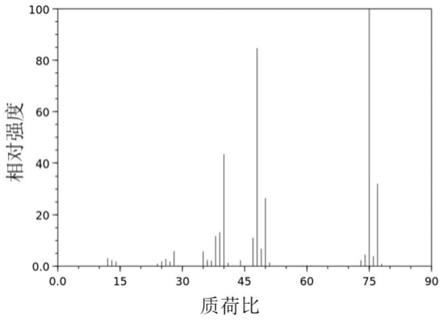
1.本发明涉及一种吡咯并嘧啶五元氮杂环衍生物及其制备方法,以及在制备与jak激酶功能有关的适应症药物中的用途。
背景技术:
2.jak激酶(janus kinase)是一类胞内非受体酪氨酸激酶家族,蛋白大约为110kda~140kda,目前发现4个家族成员:jak1、jak2、jak3和tyk2,其中jak1、jak2和tyk2广泛存在各种组织和细胞中,而jak3只在淋巴细胞和骨髓中表达。jak蛋白与不同的受体特异性结合能发挥特定的生理功能。研究表明,jak1是炎症、癌症、免疫等疾病的靶点,jak2是血液系统相关疾病的靶点,jak3是自身免疫性疾病的热门靶点。截止到目前,全球范围内共有10款jak激酶抑制剂产品获批上市。包括六款第一代jak抑制剂,分别为诺华/incyte的鲁索替尼(ruxolitinib)、辉瑞的托法替尼(tofacitinib)、礼来/incyte的巴瑞替尼(baricitinib)、辉瑞的奥拉替尼(oclacitinib)、安斯泰来(astellas)的佩非西替尼(peficitinib)、japan tobacco的迪高替尼(delgocitinib);和四款第二代的jak抑制剂,分别为新基的菲卓替尼(fedratinib),艾伯维的乌帕替尼(upadacitinib)以及吉利德的菲戈替尼(filgotinib)。其适应症包括类风湿关节炎、骨髓纤维化、银屑病关节炎、溃疡性结肠炎、移植物抗宿主病、特应性皮炎等。此外,目前还有数十款jak抑制剂处于临床开发阶段,且大多为新一代疗法,有10款产品进入iii期临床阶段。其中辉瑞的阿布罗替尼(abrocitinib)和cti biopharma的帕瑞替尼(pacritinib)已向fda提交新药上市申请(nda)。然而,无论是已经获批上市的jak抑制剂,还是正在开发的jak候选药,大多都面临安全性和耐受性方面的挑战。因此,在本领域内开发选择性高、疗效和安全性好的候选药物具有非常重要的意义和很好的应用前景。
3.
技术实现要素:
4.针对上述现有技术的不足,本发明提供了一种吡咯并嘧啶五元氮杂环衍生物及其制备方法和用途。本发明吡咯并嘧啶五元氮杂环衍生物合成方法简单高效,可用于制备包括炎性疾病、自身免疫疾病以及癌症相关疾病的治疗药物。
5.本发明吡咯并嘧啶五元氮杂环衍生物,具有下式i或式ii所示结构:
[0006][0007]
其中,x选自c或n;y选自c或n;
[0008]
r1选自
[0009]
r2选自so2r3、s(o)r3或c(o)r3;
[0010]
r3选自或c1-c6烷基,其中r3为c1-c6烷基时r2为s(o)r3;
[0011]
r4选自c3-c8环烷基,取代或未取代的苯基,六元杂环基或桥环基;
[0012]
r5为r6为so2r7或c(o)r7;r7为c1-c6烷基、c3-c8环烷基、苯基、五元或六元杂芳基、桥环基或并环基,其中所述烷基、环烷基、苯基、杂芳基、桥环基或并环基可任选地被1、2或3个独立的f、cl、cn、ph或c1-c4烷基中的取代基取代;
[0013]
*表示立体构型为s或r光学纯度或其消旋体。
[0014]
本发明中,作为优选技术方案,在式i或式ii中:
[0015]
x选自c或n;y选自c或n;
[0016]
r1选自
[0017]
r2选自so2r3、s(o)r3或c(o)r3;
[0018]
r3选自或乙基,其中r3为乙基时r2为s(o)r3;
[0019]
r4为c3-c8环烷基,取代或未取代的苯基,四氢吡喃基或桥环基;
[0020]
r5为r6为so2r7或c(o)r7;r7为c1-c6烷基、c3-c8环烷基、苯基或并环基,其中所述烷基、环烷基、苯基或并环基可任选地被1、2或3个独立的f、cl、cn、ph或c1-c4烷基中的取代基取代。
[0021]
*表示立体构型为s或r光学纯度或其消旋体。
[0022]
本发明中,作为进一步优选技术方案,在式i或式ii中,
[0023]
x选自c或n;y选自n;
[0024]
r1选自
[0025]
r2选自so2r3、s(o)r3或c(o)r3;
[0026]
r3选自或乙基,其中r3为乙基时r2为s(o)r3;
[0027]
r4为环丙基、环丁基、环戊基、环己基、苯基或4-硝基苯基、四氢-2h-吡喃-4-基、7,7-二甲基-2-氧代双环[2.2.1]庚-1-基;
[0028]
r5为r6为so2r7或c(o)r7;r7为甲基、乙基、正丙基、异丙基、环丙基、4-甲基苯基、4-叔丁基苯基、苄基、苯基、氰乙基或萘环基。
[0029]
*表示立体构型为s或r光学纯度或其消旋体。
[0030]
本发明中,作为特别优选的技术方案,所述吡咯并嘧啶五元氮杂环衍生物为下表1所示的任一结构化合物:
[0031]
表1
[0032]
[0033]
[0034]
[0035]
[0036]
[0037][0038]
本发明所用术语“c1-c6烷基”是指具有1至6个碳原子的直链或支链烷基,非限制性地包括甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、特丁基、正戊基、异戊基、正己基等。术语“c1-c4烷基”是指具有1至4个碳原子的直链或支链烷基,非限制性地包括甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基等。术语“c3-c8环烷基”是指在环上具有3至8个碳原子的环状烷基,非限制性地包括环丙基、环丁基、环戊基、环己基、环庚基、环辛基等。
[0039]
在本发明中,所述特定基团之前的c1-c6、c1-c4、c3-c8等表示基团中所含碳原子的个数,例如c1-c6表示碳原子数可以为1、2、3、4、5或6的基团、c1-c4表示碳原子数可以为1、2、3或4的基团、c3-c8表示碳原子数可以为3、4、5、6、7或8的基团。
[0040]
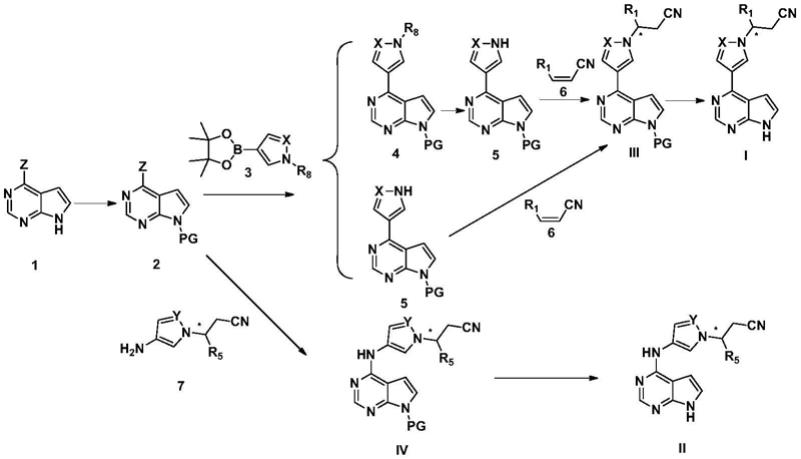
本发明还提供了吡咯并嘧啶五元氮杂环衍生物的制备方法,制备路线如下所示:
[0041][0042]
除另有注明外,制备路线中各基团定义同上文。
[0043]
其中:z选自氯、溴或碘;r8选自烷氧基氨基保护基或硅烷氨基保护基;pg表示保护基,选自x、y、r1和r5取代基的限定与上文相同,在此不再赘述。
[0044]
本发明吡咯并嘧啶五元氮杂环衍生物的制备方法,包括如下步骤:
[0045]
步骤1:式1化合物与氨基保护试剂在碱性条件下反应生成式2化合物;
[0046]
