一种
α1β
1整合素依赖增强型car巨噬细胞及其制备方法和应用
技术领域
1.本发明涉及免疫学和肿瘤治疗学技术领域,特别是涉及一种α1β1整合 素依赖增强型car巨噬细胞及其制备方法和应用。
背景技术:
2.免疫治疗在肿瘤治疗领域发挥重要作用,且不断取得重要进展。近年 来,car-t细胞疗法因其在血液系统肿瘤领域取得的很好疗效,现已被美 国fda及我国药监部门批准成为特定血液癌症治疗的临床用药。然而, car-t细胞疗法因受到t细胞对实体肿瘤浸润难度大、以及t细胞在肿瘤 微环境中遭遇免疫衰竭等问题。迄今为止,car-t细胞疗法在针对恶性实 体瘤的治疗中进展缓慢。当前探索更多类型免疫细胞用于开发过继细胞疗 法,以能开展特定有效的细胞免疫治疗已成为肿瘤免疫治疗发展的热点和 趋势。巨噬细胞先天具有肿瘤趋化性,并可深度浸润实体肿瘤,部分肿瘤 中巨噬细胞浸润度达50%,并且随着肿瘤恶性程度增加其浸润程度逐渐增 强。因此,基于巨噬细胞开展的嵌合抗体构建,进而形成的car巨噬细 胞(car-macrophage,car-m)肿瘤细胞治疗技术在今后攻克实体肿瘤 的治疗发展中被寄予厚望。
3.t细胞的活化需要表面tcr-cd3复合物与mhc分子复合物结合提供 t细胞的活化第一信号,然而肿瘤微环境中mhc分子的表达水平低下, 不能有效提刺激t细胞的第一活化信号,从而导致t细胞的免疫反应和杀 伤作用被抑制。因此研究人员创造性地设计了car-t细胞技术,使用含胞 外特异识别肿瘤抗原的scfv段换掉tcr的胞外区域,使其不依赖于mhc 分子的作用。arthur weiss等发现嵌合抗原受体含有的t细胞内信号域 cd3ζ可激活t细胞信号传导。故car-t选择了cd3ζ作为传输胞内激活 信号的基序。
4.car-m肿瘤细胞疗法作为新型肿瘤免疫治疗技术着眼于机体另一重 要免疫细胞-巨噬细胞,开拓了新的细胞治疗领域,然而,当前car-m构 建大多在沿用car-t细胞的设计单元。例如,来自宾夕法尼亚大学的saargill博士和michael klichinsky博士在nature biotechnology报道的car
‑ꢀ
巨噬细胞疗法,采用的是经典car-t中特异性发挥t细胞信号转导功能 的cd3ζ信号转导结构域。car-m若得以作为独树一帜的细胞免疫治疗机 制,首先如何构建设计真正适用于巨噬细胞信号转导功能的特异性元件亟 待深入探讨。
5.巨噬细胞的活化须经历两个阶段:致敏阶段和激活阶段。激活是由众 多基因的协同表达驱动完成,期间需要多种跨膜蛋白的参与。巨噬细胞表 面存在多种蛋白分子,均参与介导巨噬细胞活化的过程,包括α1β1整合素、 fcr、csf-1r、mr、cd36、cd163和cd206等。α1β1整合素通常表达 于巨噬细胞,并通过α1亚基胞外区的i-结构域与细胞外基质(ecm)中 的胶原分子相互作用,介导其细胞因子的分泌。α1β1整合素在巨噬细胞的 炎症反应中发挥重要作用,其在介导巨噬细胞活化方面起着重要作用。在 免疫系统中,α1β1只有在被抗原、超抗原或细胞因子激活后才在巨噬细胞 上表达。研究表明,巨噬细胞α1/β1整合素的表达被ifn-γ特异性诱导, 从而促进巨噬细胞的活化和刺激细胞因子的表达。巨噬细胞是肿瘤
免疫浸 润的重要组成部分。肿瘤细胞可以分泌白细胞介素和趋化因子等信号分子 来招募巨噬细胞,这使得巨噬细胞天然趋向并浸润至实体肿瘤内。遗憾的 是,巨噬细胞募集到肿瘤组织后,肿瘤细胞为避免被巨噬细胞免疫攻击, 其肿瘤微环境大多低表达ifn-γ等免疫诱导因子,从而无法形成对巨噬细 胞m1型极化及抗癌活化特性的有效激活,客观上促进了实体肿瘤的恶性 进展。
6.近些年,虽然也有很多研究者在构建巨噬细胞方面进行研究,但是由 于肿瘤微环境ifn-γ低表达等抑制性因素,巨噬细胞先天免疫吞噬等清除 肿瘤细胞的能力受到显著抑制,并没有真正意义上的成功构建car-m,而 car-m信号转导功能的特异性元件如何来设计,以更适宜于car-m的本 身抗癌潜能机制,从而形成真正意义的car-m技术体系,是亟需解决的 问题。
技术实现要素:
