1.本发明涉及一种含有膦酸基团的聚芳香醚的制备方法。
技术背景
2.质子交换膜燃料电池是人类未来清洁、可持续能源的重要选择之一。质子交换膜在高温无湿条件下运行可避免催化剂中毒,无需复杂的保湿系统,进而有效提高能量密度和能源效率。替代有水条件下的水合质子运输传导,质子跳跃传导成为高温无湿条件下质子交换膜燃料电池运行的理论基础。不易流失、以共价键连接的膦酸基团具有较好的氢键形成能力,具备通过质子跳跃机理传导质子的可能性。膦酸类质子交换膜材料的研究策略有两种,即分别将膦酸基团连接在脂肪烃类型和芳香类型的高分子骨架上。聚芳香醚是一类重要的高性能高分子材料,其不仅具有较高的热稳定性、化学稳定性及机械强度,而且结构多样、来源广泛,在国防军工、航空航天、高端制造等领域已经发挥了不可替代的作用,同时,聚芳香醚高分子为膦酸类质子交换膜材料的开发提供了基础。
3.膦酰化聚芳香醚的合成方法有两种:(1)膦酰化单体直接聚合方法:虽然制备可实现,但其聚合条件苛刻,反应时间长,产物分子量不高,成膜性能有待提高,例如参见cn102634010b、cn104004183b和cn104151552b等;(2)后膦酰化方法:即先聚合得到卤代的聚芳醚氧膦化合物,再通过艾伯佐夫重排反应引入膦酰基团。热法艾伯佐夫重排反应制备由于底物反应活性不够,尚无成功案例。采用高压汞灯进行的光照艾伯佐夫反应虽然可有效引入膦酰基团,但由于光源波长范围较宽,受高能量光影响而发生断链现象。
技术实现要素:
4.针对现有技术存在的不足,本发明提供一种含有膦酸酯基团的聚芳香醚的制备方法。本发明还提供了含有膦酸基团的聚芳香醚的制备方法、其制备的含有膦酸基团的聚芳香醚及应用。本发明采用后膦酰化法,通过单一波长led光源的光照艾伯佐夫重排反应,将膦酸基团引入到聚芳香醚高分子链上,制备了含有膦酸基团的聚芳香醚高分子材料。
5.为实现上述目的的,本发明提供了如下的技术方案。
6.在一个方面,本发明提供了一种含有膦酸酯基团的聚芳香醚的制备方法,其包括以下步骤:
7.s1:将碘代和/或溴代的聚芳香醚与膦酰化试剂在第一溶剂存在的条件下形成反应液体系;
8.s2:采用led光源照射所述反应液体系进行光照反应,得到含有膦酸酯基团的聚芳香醚。
9.根据本发明的一些实施方式,所述led光源的波长范围为250nm-600nm,例如可以为260nm、300nm、340nm、380nm、420nm、460nm、500nm、540nm、580nm以及它们之间的任意值。根据本发明的优选实施方式,所述led光源的波长范围为280nm-455nm。根据本发明的进一步优选实施方式,所述led光源的波长范围为365nm-395nm。根据本技术的一些具体实施例,
所述led光源的波长为365nm、395nm或455nm。
10.根据本发明的一些实施方式,所述led光源由uvled面光源照射机发生。
11.根据本发明的一些实施方式,所述第一溶剂包括所述第一溶剂包括n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮、二甲基亚砜、1,3-二甲基-2-咪唑啉酮、二苯砜、环丁砜、n-乙基吡咯烷酮或n-丁基吡咯烷酮中的一种或几种。
12.根据本发明的一些实施方式,所述膦酰化试剂包括亚磷酸三乙酯、亚磷酸三甲酯或亚磷酸三苯酯中的一种或多种。
13.根据本发明的一些实施方式,所述光照反应的温度为20℃-110℃,优选为30℃-60℃。根据本发明的一些实施方式,所述光照反应的时间为1h-10h,优选为2h-5h。
14.根据本发明的一些实施方式,所述碘代和/或溴代的聚芳香醚通过将碘代和/或溴代的二卤单体、双酚单体和任选的二卤单体在碱、分水剂和第二溶剂的存在下进行聚合反应得到。
15.根据本发明的一些实施方式,所述碘代和/或溴代的二卤单体包括如下物质中的一种或多种:
16.[0017][0018]