步骤1所述反应是在氨基保护试剂和碱的作用下进行的。所述氨基保护试剂选自2-(三甲基硅烷基)乙氧甲基氯(semcl)、二碳酸二叔丁酯(boc2o)、特戊酸氯甲酯、氯甲酸苄酯(cbzcl)、对甲苯磺酰氯、三苯基甲基溴中的任意一种或至少两种的组合;所述碱为有机碱或无机碱,所述有机碱选自三乙胺、n,n-二异丙基乙胺、n,n-二甲基苯胺、吡啶、甲醇钠、乙醇钠、叔丁醇钠或叔丁醇钾中的任意一种或至少两种的组合;所述无机碱选自氢氧化钠、氢氧化钾、碳酸钠、碳酸钾或氢化钠中的任意一种或至少两种的组合。
[0047]
优选地,式1化合物与氨基保护试剂和碱的摩尔比为1:1.0-2.0:1.0-2.0,例如1:1.0:1.0、1:1.1:1.1、1:1.2:1.2、1:1.3:1.3、1:1.4:1.4、1:1.5:1.5、1:1.6:1.6、1:1.7:1.7、1:1.8:1.8、1:1.9:1.9或1:2.0:2.0。
[0048]
优选地,步骤1的反应溶剂选自甲苯、乙腈、四氢呋喃、1,4-二氧六环、n,n-二甲基甲酰胺、二甲基亚砜、n-甲基吡咯烷酮或六甲基磷酰三胺中的任意一种或至少两种的组合。
[0049]
优选地,步骤1的反应温度为大于等于0℃且小于等于反应溶剂的沸点,例如25℃、30℃、35℃、40℃、45℃、50℃、60℃、70℃、75℃、80℃、85℃、90℃等,或者在溶剂沸点即回流状态下进行反应;反应时间为0.5-48小时,例如0.5小时、1小时、3小时、5小时、8小时、10小时、12小时、15小时、18小时、20小时、23小时、25小时、28小时、30小时、33小时、35小时、38小时、40小时、44小时或48小时。
[0050]
步骤2:式2化合物与式3化合物在催化剂和碱的条件下发生suzuki偶联反应得到式4化合物,式4化合物在酸性条件下发生水解反应得到式5化合物;或者式2化合物与式3化
合物在催化剂和碱的条件下发生suzuki偶联反应直接得到式5化合物;
[0051]
步骤2所述反应是在催化剂和碱的作用下进行的。所述催化剂选自铜催化剂、钯催化剂、钯铜合金催化剂、镍催化剂、铂催化剂中的任意一种或至少两种的组合;所述碱为有机碱或无机碱,所述有机碱选自三乙胺、n,n-二异丙基乙胺、n,n-二甲基苯胺、吡啶、甲醇钠、乙醇钠、叔丁醇钠或叔丁醇钾中的任意一种或至少两种的组合;所述无机碱选自氢氧化钠、氢氧化钾、碳酸钠、碳酸钾或氢化钠中的任意一种或至少两种的组合。
[0052]
优选地,式2化合物与式3化合物、催化剂及碱的摩尔比为1:1.0-1.5:0.01-0.1:1.0-5.0,例如1:1.0:0.01:1.0、1:1.1:0.02:2.0、1:1.2:0.04:3.0、1:1.3:0.06:4.0、1:1.4:0.08:5.0或1:1.5:0.1:5.0。
[0053]
优选地,步骤2的反应溶剂选自甲苯、乙腈、四氢呋喃、1,4-二氧六环、n,n-二甲基甲酰胺、n-甲基吡咯烷酮、水或正丁醇中的任意一种或至少两种的组合。
[0054]
优选地,步骤2的反应温度为大于等于0℃且小于等于反应溶剂的沸点,例如25℃、30℃、35℃、40℃、45℃、50℃、60℃、70℃、75℃、80℃、85℃、90℃、100℃、120℃、140℃、150℃等,或者在溶剂沸点即回流状态下进行反应;反应时间为0.5-48小时,例如0.5小时、1小时、3小时、5小时、8小时、10小时、12小时、15小时、18小时、20小时、23小时、25小时、28小时、30小时、33小时、35小时、38小时、40小时、44小时或48小时。
[0055]
此外,步骤2中,式4化合物在酸性条件下发生水解反应得到式5化合物。所述水解反应是在水、甲醇、乙醇、四氢呋喃或1,4-二氧六环中任意一种或至少两种的混合溶剂中进行的。
[0056]
优选地,所述水解反应是在酸性介质存在下进行的,所述酸性介质选自盐酸、硫酸、磷酸或氢溴酸任意一种或至少两种的组合。
[0057]
优选地,所述酸性介质的用量为式4化合物摩尔量的1-5倍,例如,1倍、1.3倍、1.5倍、2倍、2.5倍、3倍、3.5倍、4倍、4.5倍或5倍。
[0058]
优选地,所述水解反应的温度为大于等于0℃且小于等于反应溶剂的沸点,例如25℃、30℃、35℃、40℃、45℃、50℃、60℃、70℃、75℃、80℃、85℃、90℃等,或者在溶剂沸点即回流状态下进行反应。
[0059]
步骤3:式5化合物和式6化合物在碱性条件下发生加成反应得到式iii化合物;
[0060]
步骤3中所述加成反应是在碱性物质存在下进行的,所述碱性物质选自三乙胺、n,n-二异丙基乙胺、吡啶或1,8-二氮杂双环[5.4.0]十一碳-7-烯(dbu)中的任意一种或至少两种的组合。
[0061]
优选地,式5化合物与式6化合物及碱的摩尔比为1:1.0-2.0:1.0-2.0,例如1:1.0:1.0、1:1.1:1.1、1:1.2:1.2、1:1.3:1.3、1:1.4:1.4、1:1.5:1.5、1:1.6:1.6、1:1.7:1.7、1:1.8:1.8、1:1.9:1.9或1:2.0:2.0。
[0062]
优选地,步骤3中所述加成反应是在二氯甲烷、乙酸乙酯、四氢呋喃、1,4-二氧六环、乙腈、n,n-二甲基甲酰胺、二甲基亚砜或n-甲基吡咯烷酮中任意一种或至少两种的混合溶剂中进行的。
[0063]
优选地,步骤3的反应的温度为大于等于-10℃且小于等于反应溶剂的沸点,例如-10℃、-5℃、0℃、25℃、30℃、35℃、40℃、45℃、50℃、60℃、70℃、75℃、80℃、85℃、90℃等,或者在溶剂沸点即回流状态下进行反应;反应时间为0.5-48小时,例如0.5小时、1小时、3小
时、5小时、8小时、10小时、12小时、15小时、18小时、20小时、23小时、25小时、28小时、30小时、33小时、35小时、38小时、40小时、44小时或48小时。
[0064]
步骤4:式iii化合物在酸性或碱性条件下脱保护基pg得到式i化合物。
[0065]
步骤4中所述反应是在二氯甲烷、乙酸乙酯、四氢呋喃、1,4-二氧六环、乙腈、n,n-二甲基甲酰胺、水、甲醇或乙醇中任意一种或至少两种的混合溶剂中进行的。
[0066]
优选地,步骤4所述反应是在酸性或碱性物质存在下进行的,所述酸性物质优选乙酸或三氟乙酸,所述碱性物质优选三乙胺、n,n-二异丙基乙胺、吡啶、乙二胺、氨水、氢氧化钠、氢氧化锂或氢氧化钾中的任意一种或至少两种的组合。
[0067]
优选地,酸性或碱性物质的用量为式iii化合物摩尔量的1-10倍,例如,1倍、2倍、3倍、4倍、5倍、6倍、7倍、8倍、9倍或10倍。
[0068]