7.本发明的目的是提供一种α1β1整合素依赖增强型car巨噬细胞及其 制备方法和应用,以解决上述现有技术存在的问题,基于α1β1整合素与 fcγrⅰ特定基序设计构建的活化吞噬增强型car-m,car-m scfv靶向杀 伤肿瘤细胞后,激活巨噬细胞活化信号并促进巨噬细胞的吞噬,协同增强 抗肿瘤效应。
8.为实现上述目的,本发明提供了如下方案:
9.本发明提供一种α1β1整合素依赖增强型car巨噬细胞,包括胞内 α1β1整合素跨膜介导的fcγrⅰ胞内信号转导基序,所述α1β1整合素包括 α1β1整合素跨膜区及胞内信号调控基序。
10.优选的是,所述α1β1整合素跨膜区的核苷酸序列如seq id no:1所 示,所述α1β1整合素的胞内信号调控基序的核苷酸序列如seq id no:2 所示;
11.所述fcγrⅰ胞内信号转导基序的核苷酸序列如seq id no:3所示。
12.优选的是,所述car巨噬细胞还包括胞外抗原结合区。
13.优选的是,所述胞外抗原结合区为scfv,其核苷酸序列如seq id no:4 所示。
14.本发明还提供一种制备所述的α1β1整合素依赖增强型car巨噬细胞 的方法,其特征在于,包括将α1β1整合素跨膜区及胞内信号调控基序、 fcγrⅰ胞内信号转导基序、胞外抗原结合区的核苷酸序列导入car巨噬细 胞中的步骤。
15.本发明还提供一种嵌合抗原受体,包括所述的α1β1整合素跨膜区及胞 内信号调控基序、fcγrⅰ胞内信号转导基序、胞外抗原结合区的核苷酸序列。
16.本发明还提供一种载体,包括所述的α1β1整合素跨膜区及胞内信号调 控基序、fcγrⅰ胞内信号转导基序、胞外抗原结合区的核苷酸序列,或权利 要求6所述的嵌合抗原受体。
17.本发明还提供一种药物组合物,包括所述的car巨噬细胞或所述的 载体连同药学上可接受的载体。
18.本发明还提供一种所述的car巨噬细胞在制备增强巨噬细胞先天免 疫吞噬功能的药物中的应用。
19.本发明还提供一种所述的car巨噬细胞在制备治疗实体肿瘤药物中 的应用。
20.优选的是,所述实体肿瘤包括乳腺癌、卵巢癌、胃癌、肺癌、结直肠 癌等her2高表
m治疗小鼠her2 -4t1肿瘤模式图;b:actcar-m治疗小鼠肿瘤生长曲线;c:actcar-m治疗小鼠肿瘤生存曲线;d:actcar-m治疗小鼠体重;*表示不同组之间的显著性差异,***为p《0.001,**为p《0.01,*为p《0.05。
具体实施方式
33.现详细说明本发明的多种示例性实施方式,该详细说明不应认为是对本发明的限制,而应理解为是对本发明的某些方面、特性和实施方案的更详细的描述。
34.应理解本发明中所述的术语仅仅是为描述特别的实施方式,并非用于限制本发明。另外,对于本发明中的数值范围,应理解为还具体公开了该范围的上限和下限之间的每个中间值。在任何陈述值或陈述范围内的中间值以及任何其他陈述值或在所述范围内的中间值之间的每个较小的范围也包括在本发明内。这些较小范围的上限和下限可独立地包括或排除在范围内。
35.除非另有说明,否则本文使用的所有技术和科学术语具有本发明所述领域的常规技术人员通常理解的相同含义。虽然本发明仅描述了优选的方法和材料,但是在本发明的实施或测试中也可以使用与本文所述相似或等同的任何方法和材料。本说明书中提到的所有文献通过引用并入,用以公开和描述与所述文献相关的方法和/或材料。在与任何并入的文献冲突时,以本说明书的内容为准。
36.在不背离本发明的范围或精神的情况下,可对本发明说明书的具体实施方式做多种改进和变化,这对本领域技术人员而言是显而易见的。由本发明的说明书得到的其他实施方式对技术人员而言是显而易见的。本技术说明书和实施例仅是示例性的。
37.关于本文中所使用的“包含”、“包括”、“具有”、“含有”等等,均为开放性的用语,即意指包含但不限于。
38.实施例1一种α1β1整合素依赖增强型car巨噬细胞的制备方法及功能验证
39.1、活力增强型actcar-m的构建及表征
40.本研究前期参考car-t的体胞外结合理念,受益于包膜病毒的侵染出芽表达机制,采用慢病毒载体构建巨噬细胞的特异性car以及活力增强型actcar-m,以her2为模式分子成功建立活力增强型car-m平台,于细胞膜表面表达跨膜her2scfv胞外段,筛选获得具有高亲和her2结合活性的actcar-m稳转细胞系。