其中,x为卤素或羟基,优选为氟、氯或溴,y为h、溴或碘,且至少一个y为溴或碘。
[0019]
根据本发明的一些实施方式,所述二卤单体选自如下物质中的一种或多种:
[0020]
[0021][0022]
其中,x为卤素或羟基,优选为氟、氯或溴。根据本发明的一些实施方式,所述双酚单体选自如下物质中的一种或多种:
[0023]
[0024][0025]
根据本发明的一些实施方式,所述碱包括碳酸钾、碳酸氢钾、碳酸钠、碳酸氢钠、磷酸钾或磷酸钠中的一种或多种。
[0026]
根据本发明的一些实施方式,所述分水剂包括甲苯、邻二甲苯、间二甲苯、对二甲苯或氯苯中的一种或几种。
[0027]
根据本发明的一些实施方式,所述第二溶剂包括n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮、二甲基亚砜、1,3-二甲基-2-咪唑啉酮、二苯砜、环丁砜、n-乙基吡咯烷酮或n-丁基吡咯烷酮中的一种或几种。
[0028]
根据本发明的一些实施方式,所述聚合反应的温度为120℃-200℃,时间为5h-80h。
[0029]
在第二方面,本发明提供了一种含有膦酸基团的聚芳香醚的制备方法,其包括将上述的方法制备的含有膦酸酯基团的聚芳香醚进行水解反应,得到所述含有膦酸基团的聚芳香醚。
[0030]
在第三方面,本发明还提供了一种通过第二方面所述的方法制备得到的含有膦酸基团的聚芳香醚,所述含有膦酸基团的聚芳香醚的特性粘度为0.1-1.0dl/g,膦酰化度为2%-95%。
[0031]
在最后一方面,本发明还提供了根据第二方面所述的制备方法得到的含有膦酸基
团的聚芳香醚或第三方面所述的含有膦酸基团的聚芳香醚在制备质子交换膜材料中的应用。
[0032]
本发明首次将单一波长led光源用于高分子的膦酰化反应,单一波长大大降低了高分子的断链现象,实现了高分子量膦酰化高分子材料的制备。本发明制备的含有膦酸的高分子材料可以有效降低质子交换膜的溶胀现象,实现了既保持较高的离子交换容量(利于质子传导及其它应用),同时又保持了膜的机械强度。
附图说明
[0033]
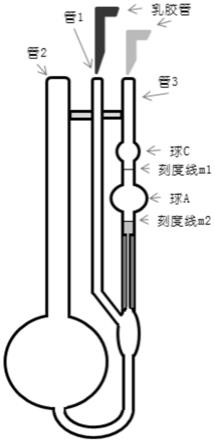
图1为根据本发明的粘度测定中采用的装置的结构示意图。
具体实施方式
[0034]
下面将通过具体实施例对本发明作进一步地说明,但本发明的范围并不限于此。
[0035]
(一)以下实施例所采用的聚芳香醚合成路线如下:
[0036][0037]
(二)适用于本发明的制备方法的碘代和/或溴代的二卤单体、二卤单体、双酚单体、碱、溶剂、膦酰化试剂和分水剂的实例分别如表1-7所示。
[0038]
表1
[0039][0040]
表2
[0041]
[0042][0043]
表3
[0044]
[0045]
[0046][0047]
表4
[0048]
代号碱(缚酸剂)的结构kck2co3khckhco3nacna2co3nahcnahco3kpk3po4napna3po4[0049]
表5
[0050]
[0051][0052]
表6
[0053][0054]
表7
[0055]
代号膦酰化试剂的结构tppip(oph)3tepip(oet)3tmpip(ome)3[0056]
(三)测试方法
[0057]
1、粘度测定方法
[0058]
采用如图1所示的装置进行粘度测试,具体步骤如下:
[0059]
1)取0.2g待测聚合物,加入20mlnmp(含有0.05m溴化锂)于锥形瓶中,室温搅拌,观察其溶解度,完全溶解后进行过滤测试。
[0060]
2)将清洁干燥的粘度计垂直地放入恒温水槽内使水面完全浸没小球。用移液管吸10ml过滤后的溶液(浓度为c1)。从管2注入b球中,于25℃恒温槽中恒温3分钟,然后进行测定。在管1套一根橡皮管,用夹子夹住,使之不通气。在管3用吸耳球将溶液从球b经毛细管、球a吸入球c,然后同时松开吸耳球和管1橡皮管,让管1通大气。此时液体即开始流回球b。用眼睛水平地注视着正在下降的液面,用秒表准确地测定液面流经刻度线1与刻度线2之间所