优选地,步骤4的反应温度为大于等于0℃且小于等于反应溶剂的沸点,例如25℃、30℃、35℃、40℃、45℃、50℃、60℃、70℃、75℃、80℃、85℃、90℃等,或者在溶剂沸点即回流状态下进行反应;反应时间为0.5-48小时,例如0.5小时、1小时、3小时、5小时、8小时、10小时、12小时、15小时、18小时、20小时、23小时、25小时、28小时、30小时、33小时、35小时、38小时、40小时、44小时或48小时。
[0069]
步骤5:式2化合物和式7化合物在酸性条件下发生取代反应得到式iv化合物;
[0070]
优选地,步骤5所述取代反应是在二氯甲烷、乙酸乙酯、四氢呋喃、1,4-二氧六环、乙腈、n,n-二甲基甲酰胺、乙酸、三氟乙酸或水中任意一种或至少两种的混合溶剂中进行的。
[0071]
优选地,式7化合物与式2化合物的摩尔比为1:1.0-1.5,例如1:1.0、1:1.1、1:1.2、1:1.3、1:1.4或1:1.5。
[0072]
优选地,步骤5的反应温度为大于等于0℃且小于等于反应溶剂的沸点,例如25℃、30℃、35℃、40℃、45℃、50℃、60℃、70℃、75℃、80℃、85℃、90℃等,或者在溶剂沸点即回流状态下进行反应;反应时间为0.5-48小时,例如0.5小时、1小时、3小时、5小时、8小时、10小时、12小时、15小时、18小时、20小时、23小时、25小时、28小时、30小时、33小时、35小时、38小时、40小时、44小时或48小时。
[0073]
步骤6:式iv化合物在酸性或碱性条件下脱保护基pg得到式ii化合物。
[0074]
步骤6所述反应是在二氯甲烷、乙酸乙酯、四氢呋喃、1,4-二氧六环、乙腈、n,n-二甲基甲酰胺、水、甲醇或乙醇中任意一种或至少两种的混合溶剂中进行的。
[0075]
优选地,步骤6所述反应是在酸性或碱性物质存在下进行的,所述酸性物质优选乙酸或三氟乙酸,所述碱性物质优选三乙胺、n,n-二异丙基乙胺、吡啶、乙二胺、氨水、氢氧化钠、氢氧化锂或氢氧化钾中的任意一种或至少两种的组合。
[0076]
优选地,酸性或碱性物质的用量为式iv化合物摩尔量的1-10倍,例如,1倍、2倍、3倍、4倍、5倍、6倍、7倍、8倍、9倍或10倍。
[0077]
优选地,步骤6的反应温度为大于等于0℃且小于等于反应溶剂的沸点,例如25℃、30℃、35℃、40℃、45℃、50℃、60℃、70℃、75℃、80℃、85℃、90℃等,或者在溶剂沸点即回流状态下进行反应;反应时间为0.5-48小时,例如0.5小时、1小时、3小时、5小时、8小时、10小时、12小时、15小时、18小时、20小时、23小时、25小时、28小时、30小时、33小时、35小时、38小时、40小时、44小时或48小时。
[0078]
本发明吡咯并嘧啶五元氮杂环衍生物的用途,是以所述吡咯并嘧啶五元氮杂环衍生物、其互变异构体、对映体、非对映体或其药学上可接受的盐用于制备预防和/或治疗与jak激酶功能有关的适应症的药物中的应用。
[0079]
所述与jak激酶功能相关的适应症包括炎性疾病、自身免疫疾病以及癌症相关疾病,其中所述炎性疾病、自身免疫疾病以及癌症疾病包括:类风湿性关节炎、强直性脊柱炎、系统性红斑狼疮、溃疡性结肠炎、银屑病、斑秃、白癜风、哮喘、过敏性鼻炎、过敏性结膜炎、特应性皮炎或神经性皮炎等适应症。
[0080]
本发明中,所述吡咯并嘧啶五元氮杂环衍生物的互变异构体、对映体、非对映体或其药学上可接受的盐同样能够发挥与所述吡咯并嘧啶五元氮杂环衍生物同样的作用效果。
[0081]
其中,药学上可接受的盐具体为式i或式ii化合物与无机酸或有机酸反应形成常规的可药用盐。例如,常规的可药用盐可通过式i或式ii化合物与无机酸或有机酸反应制得,所述无机酸包括盐酸、氢溴酸、硫酸、硝酸、胺基磺酸和磷酸等,以及所述有机酸包括柠檬酸、酒石酸、乳酸、丙酮酸、乙酸、苯磺酸、对甲苯磺酸、甲磺酸、萘磺酸、乙磺酸、萘二磺酸、马来酸、苹果酸、丙二酸、富马酸、琥珀酸、丙酸、草酸、三氟乙酸、硬酯酸、扑酸、羟基马来酸、苯乙酸、苯甲酸、水杨酸、谷氨酸、抗坏血酸、对胺基苯磺酸、2-乙酰氧基苯甲酸和羟乙磺酸等;或者式i或式ii化合物与无机碱形成的钠盐、钾盐、钙盐、铝盐或铵盐;或者式i或式ii化合物与有机碱形成的甲胺盐、乙胺盐或乙醇胺盐。
[0082]
所述药物的制备过程中可以采用药学上可以接受的载体,具体是指一种或多种相容性固体或液体填料或凝胶物质,它们适合于人使用,而且必须有足够的纯度和足够低的毒性。药学上可以接受的载体部分例子有纤维素及其衍生物(如羧甲基纤维素钠、乙基纤维素钠、纤维素乙酸酯等)、明胶、滑石、固体润滑剂(如硬脂酸、硬脂酸镁)、硫酸钙、植物油(如豆油、芝麻油、花生油、橄榄油等)、多元醇(如丙二醇、甘油、甘露醇、山梨醇等)、乳化剂润湿剂(如十二烷基硫酸钠)、着色剂、调味剂、稳定剂、抗氧化剂、防腐剂、无热原水等。
具体实施方式
[0083]
下面结合具体实施例,进一步阐述本发明。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
[0084]
合成实施例
[0085]
合成实施例1:2-(3-(4-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)-1-((环丙基甲基)磺酰基)氮杂环丁-3-基)乙腈(化合物编号i-1)的制备
[0086]
(1)(4-氯-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯的制备,具体制备方法如下:
[0087][0088]
将4-氯-7h-吡咯并[2,3-d]嘧啶(5.00g,32.56mmol,1.0eq)用n,n-二甲基甲酰胺
(50ml)溶解,室温下加入碳酸钾(4.50g,32.56mmol,1.0eq),搅拌10min后,缓慢滴入特戊酸氯甲酯(5.88g,39.07mmol,1.2eq),然后室温下反应2h,tlc监测原料反应完全时,结束反应。向体系中加入35ml水,固体析出,抽滤,滤饼烘干,得到白色固体产物(4-氯-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯(7.94g,收率:91%)。1h nmr(400mhz,dmso-d6)δ8.71(s,1h),7.83(d,j=3.7hz,1h),6.72(d,j=3.7hz,1h),6.24(s,2h),1.07(s,9h).hrms(esi)calcd for c
12h14
cln3o2[m h
]:268.0847,found:268.0893.