41.1.1慢病毒的构建和包装
42.基于慢病毒载体,通过将her2scfv、α1β1整合素跨膜区域、α1β1整合素胞内信号调控基序、fcγr1跨膜信号传导域胞内(及fcγrⅰ胞内信号转导基序)和gfp使用linker进行连接,合成嵌合抗原受体her2scfv-gfp(由生工生物工程(上海)股份有限公司进行的序列合成),将其转染巨噬细胞,构建巨噬细胞特异性car,通过将合成的her2scfv-gfp-car慢病毒质粒与rev、gag、vsv三种辅助质粒进行共转染,使其包装成为完整的her2scfv-gfp-car慢病毒。
43.具体步骤如下:
44.(1)将4
×
105个处于对数生长期的hek293t细胞均匀接种于6孔板中,在37℃、5%co2细胞培养箱中静置培养至细胞汇合度达到60%-70%,按3:1:1:1比例分别取质粒pcdh-affibody-gfp2.0μg、rev0.67μg、gag0.67μg和vsv0.67μg加入到装有400μl
opti-mem的1.5ml离心管中混匀室温孵育5min,再加入6.0μlturbofecttransfectionreagent转染试剂,枪头轻柔吹打混匀,室温孵育15-20min;
45.(2)将上述孵育好的混合液缓慢滴加到接种了293t细胞的6孔板中,边滴加边轻轻摇动培养板,置于37℃、5%co2培养16-24h后,弃掉6孔板各孔中的转染混合液,添加2-3ml包含10%胎牛血清(fbs)、1
×
cd(cdlipidconcentrategibico)、0.01mm胆固醇、4.0mml-谷氨酸(glu)、0.01mm蛋黄卵磷脂的advanceddmem完全培养基,将细胞培养板重置于37℃、5%co2培养48h,待合成为her2scfv-gfp-car慢病毒;
46.(3)为去除细胞碎片,将含有慢病毒的培养基于4℃、4000g下离心10min。用0.22μm的过滤器过滤上清液并收集,离心机4℃、15000g离心2h。弃上清,利用病毒保存液重悬病毒颗粒,再10000g离心5min后取上清于-80℃中保存备用;
47.(4)随后,检测包装出来的慢病毒滴度,向9个无菌的1.5ml离心管中加入90μl含有6.0μg/ml聚凝胺的dmem完全培养基,再将10μl已获得的重组慢病毒颗粒her2scfv-gfp-car加入第一个离心管中吹打均匀,吸出10μl混合液加入到第二个离心管中,如此连续直至最后一管,每组设置三个重复。将5
×
104个hek293t细胞均匀接种于96孔板中,细胞汇合度达到60%-70%时依次加入上述稀释好的各梯度慢病毒稀释液,同时设置不加入病毒稀释液的293t细胞作为阴性对照;培养24h后,更换新鲜的dmem完全培养基,48h后于倒置荧光显微镜下观察各孔应该情况。慢病毒滴度计算公式为:病毒滴度(tu/ml)=平均发绿色荧光细胞数
×
病毒稀释倍数/接种病毒液的体积(ml),单位为tu/ml。
48.上述构建嵌合抗原受体的各元件序列如下:
49.her2scfv序列(seqidno:4):
50.atgaatttacaaccaattttctggattggactgatcagttcagtttgctgtgtgtttgctctagcttctcaggtacaactgcagcagtctggacctgaactgaagaagcctggagagacagtcaagatctcctgcaaggcctctgggtatcctttcacaaactatggaatgaactgggtgaagcaggctccaggacagggtttaaagtggatgggctggattaacacctccactggagagtcaacatttgctgatgacttcaagggacggtttgacttctctttggaaacctctgccaacactgcctatttgcagatcaacaacctcaaaagtgaagacatggctacatatttctgtgcaagatgggaggtttaccacggctacgttccttactggggccaagggaccacggtcaccgtttcctctggcggtggcggttctggtggcggtggctccggcggtggcggttctgacatccagctgacccagtctcacaaattcctgtccacttcagtaggagacagggtcagcatcacctgcaaggccagtcaggatgtgtataatgctgttgcctggtatcaacagaaaccaggacaatctcctaaacttctgatttactcggcatcctcccggtacactggagtcccttctcgcttcactggcagtggctctgggccggatttcactttcaccatcagcagtgtgcaggctgaagacctggcagtttatttctgtcagcaacattttcgtactccattcacg。
51.linker序列(seqidno:5):
52.ggcggtggttccggcggtggatctggtggaggaactggaggaggttcaggaggtggt。
53.α1β1整合素跨膜区序列(seqidno:1):
54.ttatgggtcatcctgctgagtgcttttgccggattgttgctgttaatgctgctcattttagcactgtgg。
55.α1β1整合素胞内信号调控基序(seqidno:2):
56.aagatcggattcttcaagagacctctgaagaagaagatggagaag。
57.fcγrⅰ胞内信号转导基序(seqidno:3):
58.aagatccatagactgcagagagagaagaagtacaacctggaggtgcctctggtgagcgagcagggcaagaaggccaacagcttccagcaggtgaggagcgacggcgtgtacgaggaggtgaccgccaccgccagccagaccacccctaaggaggcccctgacggccctaggagcagcgtgggcgactgtggccctgagcagcctgagcctctgcctcctagcgacagcaccggcgcccagaccagccagagc。
59.gfp序列(seqidno:6):
60.gtgagtaagggcgaggagctgtttaccggcgtggtgcccatcctggtggagctggacggcgacgtgaacggccacaagttcagcgtgagcggcgagggggagggcgacgccacctacggcaagctgaccctgaagttcatttgcacaaccggcaagctgcccgtgccatggcctaccctggtgaccaccctgacatacggcgtccagtgttttagcaggtatcccgaccacatgaagcagcacgacttctttaagagcgccatgcccgagggctacgtgcaggagaggaccatcttcttcaaggacgacggtaactacaagaccagagccgaggtgaagttcgagggcgacaccctggtgaaccggatcgagctgaagggcatcgacttcaaggaggacggtaacatcctgggccacaagctggagtataactataatagccacaacgtgtacatcatggccgacaagcagaagaacggcattaaggtgaacttcaagatcagacataacatcgaggacggatctgtgcagctggccgaccactaccagcagaacacccctatcggagatggaccagtgctgctgcctgataatcactatctgagcacacagtctgccctgagcaaagatcctaatgaaaagagagatcacatggtgctgctggagtttgtgacagctgctggaataacactgggcatggatgagctgtataagtaa。
61.1.2筛选actcar-m稳转细胞株
62.在包装为完整的慢病毒并检测了其滴度后,使用her2scfv-gfp-car慢病毒感染巨噬细胞,并通过药物筛选获得actcar-m稳转细胞株,具体步骤如下:
63.(1)为确定药物筛选最佳浓度,取5