需的时间,并记录。重复上述操作两次,每次测定相差不大于0.1秒。取两次的平均值为t1,即为溶液的流出时间。
[0061]
3)用移液管加入5ml的nmp(含有0.05m溴化锂)对上述溶液进行稀释,稀释后的溶液的浓度c2为起始浓度c1的2/3,按上面的步骤,测定溶液(浓度为c2)的流出时间为t2。同样,依次加入nmp(含有0.05m溴化锂)5ml、10ml,使溶液浓度成为起始浓度的1/2、1/3(注意每次加入纯溶剂后,一定要混合均匀,且要等恒温后测定),分别测定其流出时间,记录为t。
[0062]
4)将粘度计中的溶液倒入回收瓶、洗涤烘干,用干净的移液管吸取纯nmp(含有0.05m溴化锂)溶液10ml,移入粘度计(注意尽量不要将溶液沾在管壁上),恒温2分钟,按上面的步骤,测定溶液的流出时间t0。
[0063]
5)根据测得的各项时间计算其特性粘度[η],计算方式如下:
[0064]
ηr:相对粘度,ηr=t/t0[0065]
η
sp
:增比粘度,η
sp
=(η-η0)/η0=(η/η0)-1=η
r-1
[0066]
η
sp
/c:比浓粘度
[0067]
[η]:特性粘度
[0068]
以η
sp
/c、lnηr/c对浓度c作图,得两条直线,外推至c
→
0得截距。经换算,即得特性粘度[η]。
[0069]
2、膦酰化度
[0070]
以溴或碘的含量计算应得膦酸酯的量为理论值,用膦酸酯实际含量除以上述理论值,即为膦酰化度。膦酸酯的实际含量通过1hnmr谱图中乙氧基的积分值与芳香环区域的积分总值的比例计算。
[0071]
(四)碘代和/或溴代的聚芳香醚的合成
[0072]
合成例1
[0073][0074]
开始反应前先将原料进行预处理,dfdps-br2、bp置于55℃真空烘箱干燥12h,碳酸钾(kc)置于120℃真空烘箱干燥12h。氮气(99.999%,流速:10-15)氛围下将4.14g(10mmol)dfdps-br2和1.86g(10mmol)bp按照等摩尔比加入到装有分水器、蛇形冷凝管、弯头、搅拌桨和导气管的100ml直三口烧瓶中,然后将36ml dmac、18mltol、4.768g kc加入其中。dmac作为溶剂,kc作为缚酸剂,tol作为分水剂,待混合物完全溶解后,将温度升至160℃(油浴温度),tol回流分水12h,分水完成后,通过分水器除去体系中的tol,再升温至180℃(油浴温度)继续反应,在该温度下继续反应4h,得到深棕色的粘稠状溶液,停止反应,将反应液缓慢倒入1000ml去离子水中,得到白色条状聚合物。105℃加热(加热盘温度)水煮12h,水煮3-4次,以便出去聚合物中包含的溶剂和无机盐,最后得到白色条状聚合物11.76g,收率:98.0%,特性粘度:1.45dl/g。
[0075]
合成例2
[0076]
[0077]
开始反应前先将原料进行预处理,dfdps-br2、bpa置于55℃真空烘箱干燥12h,kc置于120℃真空烘箱干燥12h。氮气(99.999%,流速:10-15)氛围下将2.07g(5mmol)dfdps-br2和1.14g(5mmol)bpa按照等摩尔比加入到装有分水器、蛇形冷凝管、弯头、搅拌桨和导气管的100ml直三口烧瓶中,然后将10ml dmac、5mltol、0.79g(5.75mmol)kc加入其中。dmac作为溶剂,kc作为缚酸剂,tol作为分水剂,待混合物完全溶解后,将温度升至160℃(油浴温度),tol回流分水12h,分水完成后,通过分水器除去体系中的tol,再升温至180℃(油浴温度)继续反应,在该温度下继续反应6h,得到深棕色的粘稠状溶液,停止反应,将反应液缓慢倒入1000ml去离子水中,得到白色条状聚合物。105℃加热(加热盘温度)水煮12h,水煮3-4次,以便出去聚合物中包含的溶剂和无机盐,最后得到纯白色条状聚合物3.11g,收率:97.0%,特性粘度:1.01dl/g。
