在50ml无水四氢呋喃中,然后加入s-对甲苯亚磺酰胺(5mmol),升温至回流,然后逐滴加入钛酸四乙脂(5.25mmol),利用薄层色谱法监测反应,反应完成后,降至室温,逐滴加入饱和氯化钠至产生大量沉淀,加入乙酸乙酯(20ml)后搅拌10分钟,体系在减压下过1厘米厚硅藻土抽滤,滤饼用乙酸乙酯洗涤,滤液用饱和氯化铵(3x20ml)洗涤,然后用饱和氯化钠洗涤,有机相无水硫酸钠干燥,然后抽滤,滤液拌硅胶粉在减压下脱去溶剂,快速过硅胶柱,以石油醚/乙酸乙酯(20:1)进行柱层析分离,减压脱去层析液后得到中间体i-3,该化合物为白色固体,收率为73%。
[0039]
核磁数据:1h nmr(400mhz,cdcl3)δ9.25(s,1h),7.97(dd,j=7.1,5.2hz,1h),
7.68
–
7.46(m,2h),7.46
–
7.10(m,10h),2.38(s,3h).
13
c nmr(100mhz,cdcl3)δ158.95,141.68,141.49,138.06,133.86,133.83,133.46,132.90,132.54,132.39,130.22,129.79,129.54,127.89,124.66,21.39。比旋光为[α]
d30
=239.167(c=0.1,ch2cl2)。
[0040]
实施例4:i-4的制备
[0041][0042]
在室温下,将2-氟苯甲醛(50mmol)、碳酸钾(100mmol)与2-甲基苯硫酚(50mmol)溶解于50ml二甲亚砜溶液中,加热至100℃,反应完成后抽滤,滤饼用乙酸乙酯(3x20ml)洗涤,滤液加入100ml饱和氯化铵溶液后转入分液漏斗分液,收集有机相,水相用乙酸乙酯洗涤(3x20ml),合并有机相后无水硫酸钠干燥,然后抽滤,滤液拌硅胶粉在减压下脱去溶剂,过硅胶柱,以石油醚/乙酸乙酯(50:1)进行柱层析分离,减压脱去层析液后得到中间体iv-3,该化合物为淡黄色固体,收率为81%。
[0043]
将硫醚类中间体iv-3(7.5mmol)溶解在50ml无水四氢呋喃中,s-对甲苯基亚磺酰胺(5mmol)加入上述体系,升温至回流,然后逐滴加入钛酸四乙脂(5.25mmol),利用薄层色谱法监测反应,反应完成后,降至室温,逐滴加入饱和氯化钠至产生大量沉淀,加入乙酸乙酯(20ml)后搅拌10分钟,体系在减压下过1厘米厚硅藻土抽滤,滤饼用乙酸乙酯洗涤,滤液用饱和氯化铵(3x20ml)洗涤,然后用饱和氯化钠洗涤,有机相无水硫酸钠干燥,然后抽滤,滤液拌硅胶粉在减压下脱去溶剂,快速过硅胶柱,以石油醚/乙酸乙酯(20:1)进行柱层析分离,减压脱去层析液后得到中间体i-4,该化合物为白色固体,收率为58%。
[0044]
核磁数据:1h nmr(400mhz,cdcl3)δ9.25(s,1h),7.94(dd,j=7.6,1.8hz,1h),7.60(d,j=8.3hz,2h),7.34-7.20(m,8h),7.16-7.10(m,1h),7.03(d,j=7.8hz,1h),2.39(d,j=2.5hz,6h).