×
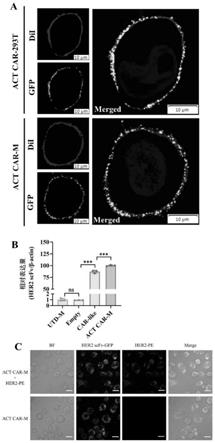
104个处于对数生长期的不经任何处理的j774a.1巨噬细胞接种于24孔板内,用dmem完全培养基于37℃、5%co2的细胞培养箱中培养。待细胞密度约60-70%时,添加含有终浓度为0.5μg/ml、1μg/ml、1.5μg/ml、2μg/ml、5μg/ml嘌呤霉素的dmem完全培养基进行最佳致死浓度的测定。24孔板置于细胞培养箱中静置培养,每天观察一次细胞形态及残留细胞数目,以第4天杀死全部巨噬细胞的嘌呤霉素终浓度作为最佳药筛浓度;
64.(2)使用慢病毒感染j774a.1巨噬细胞,取2
×
105个j774a.1巨噬细胞接种于6孔板内,待其生长到细胞汇合度至60-70%时,以每9:1的比例向培养j774a.1巨噬细胞的培养基中加入her2scfv-gfp-car慢病毒病毒颗粒,并加入聚凝胺使其终浓度为6.0μg/ml,24h后更换新鲜dmem完全培养基,48h后即完成j774a.1的慢病毒感染;
65.(3)通过嘌呤霉素最佳药筛浓度进行筛选稳转细胞株,每隔1天更换一次含有嘌呤霉素的新鲜dmem培养基,使培养基中嘌呤霉素的浓度保持不变,观察细胞生长情况及形态变化。约持续3-4次后药筛完成,胰酶消化药筛后的细胞并按0.5个/100μl的细胞浓度,将细胞接种于96孔板中,用含有嘌呤霉素的dmem完全培养基继续培养,选择仅含有单个细胞的孔进行细胞扩大培养,传代至新的培养瓶或培养皿中进行扩增或者冻存。
66.1.3qpcr检测
67.提取上述稳转细胞株的总rna,利用superrealpremixplus(sybrgreen)试剂盒检测上述的稳转细胞株中融合蛋白的表达情况,按照试剂盒说明书进行实验,qpcr实验步骤如下:
68.(1)正常溶解2
×
superrealpremixplus、50
×
roxreferencedye、模板、引物和rnase-freeddh2o,并将所有试剂在室温下平衡并彻底混匀;
69.(2)在冰上将反转得到的cdna模板以一定体系与正向引物、反向引物、mix、rox、ddh2o混合,利用移液枪将混合液添加入八连管中,20μl体系包括:10μl2
×
superrealpremixplus、0.6μl正向引物(10μm)、0.6μl反向引物(10μm)、0.1-2μlcdna模板、0.4μl50
×
roxreferencedye,使用rnase-freeddh2o补充至总体积20μl;
70.(3)盖上管盖,吹打混匀,用微量离心机离心5-10s,确保所有组分均在管底;
71.(4)将反应体系放置于rt-qpcr仪abi7300中,设置仪器参数,95℃预变性15min、95℃变性10s、60℃
±
1℃退火20s,72℃延伸31s,40个循环,运行程序,待程序结束后导出ct值,并利用2-δδct
(livak)法计算最终结果。
72.qpcr结果显示:actcar-m融合蛋白的成功表达;共聚焦显微镜结果成功验证了her2scfv的her2结合活性,如图1所示。这为后期研究提供细胞治疗模型。
73.2、actcar-m的活性检测
74.(1)cck细胞增殖试验
75.①
于96孔板中每孔加入100μl的her2阳性肿瘤靶细胞悬液,密度为1
×
105个细胞/ml,置于37℃、5%co2的培养箱中静置培养;
76.②
待her2阳性肿瘤靶细胞贴壁后,吸去孔板中的培养基,用pbs清洗细胞2-3次,分别向96孔板中分别加入100μl不同密度的j774a.1(utd-m)、empty-pcdhj774a.1(empty)、her2scfvj774a.1(car-like)、her2scfvcarj774a.1(actcar-m),设置效靶比分别为0:1、0.25:1、0.5:1、0.75:1、1:1,同时在没有靶细胞的96孔板中加入等量的utd-m、empty、car-like、actcar-m细胞作为效应细胞对照,设置全部裂解的未加效应细胞的等量her2阳性肿瘤靶细胞孔作为靶细胞阳性对照组,每组实验设置3个平行孔;
77.③