[0089]
(2)4-(1-(1-乙氧基乙基)-1h-吡唑-4-基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯的制备,具体制备方法如下:
[0090][0091]
将化合物(4-氯-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯(5.00g,18.68mmol,1.0eq)用1,4-二氧六环/水=2:1(30ml)溶解,室温下搅拌15min后,依次加入碳酸钾(7.74g,56.03mmol,3.0eq)、1-(1-乙氧基乙基)-4-吡唑硼酸频哪醇酯(5.72g,21.48mmol,1.15eq)以及四(三苯基膦)钯(431.64mg,373.54μmol,0.02eq),n2保护下,加热升温至90℃回流反应12h。tlc检测反应完全后,将体系冷却至室温,加水淬灭,用乙酸乙酯萃取(20ml*2),有机层经饱和食盐水洗涤、无水硫酸钠干燥后,减压下浓缩,残余物经柱色谱提纯(淋洗液为石油醚:乙酸乙酯=4:1),得无色油状产物(久置变为固体)(4-(1-(1-乙氧基乙基)-1h-吡唑-4-基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯(5.00g,收率:72.05%)。1h nmr(400mhz,dmso-d6)δ8.81(s,1h),8.79(s,1h),8.36(s,1h),7.73(d,j=3.8hz,1h),7.15(d,j=3.8hz,1h),6.24(s,2h),5.67(q,j=6.0hz,1h),3.55
–
3.42(m,1h),3.30
–
3.21(m,1h),1.68(d,j=6.0hz,3h),1.07(s,9h),1.06(s,3h).hrms(esi)calcd for c
19h25
n5o3[m h
]:372.2030,found:372.2124.
[0092]
(3)(4-(1h-吡唑-4-基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯的制备,具体制备方法如下:
[0093][0094]
将化合物(4-(1-(1-乙氧基乙基)-1h-吡唑-4-基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯(5.00g,13.46mmol,1.0eq)用四氢呋喃(30ml)溶解,缓慢滴入11ml 6m盐酸溶
液,室温下反应1.5h,tlc监测原料反应完全时,结束反应。用1m氢氧化钠水溶液调节体系ph值至9,此时有大量固体析出,然后蒸除大部分溶剂并将悬浮液在室温下继续搅拌1h,抽滤,滤饼用水洗涤,滤饼烘干,最后得白色固体产物(4-(1h-吡唑-4-基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯(3.63g,收率:90%)。1h nmr(400mhz,dmso-d6)δ13.41(s,1h),8.77(s,1h),8.67(s,1h),8.35(s,1h),7.70(d,j=3.9hz,1h),7.12(d,j=3.9hz,1h),6.23(s,2h),1.08(s,9h).hrms(esi)calcd for c
15h17
n5o2na[m na
]:322.1274,found:322.2517.
[0095]
(4)(4-(1-(3-(氰基甲基)-1-(环丙基甲基)磺酰基)氮杂环丁-3-基)-1h-吡唑-4-基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯的制备,具体制备方法如下:
[0096][0097]
将化合物2-(1-((环丙基甲基)磺酰基)氮杂环丁-3-亚基)乙腈(200.00mg,942.20μmol,1.0eq)用n,n-二甲基甲酰胺(20ml)溶解,室温下加入化合物(4-(1h-吡唑-4-基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯(282.03mg,942.20μmol,1.0eq),搅拌溶解15min后,降温至10-15℃,然后缓慢滴入1,8-二氮杂二环十一碳-7-烯(10.04mg,65.95μmol,0.07eq),并保持15℃左右温度反应90min,再升至25℃继续反应4h。tlc监测原料反应完毕后,结束反应,用水(30ml)淬灭反应,乙酸乙酯萃取(30ml*3),有机层经饱和食盐水洗涤、无水硫酸钠干燥后,减压浓缩,残余物经柱色谱提纯(淋洗液为石油醚:乙酸乙酯=1:1),得到白色固体产物(4-(1-(3-(氰基甲基)-1-(环丙基甲基)磺酰基)氮杂环丁-3-基)-1h-吡唑-4-基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯(272.00mg,收率:56.43%)。1h nmr(400mhz,dmso-d6)δ8.97(s,1h),8.82(s,1h),8.50(d,j=0.6hz,1h),7.77(d,j=3.8hz,1h),7.21(d,j=3.8hz,1h),6.25(s,2h),4.63(d,j=9.1hz,2h),4.26(d,j=9.2hz,2h),3.68(s,2h),3.21(d,j=7.2hz,2h),1.08(s,9h),1.07
–
0.99(m,1h),0.63
–
0.52(m,2h),0.39
–
0.31(m,2h).hrms(esi)calcd for c
24h29
n7o4s[m h
]:512.2074,found:512.2337.
[0098]
(5)2-(3-(4-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)-1-((环丙基甲基)磺酰基)氮杂环丁-3-基)乙腈的制备,具体制备方法如下:
d]嘧啶-7-基)新戊酸甲酯(512.74mg,收率:95.00%)。1h nmr(400mhz,dmso-d6)δ8.95(s,1h),8.81(s,1h),8.48(d,j=0.6hz,1h),7.77(d,j=3.8hz,1h),7.21(d,j=3.8hz,1h),6.25(s,2h),4.79(d,j=9.5hz,1h),4.49(dd,j=9.9,4.6hz,2h),4.23(d,j=10.0hz,1h),3.69(s,2h),2.09(d,j=6.9hz,2h),1.08(s,9h),0.97
–
0.86(m,1h),0.50
–
0.39(m,2h),0.15
–
0.10(m,2h).hrms(esi)calcd for c
25h29
n7o3[m h
]:476.2405,found:476.2580.