13
c nmr(100mhz,cdcl3)δ158.86,141.73,141.56,140.35,139.66,133.46,132.97,132.57,132.40,130.78,130.51,130.28,129.74,128.36,126.94,126.49,124.84,21.37,20.57。比旋光为[α]
d30
=275.967(c=0.1,ch2cl2)。
[0045]
实施例5:i-5的制备
[0046][0047]
在室温下,将2-氟苯甲醛(50mmol)、碳酸钾(100mmol)与2,6-二甲基苯硫酚(50mmol)溶解于50ml二甲亚砜溶液中,加热至100℃,反应完成后抽滤,滤饼用乙酸乙酯
(3x20ml)洗涤,滤液加入100ml饱和氯化铵溶液后转入分液漏斗分液,收集有机相,水相用乙酸乙酯洗涤(3x20ml),合并有机相后无水硫酸钠干燥,然后抽滤,滤液拌硅胶粉在减压下脱去溶剂,过硅胶柱,以石油醚/乙酸乙酯(50:1)进行柱层析分离,减压脱去层析液后得到中间体iv-4,该化合物为淡黄色固体,收率为86%。
[0048]
将硫醚类中间体iv-4(7.5mmol)溶解在50ml无水四氢呋喃中,然后加入s-对甲苯基亚磺酰胺(5mmol),升温至回流,然后逐滴加入钛酸四乙脂(5.25mmol),利用薄层色谱法监测反应,反应完成后,降至室温,逐滴加入饱和氯化钠至产生大量沉淀,加入乙酸乙酯(20ml)后搅拌10分钟,体系在减压下过1厘米厚硅藻土抽滤,滤饼用乙酸乙酯洗涤,滤液用饱和氯化铵(3x20ml)洗涤,然后用饱和氯化钠洗涤,合并有机相后无水硫酸钠干燥,然后抽滤,滤液拌硅胶粉在减压下脱去溶剂,快速过硅胶柱,以石油醚/乙酸乙酯(20:1)进行柱层析分离,减压脱去层析液后得到中间体i-5,该化合物为白色固体,收率为65%。
[0049]
核磁数据:1h nmr(400mhz,cdcl3)δ9.24(s,1h),7.86(d,j=7.3hz,1h),7.71(d,j=6.4hz,2h),7.34-7.29(m,2h),7.29-7.23(m,1h),7.21-7.16(m,3h),7.15-7.07(m,2h),6.55(dt,j=7.7,1.1hz,1h),2.39(s,3h),2.38(s,6h).
13
c nmr(100mhz,cdcl3)δ158.29,143.93,141.96,141.62,141.51,132.31,130.70,130.41,129.81,129.73,129.63,128.63,125.59,125.00,124.57,21.71,21.38。比旋光为[α]
d30
=212.083(c=0.1,ch2cl2)。
[0050]
实施例6:i-6的制备
[0051][0052]
在室温下,将2-氟苯甲醛(50mmol)、碳酸钾(100mmol)与苄硫醇(50mmol)溶解于50ml二甲亚砜溶液中,加热至100℃,反应完成后抽滤,滤饼用乙酸乙酯(3x20ml)洗涤,滤液加入100ml饱和氯化铵溶液后转入分液漏斗分液,收集有机相,水相用乙酸乙酯洗涤(3x20ml),合并有机相后无水硫酸钠干燥,然后抽滤,滤液拌硅胶粉在减压下脱去溶剂,过硅胶柱,以石油醚/乙酸乙酯(50:1)进行柱层析分离,减压脱去层析液后得到中间体iv-5,该化合物为淡黄色固体,收率为77%。
[0053]
将硫醚中间体iv-5(7.5mmol)溶解在50ml无水四氢呋喃中,然后加入s-对甲苯亚磺酰胺(5mmol),升温至回流,然后逐滴加入钛酸四乙脂(5.25mmol),利用薄层色谱法监测反应,反应完成后,降至室温,逐滴加入饱和氯化钠至产生大量沉淀,加入乙酸乙酯(20ml)后搅拌10分钟,体系在减压下过1厘米厚硅藻土抽滤,滤饼用乙酸乙酯洗涤,滤液用饱和氯化铵(3x20ml)洗涤,然后用饱和氯化钠洗涤,有机相无水硫酸钠干燥,然后抽滤,滤液拌硅胶粉在减压下脱去溶剂,快速过硅胶柱,以石油醚/乙酸乙酯(20:1)进行柱层析分离,减压脱去层析液后得到中间体i-6,该化合物为白色固体,收率为55%。
[0054]
核磁数据:1h nmr(400mhz,cdcl3)δ9.19(s,1h),7.89(d,j=7.8hz,1h),7.57-7.87(m,2h),7.48-7.40(m,1h),7.39-7.33(mz,1h),7.31-7.15(m,8h),4.06(q,j=8hz,2h),
2.38(s,3h).