细胞共培养24-48h之后,吸去96孔板中的培养基,用pbs清洗细胞2-3次,再向96孔板中加入10μl的cck8检测溶液,在37℃、5%co2的培养箱中培养3-4h;
78.④
用酶标仪测定各孔在450nm处的吸光度,靶细胞细胞活性大小=(效靶比作用组吸光度-效应细胞对照组吸光度)/(靶细胞阳性对照组吸光度-空白孔吸光度)。
79.(2)同时,分别提取各组细胞总rna,采用qpcr检测actcar-m活性,检测方法同上述的qpcr的检测方法。
80.结果如图2所示,actcar-m组细胞增殖显著增强于car-like组(图2a)。qpcr检测结果显示,actcar-m组细胞的增殖因子ccnd1和ki67的mrna表达水平均显著强于car-like组,且p53表达水平低于car-like组,见图2b-d。研究结果提示,actcar-m的基础活性显著增强,这意味着α1β1整合素和fcγrⅰ在胞内转导的信号增强了actcar-m的增殖活性。
81.3、actcar-m的体外吞噬活性检测
82.将上述构建的actcar-m,分别基于1:1的效靶比,以her2
-skov3和her2
-4t1肿瘤细胞为模型,使用actcar-m吞噬肿瘤细胞,具体操作为:
83.于6孔板中分别接种4
×
105个utd-m、empty、car-like、actcar-m、her2阳性肿瘤靶细胞,以10%fbs的dmem高糖培养基于37℃、5%co2培养箱中静置培养,待细胞长至汇合度为70-80%时,向巨噬细胞组中加入工作浓度4μm的dio染料,向her2阳性肿瘤靶细胞组中加入工作浓度4μmdii染料,孵育4-6h;吸去上清,于荧光显微镜下拍照。
84.结果如图3a和3b所示,相对于car-like组,actcar-m组的吞噬肿瘤细胞能力得
到了显著提升,这意味着α1β1整合素和fcγrⅰ在胞内转导的信号增强了actcar-m的吞噬能力,提示actcar-m活性增强,为后期各项研究奠定理论基础。
85.4、anti-her2actcar-m作用于肿瘤细胞her2靶点的抗肿瘤机制分析
86.利用上述的“2、actcar-m的活性检测”中cck细胞增殖试验培养不同组巨噬细胞后,采用her2信号通路下游相关分子(akt、pi3k),同时检测肿瘤相关活性分子p53、caspase-3、ccnd-1、ki67,进一步佐证了actcar-m对肿瘤的杀伤。具体步骤包括:
87.(1)于6孔板中每孔加入4
×
105个的her2阳性肿瘤靶细胞,置于37℃、5%co2的培养箱中静置培养;
88.(2)待her2阳性肿瘤靶细胞贴壁后,吸去孔板中的培养基,用pbs清洗细胞2-3次,分别向6孔板中分别加入不同密度的j774a.1(utd-m)、empty-pcdhj774a.1(empty)、her2scfvj774a.1(car-like)、her2scfvcarj774a.1(actcar-m),设置效靶比分别为0:1、0.25:1、0.5:1、0.75:1、1:1,同时在没有靶细胞的96孔板中加入等量的utd-m、empty、car-like、actcar-m细胞作为效应细胞对照,设置全部裂解的未加效应细胞的等量her2阳性肿瘤靶细胞孔作为靶细胞阳性对照组,每组实验设置3个平行孔;
89.(3)细胞共培养24-48h之后,吸去6孔板中的培养基,用pbs清洗细胞2-3次,再向6孔板中每孔加入1ml的trizol,提取rna,利用superrealpremixplus(sybrgreen)试剂盒,并按照试剂盒说明书进行实验。
90.(4)正常溶解2
×
superrealpremixplus、50
×
roxreferencedye、模板、引物和rnase-freeddh2o,并将所有试剂在室温下平衡并彻底混匀;
91.(5)在冰上将反转得到的cdna模板以一定体系与正向引物、反向引物、mix、rox、ddh2o混合,利用移液枪将混合液添加入八连管中,20μl体系包括:10μl2
×
superrealpremixplus、0.6μl正向引物(10μm)、0.6μl反向引物(10μm)、0.1-2μlcdna模板、0.4μl50
×
