1.本发明属于生物工程技术领域,具体涉及一种醇脱氢酶突变体及其应用,用于催化对位取代双芳基手性醇合成。
背景技术:
2.手性双芳基醇是一类重要的手性化合物,可用于合成倍他司汀、罗托沙敏等多种药物,因此,手性双芳基醇在医药领域均具有广泛的应用。
3.目前,生产手性双芳基醇的方法主要为化学不对称合成法,此方法为以潜手性双芳基酮为原料,以trans-rucl2[(r)-xylbinap][(r)-daipen]、(s)-[ru(binap)cl2]2(ne3)、(s,s)-6-choona等为催化剂或以手性binal-h等为手性还原剂,在一定的条件下(高压)进行不对称还原反应以获得手性双芳基醇(具体可见参考文献“c.y.chen,et al.,org.lett.,2003,5,5039-5042”、“赵志全等,中国医药工业杂志,2006,37,726-727”、“b.g.wang,et al,org.lett.,2017,19,2094-2097”以及公开号为cn101848893a、cn103121966a的专利申请文本)。
[0004]
但是,化学不对称合成法所使用的催化剂或手性还原剂价格昂贵、反应需要高压条件且合成得到的产物光学纯度低,既不利于工业化生产,也不能满足药物对光学纯度的要求。
[0005]
酶不对称还原法是指以潜手性双芳基酮为原料,以酶作为催化剂进行不对称还原反应以获得手性双芳基醇的方法。与化学不对称合成法相比,酶不对称还原法具有作用条件温和、成本低廉且合成得到的产物光学纯度高的优势,符合“可持续发展”、“绿色化学”、“环境友好制造”等工业发展的目标。因此,利用酶不对称还原法生产手性双芳基醇对实现手性双芳基醇的大规模工业化生产以及手性双芳基醇在医药领域的大规模应用具有重要意义。
[0006]
药物结构中不同的官能团的改变可使整个分子的理化性质、电荷密度等发生变化,很多时候可以根据取代基的特性,为药物增加相应的性质或者减弱毒副作用或不良反应。不同取代基有各自的特点,药物的改进和优化一般都是通过在不同位置上增加或者置换取代基来达成目的。
[0007]
现有的可用于不对称还原潜手性双芳基酮生产手性双芳基醇的酶主要为醇脱氢酶(alcohol dehydrogenase,adh),但是在还原不同的潜手性双芳基酮,催化活性和立体选择性差异较大,无法实现所有手性对位取代双芳基醇的大规模工业化生产以及手性对位取代双芳基醇在医药领域的大规模应用,因此,需要继续开发能够不对称还原不同对位取代基的潜手性双芳基酮生产手性双芳基醇的醇脱氢酶。
技术实现要素:
[0008]
本发明旨在提供一种醇脱氢酶突变体及其应用,针对2-吡啶基[4-(三氟甲基)苯基]甲酮和(4-甲基苯基)(2-吡啶基)甲酮,具有优良的催化活性和立体选择性。
[0009]
本发明的第一方面提供了一种醇脱氢酶的突变体,所述突变体是将氨基酸序列为seq id no.2醇脱氢酶的第214位谷氨酸、第215位苏氨酸和第237位丝氨酸进行突变得到;或
[0010]
所述突变体是将氨基酸序列为seq id no.2醇脱氢酶的第161位苯丙氨酸、第196位丝氨酸、第214位谷氨酸进行突变得到。
[0011]
进一步的,所述突变体是将氨基酸序列如seq id no.2所示的醇脱氢酶的第214位谷氨酸替换为异亮氨酸、第215位苏氨酸替换为丝氨酸,且第237位丝氨酸替换为丙氨酸,命名为m1。