[0078]
合成例3
[0079][0080]
在氮气(99.9%,流速:5-10)氛围下,将dfbn-br(0.03mol,6.54g)、bp(0.03mol,5.586g)加入装有机械搅拌的250ml的三口瓶中,加入kc(0.0345mol,4.7681g)作为缚酸剂、dmac 40ml作为溶剂、tol 20ml作为分水剂,待可溶单体完全溶解后,将温度升至169℃(油浴温度),tol回流分水3h,然后通过分水器除去体系中的tol。升温至186℃(油浴温度)继续反应3h,得到浅棕色粘稠状液体,停止反应。将反应溶液慢慢倒入600ml去离子水中,得到浅棕色条状固体,100℃(加热盘温度设定)加热水煮高分子5次,将高分子过滤(每次3-4h)。在普通烘箱烘干后,在120℃真空干燥箱中干燥过夜,得聚合物10.69g,产率98.9%,特性粘度0.88dl/g。
[0081]
合成例4
[0082][0083]
分别取适量的bfppo-i、bp、kc分别于65℃、65℃、120℃真空烘箱干燥12h,确保投料干燥且准确;准备油浴锅,搭好带有蛇形冷凝管、分水器、四氟搅拌桨和100ml直三口烧瓶的聚合装置,通入氮气,验证气密性,以保证反应过程中装置内溶剂不挥发。氮气(99.999%,流速:5-10)氛围下,按照摩尔比1:1的量称取5.62g bfppo-i、和2.38g bp加入到反应装置中,后依次加入20ml dmac溶剂、10ml tol和2.03g kc。其中,dmac做溶剂,kc做为缚酸剂,tol做分水剂,充分搅拌使其溶解,将油浴温度设置165℃使其逐渐升温。dmac回流,进行过夜分水,再将油浴温度升至170℃通过分水器蒸出反应体系中剩余tol,升温至185℃继续反应4h,液体内不断有大泡冒出,并伴有爬杆迹象,瓶内溶液呈深棕色粘稠状,停止反应,待反应液稍稍冷却后倒入800ml蒸馏水中,析出白色条状物质,即为高分子。将高分子聚合物在100℃加热盘上进行水煮,换水3次,每次间隔8h,以便除去包裹在条状聚合物内的无机盐。最后,将高分子分别于120℃普通烘箱和真空烘箱进行干燥12h,得到最终的聚合
产物,收率99.0%,特性粘度:0.90dl/g。
[0084]
合成例5
[0085][0086]
分别取适量的bfppo-i、4,4-二氯二苯砜(dcdps)、bp、kc分别于65℃、65℃、65℃、120℃真空烘箱干燥12h,确保投料干燥且准确;准备油浴锅,搭好带有蛇形冷凝管、分水器、四氟搅拌桨和250ml直三口烧瓶的聚合装置,通入氮气,验证气密性,以保证反应过程中装置内溶剂不挥发。氮气(99.999%,流速:5-10)氛围下,按照摩尔比1:1:2的量称取4.16g bfppo-i、2.72g dcdps和3.52g bp加入到反应装置中,后依次加入52ml dmac溶剂、26ml tol和3.01g kc。其中,dmac做溶剂,kc做为缚酸剂,tol做分水剂,充分搅拌使其溶解,将油浴温度设置165℃使其逐渐升温。dmac回流,进行过夜分水,再将油浴温度升至170℃通过分水器蒸出反应体系中剩余的tol,升温至185℃继续反应8h,液体内不断有大泡冒出,并伴有爬杆迹象,瓶内溶液呈粉棕色粘稠状,停止反应,待反应液稍稍冷却后倒入800ml蒸馏水中,析出白色条状物质,即为高分子。将高分子聚合物在100℃加热盘上进行水煮,换水3次,每次间隔8h,以便除去包裹在条状聚合物内的无机盐。最后,将高分子分别于120℃普通烘箱和真空烘箱进行干燥12h,得到最终的聚合产物,收率99.0%,特性粘度:0.98dl/g。
[0087]
合成例6
[0088][0089]
分别取适量的dfdps-br2、br