roxreferencedye,使用rnase-freeddh2o补充至总体积20μl;
92.(6)盖上管盖,吹打混匀,用微量离心机离心5-10s,确保所有组分均在管底;
93.(7)将反应体系放置于rt-qpcr仪abi7300中,设置仪器参数,95℃预变性15min、95℃变性10s、60
±
1℃退火20s,72℃延伸31s,40个循环,运行程序,待程序结束后导出ct值,并利用2-δδct
(livak)法计算最终结果。
94.结果如图4和图5所示,与car-like组相比,actcar-m组akt、pi3k表达发生了显著下调,说明actcar-m阻断了her2信号的传导,从而诱导p53和caspase-3的表达均发生了显著上调,最终导致了对肿瘤的杀伤抑制,其体现在ki67和ccnd-1的表达均发生了显著下调,提示actcar-m细胞阻断了her2
肿瘤细胞her2信号通路的传导,抑制her2
肿瘤细胞周期进展,导致肿瘤凋亡事件的发生。这为actcar-m的体外肿瘤杀伤效应研究奠定基础,也为后期肿瘤调控的活力增强型car-m的设计提供可靠理论支撑。
95.6、anti-her2actcar-m对多种肿瘤细胞系的体外杀伤检测
96.本研究前期利用cck-8技术(同上述“2、actcar-m的活性检测中cck细胞增殖试验”)在细胞水平对anti-her2actcar-m的体外杀伤能力进行检测,使用her2 -4t1细胞和her2
-skov3细胞,明确anti-her2actcar-m对her2靶点杀伤的有效性,确定其有效的效靶比。
97.如图6所示,相对于car-like组,actcar-m组对her2
的肿瘤细胞有显著杀伤作
用,说明act car-m抑瘤效果优于car-like,提示α1β1 整合素和fcγrⅰ在胞内转导的信号增强了act car-m的杀伤肿瘤能力。
98.7、act car-m体内治疗肿瘤效果验证
99.本研究前期初探act car-m在体内靶向肿瘤能力的验证,使用小鼠 her2
-4t1肿瘤模型验证act car-m的抗肿瘤效果,her2
-4t1肿瘤模 型的建立包括如下步骤:
100.(1)小鼠乳腺癌细胞系her2
-4t1利用1640培养基(hyclone)于 37℃、5%co2细胞培养箱中培养;
101.(2)4-6周龄的磁性balb/c小白鼠购买后需于恒温无菌通风小鼠饲 养间饲养一周以上后再进行小鼠实验;
102.(3)将her2
-4t1细胞扩大培养,待小鼠饲养约一周后,消化 her2
-4t1细胞,用pbs清洗2-3次,并利用pbs重悬,稀释密度为107个细胞/ml,将100μl细胞重悬液以皮下注射的方式注射到小鼠右肢上方 皮下;
103.(4)将utd-m、empty、car-like和act car-m细胞均利用dmem 高糖培养基于37℃、5%co2细胞培养箱中培养;
104.(5)待her2
-4t1细胞成瘤后,每组设置4只小鼠的平行实验,分 别将100μl pbs或细胞重悬液通过尾静脉方式进行注射,utd-m、empty、 car-like和act car-m组分别注射3
×
106个细胞,在注射第一次后的第 5d,进行第二次注射,每组注射量为3
×
106个细胞,总共注射了两次。
105.(6)用电子游标卡尺对小鼠肿瘤的长和宽分别进行测量,利用电子天 平对小鼠体重进行测量,小鼠肿瘤体积=(长*宽2)/2,每两天测量一次, 最后计算小鼠生存曲线。
106.结果见图7,相对于car-like组,act car-m组肿瘤得到了明显抑 制,生存期延长,取得了显著的治疗效果。另外,相对于car-like组, act car-m组体重无明显变化,提示其具有较好安全性。
107.以上所述的实施例仅是对本发明的优选方式进行描述,并非对本发明 的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通技术人 员对本发明的技术方案做出的各种变形和改进,均应落入本发明权利要求 书确定的保护范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。