[0105]
(2)2-(3-(4-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)-1-(2-环丙基乙酰基)氮杂环丁-3-基)乙腈的制备,具体制备方法如下:
[0106][0107]
将化合物(4-(1-(3-(氰基甲基)-1-(2-环丙基乙酰基)氮杂环丁烷-3-基)-1h-吡唑-4-基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯(350.00mg,736.00μmol,1.0eq)用甲醇(20ml)溶解,室温下加入1m氢氧化钠水溶液(44.16mg,1.10mmol,1.5eq)室温搅拌2h。tlc监测原料反应完毕后,结束反应。加水淬灭反应,用1m盐酸调节体系ph值至7左右,白色固体析出,旋掉大部分溶剂,抽滤,滤饼烘干,得到白色固体产物2-(3-(4-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)-1-(2-环丙基乙酰基)氮杂环丁-3-基)乙腈(148.95mg,收率:56.00%)。
[0108]
化合物i-2的1h nmr(400mhz,dmso-d6)δ12.14(s,1h),8.91(d,j=0.7hz,1h),8.70(s,1h),8.46(d,j=0.6hz,1h),7.62(dd,j=3.6,2.2hz,1h),7.08(dd,j=3.7,1.5hz,1h),4.79(d,j=9.5hz,1h),4.49(dd,j=9.9,5.4hz,2h),4.23(d,j=10.5hz,1h),3.69(s,2h),2.09(d,j=6.8hz,2h),0.99
–
0.90(m,1h),0.48
–
0.41(m,2h),0.15
–
0.10(m,2h).hrms(esi)calcd for c
19h19
n7o[m h
]:362.1724,found:362.1949.
[0109]
合成实施例3:2-(3-(4-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)-1-(乙基亚磺酰基)氮杂环丁-3-基)乙腈(化合物编号i-3)的制备
[0110]
(1)(4-(1-(3-(氰基甲基)-1-(乙基亚磺酰基)氮杂环丁-3-基)-1h-吡唑-4-基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯的制备,具体制备方法如下:
[0111][0112]
将化合物化合物(4-(1h-吡唑-4-基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯(193.42mg,646.18μmol,1.1eq)用乙腈(30ml)溶解,氮气保护-5℃下缓慢滴入2-(1-(乙基亚磺酰基)氮杂环丁-3-亚基)乙腈(100.00mg,587.43μmol,1.0eq)和1,8-二氮杂二环十一碳-7-烯(89.43mg,587.43μmol,1.0eq),加完原料体系-5℃下继续反应2h。tlc监测原料反应完毕后,结束反应,用水(30ml)淬灭反应,乙酸乙酯萃取(20ml*3),有机层经饱和食盐水洗涤、无水硫酸钠干燥后,减压浓缩,残余物经柱色谱提纯(淋洗液为石油醚:乙酸乙酯=1:2),得到(4-(1-(3-(氰基甲基)-1-(乙基亚磺酰基)氮杂环丁-3-基)-1h-吡唑-4-基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯(55.17mg,收率:20.00%)。1h nmr(600mhz,dmso-d6)δ8.94(s,1h),8.82(s,1h),8.48(s,1h),7.78(d,j=3.8hz,1h),7.20(d,j=3.8hz,1h),6.26(s,2h),4.55(d,j=9.1hz,1h),4.35(d,j=9.0hz,1h),4.13(d,j=9.2hz,1h),3.92(d,j=9.0hz,1h),3.66(s,2h),2.65
–
2.60(m,2h),1.12(t,j=7.5hz,3h),1.09(s,9h).hrms(esi)calcd for c
22h27
n7o3s[m h
]:470.1969,found:470.1977.
[0113]
(2)2-(3-(4-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)-1-(乙基亚磺酰基)氮杂环丁-3-基)乙腈的制备,具体制备方法如下:
[0114][0115]
将化合物(4-(1-(3-(氰基甲基)-1-(乙基亚磺酰基)氮杂环丁-3-基)-1h-吡唑-4-基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯(50.00mg,106.48μmol,1.0eq)用甲醇(20ml)溶解,室温下加入1m氢氧化钠水溶液(6.39mg,159.72μmol,1.5eq)室温搅拌2h。tlc监测原料反应完毕后,结束反应。加水淬灭反应,用1m盐酸调节体系ph值至7左右,淡黄色固体析出,旋掉大部分溶剂,抽滤,滤饼烘干,得到淡黄色固体产物2-(3-(4-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)-1-(乙基亚磺酰基)氮杂环丁-3-基)乙腈(29.41mg,收率:77.70%)。
[0116]
化合物i-3的1h nmr(400mhz,dmso-d6)δ12.14(s,1h),8.89(s,1h),8.70(s,1h),
8.45(s,1h),7.62(dd,j=3.6,2.3hz,1h),7.06(dd,j=3.6,1.7hz,1h),4.54(d,j=9.0hz,1h),4.35(d,j=9.0hz,1h),4.12(d,j=9.0hz,1h),3.91(d,j=9.0hz,1h),3.65(s,2h),2.62(qd,j=7.1,6.5,1.4hz,2h),1.11(t,j=7.5hz,3h).hrms(esi)calcd for c
16h17
n7os[m h
]:356.1288,found:356.1296.
[0117]
合成实施例4:2-(3-(3-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡咯-1-基)-1-((环丙基甲基)磺酰基)氮杂环丁-3-基)乙腈(化合物编号i-4)的制备
[0118]
(1)4-氯-7-((2-(三甲硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶的制备,具体制备方法如下:
[0119][0120]
将氢化钠(1.56g,65.12mmol,2.0eq)用n,n-二甲基甲酰胺(40ml)溶解,0℃下缓慢滴入4-氯-7h-吡咯并[2,3-d]嘧啶(5.00g,32.56mmol,1.0eq)的dmf(20ml)溶液,保温反应1.5h后,缓慢加入2-(三甲基硅烷基)乙氧甲基氯(6.51g,39.07mmol,1.2eq),加完原料体系室温下反应1h,tlc监测原料反应完全时,结束反应。向体系中加入30ml水淬灭反应,乙酸乙酯萃取(30ml*3),有机层经饱和食盐水洗涤、无水硫酸钠干燥后,减压浓缩,残余物经柱色谱提纯(淋洗液为石油醚:乙酸乙酯=4:1),得到淡黄色油状产物4-氯-7-((2-(三甲硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶(6.45g,收率:69.80%)。1h nmr(400mhz,cdcl3)δ8.64(s,1h),7.37(d,j=3.7hz,1h),6.64(d,j=3.7hz,1h),5.63(s,2h),3.53
–
3.49(m,2h),0.91
–
0.87(m,2h),0.08(s,9h).hrms(esi)calcd for c
12h18
cln3osi[m h
]:284.0980,found:284.2168.