13
c nmr(100mhz,cdcl3)δ159.05,141.83,141.49,138.99,136.42,134.08,132.25,131.94,129.71,129.52,128.92,128.46,127.31,126.99,124.79,40.08,21.36。比旋光为[α]
d30
=247.683(c=0.1,ch2cl2)。
[0055]
实施例7:i-7的制备
[0056][0057]
在室温下,将2-氟苯甲醛(50mmol)、碳酸钾(100mmol)与叔丁基硫醇(50mmol)溶解于50ml二甲亚砜溶液中,加热至100℃,反应完成后抽滤,滤饼用乙酸乙酯(3x20ml)洗涤,滤液加入100ml饱和氯化铵溶液后转入分液漏斗分液,收集有机相,水相用乙酸乙酯洗涤(3x20ml),合并有机相后无水硫酸钠干燥,然后抽滤,滤液拌硅胶粉在减压下脱去溶剂,过硅胶柱,以石油醚/乙酸乙酯(50:1)进行柱层析分离,减压脱去层析液后得到中间体iv-6,该化合物为淡黄色液体,收率为88%。
[0058]
将硫醚中间体iv-6(7.5mmol)溶解在50ml无水四氢呋喃中,然后加入s-对甲苯亚磺酰胺(5mmol),升温至回流,然后逐滴加入钛酸四乙脂(5.25mmol),利用薄层色谱法监测反应,反应完成后,降至室温,逐滴加入饱和氯化钠至产生大量沉淀,加入乙酸乙酯(20ml)后搅拌10分钟,体系在减压下过1厘米厚硅藻土抽滤,滤饼用乙酸乙酯洗涤,滤液用饱和氯化铵(3x20ml)洗涤,然后用饱和氯化钠洗涤,有机相无水硫酸钠干燥,然后抽滤,滤液拌硅胶粉在减压下脱去溶剂,快速过硅胶柱,以石油醚/乙酸乙酯(20:1)进行柱层析分离,减压脱去层析液后得到中间体i-7,该化合物为白色固体,收率为68%。
[0059]
核磁数据:1h nmr(400mhz,cdcl3)δ9.63(s,1h),8.11(d,j=7.1hz,1h),7.70
–
7.54(m,3h),7.49-7.38(m,2h),7.34-7.22(m,2h),2.38(s,3h),1.27(s,9h).
13
c nmr(100mhz,cdcl3)δ161.17,141.83,141.55,139.70,138.29,135.67,131.81,129.74,129.35,128.48,124.64,47.84,30.91,21.37.比旋光为[α]
d30
=313.250(c=0.1,ch2cl2)。
[0060]
实施例9:不对称烯丙基化反应
[0061]
在氮气保护下,将0.01mmol的烯丙基氯化钯二聚体和0.022mmol的新型手性硫醚-亚磺酰亚胺配体i溶于2ml乙腈中并在室温下强烈搅拌一个小时,再向体系中加入0.2mmol烯丙基醋酸酯、0.6mmol的碳酸铯,升温至40℃搅拌10分钟,0.3mmol丙二酸二甲酯加入体系反应,搅拌直至反应完全,加入10ml饱和氯化铵溶液终止反应后,用二氯甲烷萃取、无水硫酸钠干燥,抽滤,滤液拌硅胶粉脱溶,以石油醚/乙酸乙酯(5:1)进行柱层析分离,减压脱去层析液即得烯丙基烷基化产物。部分配体在钯催化的不对称烯丙基化反应的结果见表1。
[0062]
化学反应式如下:
[0063][0064]
表1配体效应对烯丙基化反应的影响a[0065][0066]
由实验结果可以看出,本发明的新型手性硫醚-亚磺酰亚胺配体i化合物与烯丙基氯化钯二聚体形成的络合物对催化不对称烯丙基烷基化反应具有良好的催化活性,并且对各类底物都具有很好的催化效果,其中以配体化合物i-4效果最好。本发明的新型手性硫醚-亚磺酰亚胺作为钯催化的不对称烯丙基烷基化反应的配体,可直接用来制备各种烯丙基烷基化类化合物。
[0067]
尽管本发明的实施方案已公开如上,但其并不仅仅限于说明书和实施方式中所列运用,它完全可以被适用于各种适合本发明的领域,对于熟悉本领域的人员而言,可容易地实现另外的修改,因此在不背离权利要求及等同范围所限定的一般概念下,本发明并不限于特定的细节和这里示出与描述的实施例。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