[0012]
进一步的,所述突变体是将氨基酸序列如seq id no.2所示的醇脱氢酶的第161位苯丙氨酸替换为缬氨酸、第196位丝氨酸替换为甘氨酸,且第214位谷氨酸替换为甘氨酸,命名为m2。
[0013]
本发明的第二方面提供了编码上述醇脱氢酶的突变体的基因。
[0014]
本发明的第三方面提供了携带上述基因的载体。
[0015]
进一步的,所述载体为pet28a( )质粒、pet28b( )质粒或pet20b( )质粒,优选为pet28a( )质粒。
[0016]
本发明的第四方面提供了含上述载体的宿主细胞(重组菌)。
[0017]
进一步的,所述宿主细胞在重组大肠杆菌;具体的,其出发菌为e.coli bl21(de3)。
[0018]
进一步的,所述重组大肠杆菌的构建方法:将编码所述突变体的基因(核酸分子)克隆到载体(pet28a( )质粒)中,得到重组载体;将所得重组载体转化到大肠杆菌中,得到重组大肠杆菌。
[0019]
通过培养所得重组大肠杆菌,即可分离纯化获得所述醇脱氢酶的突变体。
[0020]
具体的,利用上述重组菌生产醇脱氢酶的突变体的方法,包括以下步骤:将重组菌接种至含有40-60μg/ml硫酸卡那霉素的lb培养基中,30-40℃,100-200rpm摇床培养,培养液的吸光度od
600
达到0.5-1.0时,加入0.05-1.0mm的异丙基-β-d-六代呋喃半乳糖苷(iptg)进行诱导,诱导温度为16-30℃,诱导5-10h即可获得高效表达重组醇脱氢酶的突变体。
[0021]
本发明的第四方面提供了上述醇脱氢酶的突变体、基因、载体或宿主细胞在催化对位取代双芳基手性醇合成中的应用。
[0022]
进一步的,所述双芳基手性醇为2-吡啶基[4-(三氟甲基)苯基]甲醇或(4-甲基苯基)(2-吡啶基)甲醇;其底物分别为2-吡啶基[4-(三氟甲基)苯基]甲酮和(4-甲基苯基)(2-吡啶基)甲酮。
[0023]
底物取代基团的不同则会改变底物一侧的大小或性质,基团过大则可能导致存在空间位阻使得底物无法形成催化的构象,而性质的改变则会影响底物与酶之间的结合作用力,如增加疏水作用力等,从而降低km值,提高催化效率。
[0024]
本发明的第五方面提供了一种双芳基手性醇的合成方法,包括以下步骤:将上述醇脱氢酶的突变体添加至含有潜手性羰基化合物的反应体系中进行反应,得到所述双芳基手性醇。
[0025]
进一步的,所述潜手性羰基化合物为2-吡啶基[4-(三氟甲基)苯基]甲酮或(4-甲基苯基)(2-吡啶基)甲酮。
[0026]
进一步的,所述反应体系中潜手性羰基化合物的浓度为10-100mm;以所述反应体系为参考,所述醇脱氢酶的突变体的添加量为1-10ku/l。
[0027]
进一步的,所述反应体系中还包括辅酶循环系统,所述辅酶循环系统中含有以下组合中的一种:葡萄糖脱氢酶gdh和d-葡萄糖;亚磷酸盐脱氢酶(ftdh)和亚磷酸盐;甲酸脱氢酶(fdh)和甲酸;乳酸脱氢酶(ldh)和乳酸;或者甘油脱氢酶和甘油。
[0028]
具体的,所述合成方法包括以下步骤:构建反应体系,2-吡啶基[4-(三氟甲基)苯基]甲酮或(4-甲基苯基)(2-吡啶基)甲酮浓度为10-100mm,上述醇脱氢突变体用量为1-10ku/l,nadp