[0121]
(2)4-(1h-吡咯-3-基)-7-(2-(三甲基甲硅烷基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶的制备,具体制备方法如下:
[0122][0123]
将化合物4-氯-7-((2-(三甲硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶(5.00g,17.62mmol,1.0eq)用正丁醇/水=1:1(50ml)溶解,室温下依次加入3-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1-(三异丙基甲硅烷基)-1h-吡咯(7.36g,21.14mmol,1.2eq)、碳酸钾(6.09g,44.04mmol,2.5eq)以及四(三苯基膦)钯(1.02g,880.81μmol,0.05eq),n2保护下,加热升温至100℃回流反应12h。tlc检测反应完全后,将体系冷却至室温,加水淬灭,用乙酸乙酯萃取(30ml*3),有机层经饱和食盐水洗涤、无水硫酸钠干燥后,减压下浓缩,残余物经柱色谱提纯(淋洗液为石油醚:乙酸乙酯=2:1),得到淡黄色固体产物4-(1h-吡咯-3-基)-7-(2-(三甲基甲硅烷基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶(3.77g,收率:68.05%)。1h nmr(400mhz,dmso-d6)δ11.40(s,1h),8.66(s,1h),7.78
–
7.76(m,1h),7.64(d,j=3.7hz,1h),6.99(d,j=3.7hz,1h),6.93
–
6.89(m,2h),5.60(s,2h),3.55
–
3.50(m,2h),0.85
–
0.80
(m,2h),0.10(s,9h).hrms(esi)calcd for c
16h22
n4osi[m h
]:315.1636,found:315.1818.
[0124]
(3)2-(1-((环丙基甲基)磺酰基)-3-(3-(7-((2-(三甲基甲硅烷基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡咯-1-基)氮杂环丁-3-基)乙腈的制备,具体制备方法如下:
[0125][0126]
将化合物4-(1h-吡咯-3-基)-7-(2-(三甲基甲硅烷基)乙氧基)甲基)-7h-吡咯[2,3-d]嘧啶(1.00g,3.18mmol,1.0eq)用乙腈溶解(50ml),室温下依次加入2-(1-((环丙基甲基)磺酰基)氮杂环丁-3-亚基)乙腈(810.04mg,3.82mmol,1.2eq)和1,8-二氮杂二环十一碳-7-烯(726.19mg,4.77mmol,1.5eq),加完原料体系70℃下反应5h。tlc监测原料反应完毕后,结束反应,用水(30ml)淬灭反应,乙酸乙酯萃取(30ml*3),有机层经饱和食盐水洗涤、无水硫酸钠干燥后,减压浓缩,残余物经柱色谱提纯(淋洗液为石油醚:乙酸乙酯=1:1),得到淡黄色固体产物2-(1-((环丙基甲基)磺酰基)-3-(3-(7-((2-(三甲基甲硅烷基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡咯-1-基)氮杂环丁-3-基)乙腈(777.34mg,收率:46.27%)。1h nmr(400mhz,dmso-d6)δ8.71(s,1h),7.97(s,1h),7.71(d,j=3.7hz,1h),7.20(t,j=2.6hz,1h),7.10(d,j=3.7hz,1h),7.00(dd,j=3.0,1.6hz,1h),5.62(s,2h),4.49(d,j=9.1hz,2h),4.23(d,j=8.8hz,2h),3.58(s,2h),3.53(t,j=8.0hz,2h),3.19(d,j=7.1hz,2h),1.06
–
0.99(m,1h),0.83(t,j=8.0hz,2h),0.61
–
0.55(m,2h),0.36
–
0.32(m,2h),0.10(s,9h).hrms(esi)calcd for c
25h34
n6o3ssi[m h
]:527.2255,found:527.2559.
[0127]
(4)2-(3-(3-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡咯-1-基)-1-((环丙基甲基)磺酰基)氮杂环丁-3-基)乙腈的制备,具体制备方法如下:
[0128][0129]
向50ml圆底烧瓶中加入7.9ml三氟乙酸,0℃下缓慢加入2-(1-((环丙基甲基)磺酰基)-3-(3-(7-((2-(三甲基甲硅烷基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡
咯-1-基)氮杂环丁-3-基)乙腈(200.00mg,379.70μmol,1.0eq)的二氯甲烷(30ml)溶液,加完原料体系移至室温下反应1h,将体系减压下浓缩,用甲醇(30ml)溶解,室温下加入乙二胺(1.58ml)并保温反应2h,tlc监测原料反应完毕后,加3m盐酸调节体系ph为7左右,用水(30ml)淬灭反应,乙酸乙酯萃取(30ml*3),有机层经饱和食盐水洗涤、无水硫酸钠干燥后,减压下浓缩,残余物经柱色谱提纯(淋洗液为乙酸乙酯),得到白色固体产物2-(3-(3-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡咯-1-基)-1-((环丙基甲基)磺酰基)氮杂环丁-3-基)乙腈(86.14mg,收率:57.22%)。
[0130]
化合物i-4的1h nmr(400mhz,dmso-d6)δ11.99(s,1h),8.64(s,1h),7.94(t,j=2.0hz,1h),7.53(dd,j=3.6,2.3hz,1h),7.18(dd,j=3.0,2.3hz,1h),6.99(dd,j=3.2,1.7hz,2h),4.49(d,j=9.1hz,2h),4.23(d,j=9.1hz,2h),3.58(s,2h),3.20(d,j=7.1hz,2h),1.10
–
0.96(m,1h),0.62
–
0.53(m,2h),0.37
–
0.32(m,2h).hrms(esi)calcd for c
19h20
n6o2s[m h
]:397.1441,found:397.1904.
[0131]
合成实施例5:2-(3-(3-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡咯-1-基)-1-(2-环丙基乙酰基)氮杂环丁-3-基)乙腈(化合物编号i-5)的制备
[0132]
(1)2-(1-(2-环丙基乙酰基)-3-(3-(7-(2-(三甲基甲硅烷基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡咯-1-基)氮杂环丁-3-基)乙腈的制备,具体制备方法如下:
[0133][0134]
将化合物4-(1h-吡咯-3-基)-7-(2-(三甲基甲硅烷基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶(200.00mg,636.02μmol,1.0eq)用乙腈溶解(50ml),室温下依次加入2-(1-(2-环丙基乙酰基)氮杂环丁-3-亚基)乙腈(168.11mg,954.02μmol,1.5eq)和1,8-二氮杂二环十一碳-7-烯(145.24mg,954.02μmol,1.5eq),加完原料体系70℃下反应4h。tlc监测原料反应完毕后,结束反应,用水(30ml)淬灭反应,乙酸乙酯萃取(30ml*3),有机层经饱和食盐水洗涤、无水硫酸钠干燥后,减压浓缩,残余物经柱色谱提纯(淋洗液为石油醚:乙酸乙酯=1:1),得到淡黄色固体产物2-(1-(2-环丙基乙酰基)-3-(3-(7-(2-(三甲基甲硅烷基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡咯-1-基)氮杂环丁-3-基)乙腈(196.05mg,收率:62.82%)。1h nmr(400mhz,dmso-d6)δ8.70(s,1h),7.95(t,j=2.0hz,1h),7.71(d,j=3.7hz,1h),7.19(dd,j=3.0,2.3hz,1h),7.09(d,j=3.7hz,1h),6.99(dd,j=3.1,1.7hz,1h),5.62(s,2h),4.71(d,j=9.5hz,1h),4.46(d,j=9.5hz,1h),4.38(d,j=10.5hz,1h),4.22(d,j=10.5hz,1h),3.57(s,2h),3.57
–
3.47(m,2h),2.08(d,j=6.9hz,2h),1.00
–
0.88(m,1h),0.88
–
0.78(m,2h),0.50
–
0.39(m,2h),0.15
–
0.10(m,2h),0.10(s,9h).hrms(esi)
calcd for c
26h34
n6o2si[m h
]:491.2585,found:491.2732.