2-bpa和kc作为原料,氮气保护下(一进一出式),在装有温度计,分水器和冷凝管的100ml的四口反应瓶中加入所有原料,油浴加热至165℃(油浴温度),内温132℃开始分水,分水12h,分出水0.3ml,油浴温度165℃-188℃蒸出甲苯,共蒸出23ml,油浴温度升温到188℃开始聚合,内温166-167℃,聚合57h,反应液颜色由白-黄绿-棕-深棕,反应结束后,降温至50℃倒入蒸馏水中,呈棕色条状,80℃加热搅拌,500ml水洗四次,常压鼓风烘箱110℃,干燥12h,真空烘箱120℃,干燥12h,干燥后呈浅棕色粉末,特性粘度:0.32dl/g;产量:12.48g,收率:96.9%。
[0090]
(五)含有膦酸基团的聚芳香醚的制备
[0091]
实施例1-3
[0092][0093]
在氮气(99.999%,流速:10-15)氛围下,将3g溴代高分子聚合物(合成例1),加入装有磁力搅拌的石英四口瓶中,然后加入120mldmi80℃(加热盘)加热搅拌,使原料完全溶解,用恒压滴液漏斗加入80ml的tepi,待完全溶解为澄清透明溶液时,用uvled面光源照射机按照表1的条件进行光照反应,减压蒸馏除溶剂,用氯仿溶解,将其滴入20ml异丙醇中,产物析出,异丙醇洗涤3次除去反应中的低分子量有机物,过滤,取滤饼普通烘箱120℃干燥12小时,真空烘箱120℃干燥12h,进行粘度测试和膦酰化度测试,结果如表8所示。根据表8,可得最佳条件为:dmi为溶剂,50℃条件下,tepi为膦酰化试剂,以365nmled面光源照射2.5h,聚合物膦酰化度为32%,特性粘度为0.56dl/g,产率为96.5%。
[0094]
表8
[0095][0096]
实施例4-6
[0097][0098]
采用合成例2制备的聚合物,实验步骤同实施例1,溶剂和膦酰化试剂用量如下表9所示,结果如下:
[0099]
表9
[0100][0101]
实施例7
[0102][0103]
在氩气(流速:10-15)氛围下,将3-溴-2,6-二氟苯腈聚合物(合成例3)(3g,8.2mmol),dmac(75ml)投入到装有磁力搅拌的光照反应器中进行加热(油温40℃)溶解,反应过夜至高分子完全溶解。用恒压滴液漏斗匀速加入60mltepi,待溶液搅拌至均相,使用隔膜泵抽真空,充放惰性气体三次,使体系中残存的低沸点的物质和空气完全除去,保证体系达到无水无氧环境。光照反应器中间口处接入冷凝循环以及尾接管收集反应中副产物溴乙烷,一边通入氩气,另外一边出惰性气体。然后利用365nm面光源紫外灯进行光照反应,控制反应温度在60-90℃,反应过程中取样观察,反应时间达到240min中,停止光照,将产物减压蒸馏除去溶剂,异丙醇洗涤6次除去反应中的剩余有机物,后水洗3次。将生成物在普通烘箱120℃干燥12小时,真空烘箱120℃干燥12h,产率98.0%,膦酰化度68%,特性粘度0.83dl/g。
[0104]
实施例8
[0105][0106]
氩气(99.999%,流速:5-10)氛围下,将含碘代高分子聚合物(合成例4)加入装有电磁搅拌的石英三口瓶(特制光照反应器)中,然后加入dmi,将加热盘设定约50℃的条件下进行加热搅拌,使其完全溶解。随后,向溶液中加入最大可溶解量的tepi,待其充分混合均匀后,液氮冷却,使用隔膜泵抽真空,并连续充放惰性气体,排除体系中残存低沸点物质和空气,确保反应体系达到无水无氧状态。随后,使用(365nm、395nm)uvled面光源照射机进行照射,照射一定时间后,取反应液进行核磁测试,监测tepi的消耗情况。待反应液中的tepi消耗完,对反应液进行减压蒸馏处理,除去溶剂及反应副产物,测核磁监测产物生成情况。
若碘代高分子原料未反应完,则继续重复上述操作,核磁监测直至原料彻底反应完,结束反应,减压蒸馏除溶剂,高分子倒入蒸馏水中,在100℃的加热盘上进行水煮,换水重复3次,每次间隔8h。后依次将高分子放入120℃普通烘箱和真空烘箱干燥12h。膦酰化度:64%,特性粘度:0.80dl/g,收率:95.4%。