(烟酰胺腺嘌呤二核苷磷酸)用量为0.1-1.0mm,加入辅酶循环系统,辅酶循环系统中含有葡萄糖脱氢酶gdh和d-葡萄糖,其中葡萄糖脱氢酶gdh用量为1-10ku/l,d-葡萄糖用量为20-1000mm,磷酸盐缓冲液的浓度为0.1-0.2m;在30-35℃,ph6-8的条件下反应1-24h,不对称还原反应结束后,可按照有机溶剂萃取方法从反应液中提取手性2-吡啶基[4-(三氟甲基)苯基]甲醇或(4-甲基苯基)(2-吡啶基)甲醇。
[0029]
进一步的,所述辅酶循环系统还可以是亚磷酸盐/亚磷酸盐脱氢酶(ftdh)、甲酸/甲酸脱氢酶(fdh)、乳酸/乳酸脱氢酶(ldh)或甘油/甘油脱氢酶。
[0030]
进一步的,所述(r)-和(s)-产物的色谱分析方法为:取100μl反应液,加入500μl乙酸乙酯,震荡1-2min,12000rpm离心2-5min,取上清到离心管中,待有机自然挥发完全,加入500μl色谱纯乙醇,进行手性液相色谱分析转化率和ee值。色谱条件具体如下:daicel chiralcel ad-h(5μm,250mm
×
4.6mm)液相色谱柱,流动相为正己烷:异丙醇(90:10,v/v),流速1ml/min,柱温30℃,紫外检测波长254nm,进样量10μl,(s)-和(r)-(4-甲基苯基)(2-吡啶基)甲醇保留时间分别为12.6min和14.7min,2-吡啶基[4-(三氟甲基)苯基]甲醇保留时间分别为9.4min和12.1min。
[0031]
本发明的技术方案相比现有技术具有以下优点:
[0032]
(1)本发明得到的醇脱氢酶突变对多种羰基化合物具有较高的活力,可以催化还原多种脂肪族或芳基取代的酮底物,尤其是空间位阻较大的双芳基酮底物。通过组合突变手段对野生型醇脱氢酶(kpadh,来源于kluyveromyces polyspora)进行分子改造,提高该酶的立体选择性,这将使其具有更高的工业应用价值;
[0033]
(2)与野生型醇脱氢酶kpadh相比,本发明所述醇脱氢酶突变体m1,m2对底物2-吡啶基[4-(三氟甲基)苯基]甲酮及(4-甲基苯基)(2-吡啶基)甲酮提高了立体选择性,其产物2-吡啶基[4-(三氟甲基)苯基]甲醇及(4-甲基苯基)(2-吡啶基)甲醇的ee值由野生型的49.4%(s)、82.6%(r)提高至》99.9%(s)(m1),》99.9%(r)(m2);本发明所获得的醇脱氢酶突变体特别适合于对位取代双芳基酮的不对称还原,具有良好的工业应用前景。
附图说明
[0034]
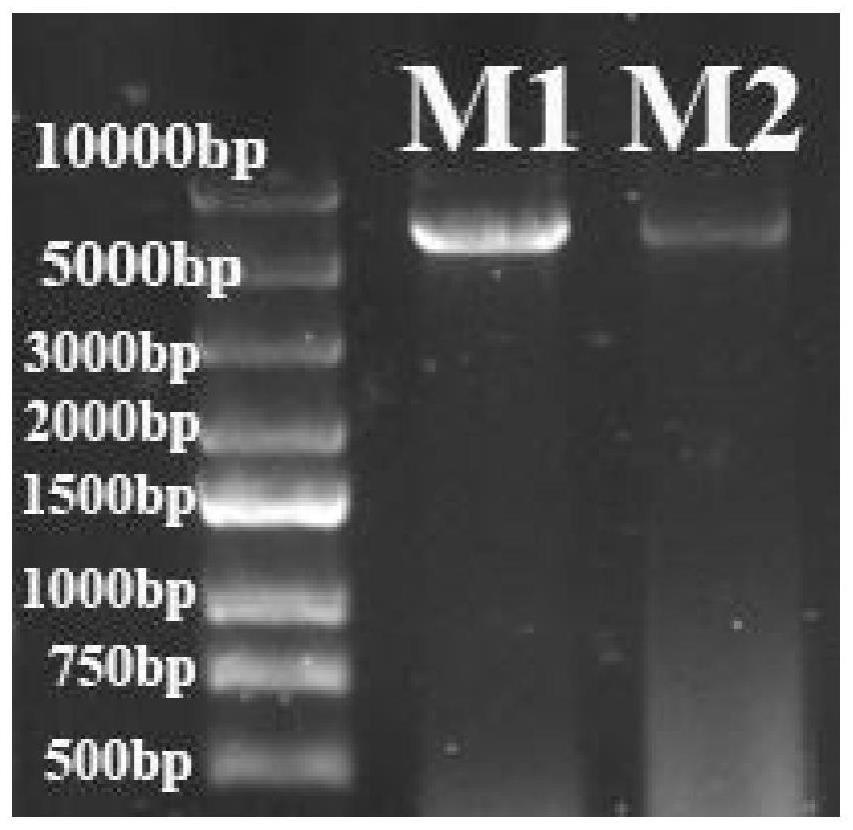
图1为醇脱氢酶突变体m1和m2的全质粒pcr核酸电泳图;
[0035]
图2为醇脱氢酶突变体梯度洗脱蛋白电泳图;
[0036]
图3为醇脱氢酶突变体m2催化还原2-吡啶基[4-(三氟甲基)苯基]甲酮产物手性色谱图;
[0037]
图4为醇脱氢酶突变体m2催化还原(4-甲基苯基)(2-吡啶基)甲酮产物手性色谱图。
具体实施方式
[0038]
下面结合附图和具体实施例对本发明作进一步说明,以使本领域的技术人员可以更好地理解本发明并能予以实施,但所举实施例不作为对本发明的限定。
[0039]
下述实施例中,醇脱氢酶的活力和产物光学纯度的测定方法如下:
[0040]
总反应体系为200μl,包括:1.0mm nadph(还原型烟酰胺腺嘌呤二核苷酸磷酸),1.0mm底物2-吡啶基[4-(三氟甲基)苯基]甲酮或(4-甲基苯基)(2-吡啶基)甲酮,磷酸钠缓冲液(pbs,100mm,ph7.0),充分混匀,30℃保温2min,加入适量的酶液,检测340nm下光吸收值的变化。