[0135]
(2)2-(3-(3-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡咯-1-基)-1-(2-环丙基乙酰基)氮杂环丁-3-基)乙腈的制备,具体制备方法如下:
[0136][0137]
向100ml圆底烧瓶中加入15.4ml三氟乙酸,0℃下缓慢加入2-(1-(2-环丙基乙酰基)-3-(3-(7-(2-(三甲基甲硅烷基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡咯-1-基)氮杂环丁-3-基)乙腈(150.00mg,305.70μmol,1.0eq)的二氯甲烷(30ml)溶液,加完原料体系移至室温下反应1h,将体系减压下浓缩,用甲醇(30ml)溶解,室温下加入乙二胺(1.28ml)并保温反应2h,tlc监测原料反应完毕后,加3m盐酸调节体系ph为7左右,用水(30ml)淬灭反应,乙酸乙酯萃取(30ml*3),有机层经饱和食盐水洗涤、无水硫酸钠干燥后,减压下浓缩,残余物经柱色谱提纯(淋洗液为乙酸乙酯),得到白色固体产物2-(3-(3-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡咯-1-基)-1-(2-环丙基乙酰基)氮杂环丁-3-基)乙腈(61.83mg,收率:56.12%)。
[0138]
化合物i-5的1h nmr(400mhz,dmso-d6)δ11.98(s,1h),8.64(s,1h),7.93(t,j=2.0hz,1h),7.53(dd,j=3.6,2.3hz,1h),7.18(t,j=2.7hz,1h),7.02
–
6.95(m,2h),4.71(d,j=9.5hz,1h),4.46(d,j=9.5hz,1h),4.37(d,j=10.5hz,1h),4.23(d,j=10.5hz,1h),3.57(s,2h),2.09(d,j=6.9hz,2h),1.00
–
0.90(m,1h),0.49
–
0.41(m,2h),0.17
–
0.09(m,2h).hrms(esi)calcd for c
20h20
n6o[m h
]:361.1771,found:361.2097.
[0139]
合成实施例6:2-(4-(4-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)-1-(2-环丙基乙酰基)哌啶-4-基)乙腈(化合物编号i-6)的制备
[0140]
(1)(4-(1-(4-(氰基甲基)-1-(2-环丙基乙酰基)哌啶-4-基)-1h-吡唑-4-基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯的制备,具体制备方法如下:
[0141][0142]
将化合物2-(1-(2-环丙基乙酰基)哌啶-4-亚基)乙腈(204.73mg,1.00mmol,
1.5eq)用乙腈(30ml)溶解,室温下依次加入化合物(4-(1h-吡唑-4-基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯(200.00mg,668.16μmol,1.0eq)和1,8-二氮杂二环十一碳-7-烯(152.58mg,1.00mmol,1.5eq),加完原料体系70℃左右反应4h。tlc监测原料反应完毕后,结束反应,用水(30ml)淬灭反应,乙酸乙酯萃取(30ml*3),有机层经饱和食盐水洗涤、无水硫酸钠干燥后,减压浓缩,残余物经柱色谱提纯(淋洗液为石油醚:乙酸乙酯=1:1),得到白色固体产物(4-(1-(4-(氰基甲基)-1-(2-环丙基乙酰基)哌啶-4-基)-1h-吡唑-4-基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯(201.89mg,收率:60.00%)。1h nmr(400mhz,dmso-d6)δ8.87(s,1h),8.83(s,1h),8.48(s,1h),7.77(d,j=3.8hz,1h),7.22(d,j=3.8hz,1h),6.27(s,2h),4.09
–
4.02(m,1h),3.75(d,j=14.0hz,1h),3.29(s,2h),3.18(t,j=12.0hz,1h),2.95(t,j=11.7hz,1h),2.70(t,j=15.7hz,2h),2.30(d,j=6.7hz,2h),2.08
–
1.97(m,2h),1.10(s,9h),0.98
–
0.94(m,1h),0.48
–
0.43(m,2h),0.15
–
0.10(m,2h).hrms(esi)calcd for c
27h33
n7o3[m h
]:504.2718,found:504.2935.
[0143]
(2)2-(4-(4-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)-1-(2-环丙基乙酰基)哌啶-4-基)乙腈的制备,具体制备方法如下:
[0144][0145]
将化合物(4-(1-(4-(氰基甲基)-1-(2-环丙基乙酰基)哌啶-4-基)-1h-吡唑-4-基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯(150.00mg,297.86μmol,1.0eq)用甲醇(30ml)溶解,室温下加入1m氢氧化钠水溶液(17.87mg,446.79μmol,1.5eq)室温搅拌2h。tlc监测原料反应完毕后,结束反应。加水淬灭反应,用1m盐酸调节体系ph值至7左右,淡黄色固体析出,旋掉大部分溶剂,抽滤,滤饼烘干,得到淡黄色固体产物2-(4-(4-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)-1-(2-环丙基乙酰基)哌啶-4-基)乙腈(83.80mg,收率:72.24%)。
[0146]
化合物i-6的1h nmr(400mhz,dmso-d6)δ12.13(s,1h),8.81(s,1h),8.70(s,1h),8.44(s,1h),7.64
–
7.56(m,1h),7.16
–
6.99(m,1h),4.10
–
3.98(m,1h),3.73(d,j=14.0hz,1h),3.28(s,2h),3.17(t,j=12.6hz,1h),3.00
–
2.87(m,1h),2.69(t,j=16.1hz,2h),2.29(d,j=6.7hz,2h),2.07
–
1.91(m,2h),1.41
–
1.33(m,1h),0.90
–
0.83(m,2h),0.44(d,j=7.9hz,2h).hrms(esi)calcd for c
21h23
n7o[m h
]:390.2037,found:390.2386.
[0147]
合成实施例7:2-(4-(4-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)-1-((环丙基甲基)磺酰基)哌啶-4-基)乙腈(化合物编号i-7)的制备
[0148]
(1)(4-(1-(4-(氰基甲基)-1-(环丙基甲基)磺酰基)哌啶-4-基)-1h-吡唑-4-基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯的制备,具体制备方法如下:
[0149][0150]
将化合物2-(1-((环丙基甲基)磺酰基)哌啶-4-亚基)乙腈(240.86mg,1.00mmol,1.5eq)用n,n-二甲基甲酰胺(30ml)溶解,室温下加入化合物(4-(1h-吡唑-4-基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯(200.00mg,668.16μmol,1.0eq),搅拌溶解15min后,降温至10-15℃,然后缓慢滴入1,8-二氮杂二环十一碳-7-烯(101.72mg,668.16μmol,1.0eq),并保持15℃左右温度反应90min,再升至25℃继续反应12h。tlc监测原料反应完毕后,结束反应,用水(30ml)淬灭反应,乙酸乙酯萃取(30ml*3),有机层经饱和食盐水洗涤、无水硫酸钠干燥后,减压浓缩,残余物经柱色谱提纯(淋洗液为石油醚:乙酸乙酯=1:1),得到白色固体产物(4-(1-(4-(氰基甲基)-1-(环丙基甲基)磺酰基)哌啶-4-基)-1h-吡唑-4-基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯(87.08mg,收率:24.15%)。1h nmr(400mhz,dmso-d6)δ8.85(s,1h),8.81(s,1h),8.47(s,1h),7.76(d,j=3.8hz,1h),7.22(d,j=3.8hz,1h),6.26(s,2h),3.59
–
3.52(m,2h),3.28(s,2h),2.97(d,j=7.0hz,2h),2.96
–
2.86(m,2h),2.83(d,j=14.1hz,2h),2.14
–
2.06(m,2h),1.09(s,9h),0.94
–
0.89(m,1h),0.51
–
0.45(m,2h),0.24
–
0.18(m,2h).hrms(esi)calcd for c
26h33
n7o4s[m h
]:540.2387,found:540.2797.