[0107]
实施例9
[0108][0109]
氩气(99.999%,流速:5-10)氛围下,将含碘代高分子聚合物(合成例5)加入装有电磁搅拌的石英三口瓶(特制光照反应器)中,然后加入dmi,将加热盘设定约50℃的条件下进行加热搅拌,使其完全溶解。随后,向溶液中加入最大可溶解量的tepi,待其充分混合均匀后,液氮冷却,使用隔膜泵抽真空,并连续充放惰性气体,排除体系中残存的低沸点物质和空气,确保反应体系达到无水无氧状态。随后,使用(365nm、395nm)uvled面光源照射机进行照射,照射一定时间后,取反应液进行核磁测试,监测tepi的消耗情况。待反应液中的tepi消耗完,取少量反应液进行减压蒸馏处理,除去溶剂及反应副产物,测核磁监测产物生成情况。反应结束后,减压蒸馏除溶剂,高分子倒入蒸馏水中,在100℃的加热盘上进行水煮,换水重复3次,每次间隔8h。后依次将高分子放入120℃普通烘箱和真空烘箱干燥12h。膦酰化度:70.0%,特性粘度:0.87dl/g,收率,96.7%。
[0110]
实施例10
[0111][0112]
在氮气(99.999%,流速:10-15ml/min)氛围下,将3.15g溴代高分子聚合物(合成例6),加入装有磁力搅拌的石英四口瓶中,然后加入50mldmac和60mltepi,室温搅拌,待完全溶解为澄清透明溶液时,用(365nm)uvled面光源照射机进行光照反应,反应温度为90℃,光照时间8h,将反应液减压蒸馏后滴入100ml蒸馏水中,析出固体,100ml蒸馏水洗涤三次,抽滤,取滤饼真空烘箱55℃干燥12h,得到固体3.34g,产率82.2%,膦酰化度:80.%%,特性
粘度0.18dl/g。
[0113]
实施例11
[0114][0115]
在氮气(99.999%,流速:10-15)氛围下,将0.50g实施例1制备的聚合物溶解在5ml氯仿中,待完全溶解为澄清透明溶液后,在冰水浴0℃条件下,用恒压滴液漏斗缓慢滴加三甲基溴硅烷1ml,待滴加至恰好无沉淀析出时,再40℃(加热盘)加热搅拌24h,反应完成后,用水泵减压蒸干溶剂得到固体残留物,再向其中加入5ml甲醇,室温搅拌18h,蒸走甲醇,获得最终产品。将得到的产品在50ml去离子水中洗涤,80℃加热(加热盘温度)水煮12h,水煮3-4次,以便出去聚合物中包含的溶剂和无机盐,在真空50℃下烘干24h,最后得到灰色块状固体0.41g,收率:82.0%,膦酰化度:32%,特性粘度:0.51dl/g。
[0116]
实施例12
[0117][0118]
在氮气(99.9%,流速:10-15)氛围下,将实施例7制备的膦酰化聚芳醚腈聚合物(2.4mmol,1.00g)、18ml dmac加入装有磁力搅拌的100ml的单口瓶中进行加热(油温30℃)溶解,反应过夜至高分子完全溶解。在冰浴(2℃)状态下用恒压滴液漏斗匀速加入10ml三甲基溴硅烷,在2℃左右进行酸化反应12h,后常温反应48h,将产物减压蒸馏除去溶剂。在室温下,重新加入18ml dmac,待反应液呈现完全澄清透明状态后,用恒压滴液漏斗匀速加入18ml甲醇充分淬灭,反应时间48h。待反应完成,将产物旋蒸(水温:32℃)除去甲醇,再进行减压蒸馏(油温:60℃,蒸汽温度:30℃)除去多余的dmac,后将固体产物沉入h2o中,固定加热盘温度100℃水洗6次,每次6h。将产物在50℃真空烘箱干燥12h,得产物0.76g,产率85.0%,膦酰化度:68%,特性粘度0.63dl/g。
[0119]
实施例13
[0120][0121]
氮气(99.999%,流速:5-10)氛围下,将2.60g实施例8制备的高分子聚合物完全溶解在53ml的n,n-二甲基乙酰胺(dmac)中,配成5%的溶液;0℃环境下逐滴加入三甲基溴硅烷,冰浴(0-5℃)搅拌24h,减压蒸馏除剩余三甲基溴硅烷;加入dmac重新溶解固体,逐滴加入36ml甲醇,常温搅拌6h,旋蒸、减压蒸馏除溶剂,倒入水中,80℃煮2-3次,真空烘箱50℃干燥24h,称重:2.05g,收率:87.6%,膦酰化度:65%,特性粘度0.80dl/g。