[0041]
酶活力的计算公式如下:
[0042]
酶活力(u)=ew
×v×
103/(6220
×
1);
[0043]
式中,ew为1min内340nm处吸光度的变化;v为反应液的体积,单位为ml;6220为nadph的摩尔消光系数,单位为l/(mol
·
cm);1为光程距离,单位为cm。1个活力单位(u)对应于上述条件下每分钟催化氧化1μmol nadph所需的酶量。
[0044]
光学纯度ee的测定方法:
[0045]
as:液相色谱获得的(s)-产物的摩尔浓度;ar:液相色谱获得的(r)-产物的摩尔浓度;
[0046]
实施例1:醇脱氢酶突变体基因和重组表达转化体的构建
[0047]
采用全质粒pcr方法对位于214位、215位、237位氨基酸(第161位、第196位、第237位氨基酸)残基进行定点突变,构建迭代组合突变体。其引物设计如下(均按5
’‑3’
方向描述,下划线代表突变位点):
[0048]
m1(以pet28a-kpadh重组质粒为模板)
[0049]
e214i-f:aagaaactaaatattacttgtgaaatt
[0050]
e214i-r:aatttcacaagtaatatttagtttctt
[0051]
t215s-f:aaactaaatgaaagctgtgaaattatc
[0052]
e215s-r:gataatttcacagctttcatttagttt
[0053]
s237a-f:aagactcacttcgcacaattcattgat
[0054]
s237a-r:atcaatgaattgtgcgaagtgagtctt
[0055]
m2(以pet28a-kpadh重组质粒为模板)
[0056]
f161v-f tatgaaaatgtcgttactgcttattgt
[0057]
f161v-r acaataagcagtaacgacattttcata
[0058]
s196g-f actatccacccaggtttcgttttcgga
[0059]
s196g-r tccgaaaacgaaacctgggtggatagt
[0060]
e214g-f aagaaactaaatggtacttgtgaaatt
[0061]
e214g-r aatttcacaagtaccatttagtttctt
[0062]
pcr反应体系为:pcr反应体系(50μl)包括kod酶(2.5u/μl)1.0μl,模板(5-50ng)1.0μl,dntp 4.0μl,10
×
reaction buffer 5.0μl,上下游引物各1.0μl,ddh2o补足至50μl。
[0063]
pcr扩增程序为:(1)94℃变性3min,(2)94℃变性30sec,(3)54℃退火30sec,(4)72℃延伸150sec,重复步骤(2)-(4)进行10-15个循环,最后72℃延伸10min,4℃保存pcr扩增
产物。
[0064]
pcr结束后,添加dpni(甲基化模板消化酶)限制性内切酶于反应混合物中并置于37℃孵育1h,用cacl2热转化法将10μl消化后pcr反应液转入50μl e.coli bl21(de3)感受态细胞,并均匀涂布于含有50μg/ml硫酸卡那霉素的lb琼脂平板,37℃倒置培养12h。
[0065]
实施例2:醇脱氢酶及其突变体的表达及纯化
[0066]
将携带立体选择性改善突变体的重组大肠杆菌按2%的转接量接种至含有硫酸卡那霉素(50μg/ml)的lb培养基中,37℃,200rpm摇床培养,培养液的吸光度od
600
达到0.8时,加入0.2mm的异丙基-β-d-六代呋喃半乳糖苷(iptg)进行诱导,诱导温度为25℃,诱导进行8h后,8000rpm离心10min获得高效表达重组醇脱氢酶突变体的菌体,将收集的菌体悬浮于磷酸钾缓冲液(100mm,ph 6.0)中,超声破碎。
[0067]
纯化所使用的柱子为镍亲和柱histrap ff crude,利用重组蛋白上的组氨酸标签进行亲和层析来完成。首先使用a液将镍柱平衡,粗酶液上样,继续使用a液将穿透峰洗脱下来,待平衡后用b液(20mm磷酸钠,500mm nacl,1000mm咪唑,ph 7.4)进行梯度洗脱,将结合到镍柱上的重组蛋白洗脱下来,获得重组醇脱氢酶突变体。对纯化后的蛋白进行活力测定(nadph为辅酶)及sds-page分析。镍柱纯化后,在45kda左右显示单条带,且杂蛋白较少,说明柱纯化效果较好。之后使用hi-trap desalting脱盐柱(ge healthcare)将纯化后的醇脱氢酶蛋白置换到tris-hcl(100mm,ph 7.0)缓冲液中。
[0068]
实施例3:醇脱氢酶突变体还原潜手性羰基化合物的情况
[0069]
所考察的潜手性羰基化合物包括4-氯苯基-吡啶-2-基-甲酮((4-chlorophenyl)-(pyridin-2-yl)-methanone,cpmk),(4-甲基苯基)(2-吡啶基)甲酮(2-(4-methylbenzoyl)pyridine),2-吡啶基[4-(三氟甲基)苯基]甲酮(2-pyridinyl[4-(trifluoromethyl)phenyl]methanone)。由表1可以看出,对于底物底物2-吡啶基[4-(三氟甲基)苯基]甲酮及(4-甲基苯基)(2-吡啶基)甲酮,m1,m2的还原产物的ee值达到97%以上。立体选择性的显著提升是由于突变位点的引入,增强了酶与底物的结合力,使得底物可以以从而有利于底物的稳定和催化。
[0070]
表1
[0071][0072]
实施例4:醇脱氢酶突变体的动力学和立体选择性分析
[0073]
测定kpadh在不同底物浓度和辅酶浓度情况下的活力,并根据活力和底物浓度的倒数做出双倒数曲线,计算动力学参数。
[0074]
由表2可知kpadh对底物2-吡啶基[4-(三氟甲基)苯基]甲酮的k
cat
/km为2.41s-1
·
mm-1
,其还原产物构型为s构型,ee值为49.4%(s)。突变体m1,m2合成(s)-,(r)-2-吡啶基[4-(三氟甲基)苯基]甲醇立体选择性提高,达到98%以上,产物ee值分别为99.0%(s)(m1),98.7%(r)(m2)。
[0075]
由表3可知kpadh对底物(4-甲基苯基)(2-吡啶基)甲酮的k
cat
/km为4.58s-1
·
mm-1
,其还原产物构型为s构型,ee值为82.6%(s)。突变体m1,m2合成(s)-,(r)-(4-甲基苯基)(2-吡啶基)甲醇立体选择性提高,达到97%以上,产物ee值分别为99.9%(s)(m1),97.7%(r)(m2)。