[0122]
实施例14
[0123]
制膜步骤:将真空干燥过的表10中的聚合物样品溶于10ml dmac中,60℃(加热盘温度设定)加热搅拌使样品完全溶解,溶液浓度为5g/dl(dl=100ml),用中速定性滤纸除去其中的不溶物。将制膜时所需10
×
10cm的玻璃板清洗干净,平放在已经调平的调平板上,将滤液均匀倒在玻璃板上,用毛细管充分均匀的延展使高分子平铺在整个玻璃板表面。通过调节红外灯的高度和功率使玻璃板表面温度保持在40℃,待完全干燥。
[0124]
膜性能测试:将干燥后的膜冷却至室温,将带有膜的玻璃板浸泡在1000ml去离子水中,105℃(加热盘温度设定)水煮24小时,将膜与玻璃板分离,得到透明柔韧性较好的聚合物薄膜,用擦镜纸拭去膜表面的水迅速称重并测量膜的长宽和厚度,记录数据,在真空干燥箱120℃下干燥8小时以上,测量烘干后的膜的质量、长宽和厚度,记录数据,计算膜的溶胀度和吸水率,并测试质子传导率。
[0125]
表10膜的溶胀率和吸水率测试结果
[0126][0127]
表11质子传导率测试结果
[0128][0129][0130]
对比例1:热法后膦酰化
[0131][0132]
将二氯亚砜和六水氯化镍(1:15)加入到二颈瓶内,在油温80℃回流反应4h,回流结束后更换蒸馏装置,蒸出过量的溶剂。在回流装置和蒸馏装置的尾接口连上无水氯化钙和碱吸收装置,防止水汽进入并中和产生的氯化氢和二氧化硫气体。最后换用真空泵抽走剩余的二氯亚砜。得到砖粉色固体粉末;亚磷酸三乙酯(tepi)重蒸使用;氮气(99.999%,流速:10-15)氛围下,将1g(1.79mmol)聚合物(合成例1)、1.17g(0.071mmol)nicl2加入到装有常压蒸馏装置、蛇形冷凝管、恒压滴液漏斗和导气管的50ml直三口烧瓶中,然后升温至200℃2h,加入5ml dmi,磁力搅拌使高聚物完全溶解,从恒压滴液漏斗中缓慢滴加tepi约每秒1-2滴,反应3小时后将反应温度降低到170℃再反应8h,将反应物用thf溶解,然后倒入甲醇和异丙醇1:1(v/v)的混合溶剂中沉降洗涤3-4次,然后分别用普通烘箱,真空烘箱120℃12h。反应得到棕灰色粉末状物质。特性粘度:0.20dl/g,产物发生断链情况,无法进行下一步实验。
[0133]
对比例2:高压汞灯后膦酰化
[0134][0135]
在氮气(99.999%,流速:10-15)氛围下,将3g溴代高分子聚合物(合成例1),加入装有磁力搅拌的石英四口瓶中,然后加入120ml dmi 80℃(加热盘)加热搅拌,使原料完全溶解,用恒压滴液漏斗加入80ml的tepi,待完全溶解为澄清透明溶液时,用高压汞灯进行光照反应,减压蒸馏除溶剂,用氯仿溶解,将其滴入20ml异丙醇中,产物析出,异丙醇洗涤3次除去反应中的低分子有机物,过滤,取滤饼普通烘箱120℃干燥12小时,真空烘箱120℃干燥12h,粘度测试。在dmac和dmf中发生断链现象,在四乙二醇二甲醚中的转化率非常低。
[0136]
表12
[0137][0138]
应当注意的是,以上所述的实施例仅用于解释本发明,并不构成对本发明的任何限制。通过参照典型实施例对本发明进行了描述,但应当理解为其中所用的词语为描述性和解释性词汇,而不是限定性词汇。可以按规定在本发明权利要求的范围内对本发明作出修改,以及在不背离本发明的范围和精神内对本发明进行修订。尽管其中描述的本发明涉及特定的方法、材料和实施例,但是并不意味着本发明限于其中公开的特定例,相反,本发明可扩展至其他所有具有相同功能的方法和应用。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。