[0076]
表2醇脱氢酶突变体对底物2-吡啶基[4-(三氟甲基)苯基]甲酮的动力学参数及立体选择性
[0077][0078]
表3醇脱氢酶突变体对底物(4-甲基苯基)(2-吡啶基)甲酮的动力学参数及立体选择性
[0079][0080]
实施例5:醇脱氢酶突变体不对称还原制备高光学纯度的(s)-(4-甲基苯基)(2-吡啶基)甲醇
[0081]
建立了20ml的生物催化体系:100mm磷酸钾缓冲液(ph7.0),加入实例2获得的醇脱氢酶突变体m1细胞10g/l,(4-甲基苯基)(2-吡啶基)甲酮50mm。在30℃和200rpm条件下反应24h,恒定ph 7.0。反应过程中的转化率分析结果见表4。当底物浓度为50mm,醇脱氢酶突变体m1均能够在24h内达到99%以上的底物转化率,它们的还原产物均为(s)-(4-甲基苯基)(2-吡啶基)甲醇,其中野生型kpadh还原产物的ee值仅为82.6%,突变体m1还原产物的ee值可达到99%。将所得(s)-(4-甲基苯基)(2-吡啶基)甲醇粗品重新溶解于乙醇中,加入相应的产物纯品为晶种于4℃重结晶,最终获得光学纯度均为》99.9%ee的产品。
[0082]
表4醇脱氢酶突变体m1催化50mm(4-甲基苯基)(2-吡啶基)甲酮不对称还原
[0083][0084]
实施例6:醇脱氢酶突变体不对称还原制备高光学纯度的(r)-2-吡啶基[4-(三氟甲基)苯基]甲醇
[0085]
建立了20ml的生物催化体系:100mm磷酸钾缓冲液(ph7.0),加入实例2获得的醇脱氢酶突变体m2细胞10g/l,2-吡啶基[4-(三氟甲基)苯基]甲酮50mm。在30℃和200rpm条件下反应24h,恒定ph7.0。反应过程中的转化率分析结果见表5。当底物浓度为50mm,醇脱氢酶突变体m2均能够在24h内达到99%以上的底物转化率,它们的还原产物均为(r)-2-吡啶基[4-(三氟甲基)苯基]甲醇,其中野生型kpadh还原产物的ee值为49.4%(s),突变体m2还原产物的ee值可达到98.7%(r)。将所得(r)-2-吡啶基[4-(三氟甲基)苯基]甲醇粗品重新溶解于乙醇中,加入相应的产物纯品为晶种于4℃重结晶,最终获得光学纯度均为》99%ee的产品。
[0086]
表5醇脱氢酶突变体m2催化50mm 2-吡啶基[4-(三氟甲基)苯基]甲酮不对称还原
[0087][0088][0089]
综上,本发明的醇脱氢酶突变体具有优良的催化活性和立体选择性,可高效催化制备一系列r-和s-构型的具有对位取代基的手性双芳基醇;将本发明醇脱氢酶突变体与葡萄糖脱氢酶或甲酸脱氢酶进行偶联,可用于多种抗组胺药物手性双芳基醇中间体的合成;本发明的醇脱氢酶不对称催化还原制备双芳基手性醇的方法具有操作简便、底物浓度高、反应完全、产品纯度高的优势,具有很强的工业应用前景。
[0090]
显然,上述实施例仅仅是为清楚地说明所作的举例,并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引申出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
[0091]
[0092]
[0093]
[0094]
[0095]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。