[0151]
(2)2-(4-(4-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)-1-((环丙基甲基)磺酰基)哌啶-4-基)乙腈的制备,具体制备方法如下:
[0152][0153]
将化合物(4-(1-(4-(氰基甲基)-1-(环丙基甲基)磺酰基)哌啶-4-基)-1h-吡唑-4-基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯(80.00mg,148.24μmol,1.0eq)用甲醇(20ml)溶解,室温下加入1m氢氧化钠水溶液(8.89mg,222.37μmol,1.5eq)室温搅拌2h。tlc监测原料反应完毕后,结束反应。加水淬灭反应,用1m盐酸调节体系ph值至7左右,淡黄色固体析出,旋掉大部分溶剂,抽滤,滤饼烘干,得到淡黄色固体产物2-(4-(4-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)-1-((环丙基甲基)磺酰基)哌啶-4-基)乙腈(56.77mg,收率:90.00%)。
[0154]
化合物i-7的1h nmr(400mhz,dmso-d6)δ12.12(s,1h),8.81(s,1h),8.70(s,1h),8.45(s,1h),7.61(dd,j=3.6,2.4hz,1h),7.08(dd,j=3.6,1.8hz,1h),3.62
–
3.51(m,2h),3.27(s,2h),2.97(d,j=7.0hz,2h),2.96
–
2.89(m,2h),2.83(d,j=14.1hz,2h),2.15
–
2.06(m,2h),0.93
–
0.88(m,1h),0.52
–
0.44(m,2h),0.24
–
0.18(m,2h).hrms(esi)calcd for c
20h23
n7o2s[m h
]:426.1707,found:426.2116.
[0155]
合成实施例8:2-(3-(4-((7h-吡咯并[2,3-d]嘧啶-4-基)氨基)-1h-吡唑-1-基)-1-(甲磺酰基)氮杂环丁-3-基)乙腈(化合物编号ii-1)的制备
[0156]
(1)(4-((1-(3-(氰甲基)-1-(甲基磺酰基)氮杂环丁-3-基)-1h-吡唑-4-基)氨基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯的制备,具体制备方法如下:
[0157][0158]
将(4-氯-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯(500.00mg,1.87mmol,1.0eq)用乙酸/水(20ml:20ml)溶解,室温下加入2-(3-(4-氨基-1h-吡唑-1-基)-1-(甲磺酰基)氮杂环丁烷-3-基)乙腈(476.81mg,1.87mmol,1.0eq),加完原料体系70℃下反应5-7h,tlc监测原料反应完全时,结束反应。加入10%naoh水溶液调节体系ph=8-9,酸乙酯萃取(30ml*3),有机层经饱和食盐水洗涤、无水硫酸钠干燥后,减压浓缩,残余物经柱色谱提纯(淋洗液为乙酸乙酯),得到浅褐色油状产物(4-((1-(3-(氰甲基)-1-(甲基磺酰基)氮杂环丁-3-基)-1h-吡唑-4-基)氨基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯(426.46mg,收率:46.93%)。1h nmr(400mhz,dmso-d6)δ9.73(s,1h),8.44(s,1h),8.38(s,1h),7.85(s,1h),7.36(d,j=3.6hz,1h),6.75(s,1h),6.15(s,2h),4.46(d,j=9.2hz,2h),4.22(d,j=9.1hz,2h),3.61(s,2h),3.11(s,3h),1.09(s,9h).hrms(esi)calcd for c
21h26
n8o4s[m h
]:487.1870,found:487.2060.
[0159]
(2)2-(3-(4-((7h-吡咯并[2,3-d]嘧啶-4-基)氨基)-1h-吡唑-1-基)-1-(甲磺酰基)氮杂环丁-3-基)乙腈的制备,具体制备方法如下:
[0160][0161]
将化合物(4-((1-(3-(氰甲基)-1-(甲基磺酰基)氮杂环丁-3-基)-1h-吡唑-4-基)氨基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯(200.00mg,411.06μmol,1.0eq)用甲醇
(20ml)溶解,室温下加入1m氢氧化钠水溶液(24.66mg,616.59μmol,1.5eq)室温搅拌2h。tlc监测原料反应完毕后,结束反应。加水淬灭反应,用1m盐酸调节体系ph值至7左右,淡黄色固体析出,旋掉大部分溶剂,抽滤,滤饼烘干,得到淡黄色固体产物2-(3-(4-((7h-吡咯并[2,3-d]嘧啶-4-基)氨基)-1h-吡唑-1-基)-1-(甲磺酰基)氮杂环丁-3-基)乙腈(77.95mg,收率:50.92%)。
[0162]
化合物ii-1的1h nmr(400mhz,dmso-d6)δ11.70(s,1h),9.60(s,1h),8.44(s,1h),8.28(s,1h),7.85(s,1h),7.19(t,j=2.9hz,1h),6.69(s,1h),4.46(d,j=9.1hz,2h),4.23(d,j=9.1hz,2h),3.61(s,2h),3.12(s,3h).hrms(esi)calcd for c
15h16
n8o2sna[m na
]:395.1009,found:395.1332.
[0163]
合成实施例9:2-(3-(4-((7h-吡咯并[2,3-d]嘧啶-4-基)氨基)-1h-吡唑-1-基)-1-苯甲酰基氮杂环丁烷-3-基)乙腈(化合物编号ii-10)的制备
[0164]
(1)(4-((1-(1-苯甲酰基-3-(氰甲基)氮杂环丁烷-3-基)-1h-吡唑-4-基)氨基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯的制备,具体制备方法如下:
[0165][0166]
将(4-氯-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯(500.00mg,1.87mmol,1.0eq)用乙酸/水(20ml:20ml)溶解,室温下加入2-(3-(4-氨基-1h-吡唑-1-基)-1-苯甲酰基氮杂环丁烷-3-基)乙腈(525.40mg,1.87mmol,1.0eq),加完原料体系70℃下反应5-7h,tlc监测原料反应完全时,结束反应。加入10%naoh水溶液调节体系ph=8-9,酸乙酯萃取(30ml*3),有机层经饱和食盐水洗涤、无水硫酸钠干燥后,减压浓缩,残余物经柱色谱提纯(淋洗液为二氯甲烷:甲醇=10:1),得到淡黄色油状产物(4-((1-(1-苯甲酰基-3-(氰甲基)氮杂环丁烷-3-基)-1h-吡唑-4-基)氨基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯(880.52mg,收率:91.98%)。1h nmr(400mhz,dmso-d6)δ9.73(s,1h),8.46(s,1h),8.38(s,1h),7.84(s,1h),7.72
–
7.69(m,2h),7.57
–
7.53(m,1h),7.51
–
7.46(m,2h),7.36(d,j=3.6hz,1h),6.76(s,1h),6.15(s,2h),4.91(d,j=9.6hz,1h),4.63(d,j=9.5hz,1h),4.57(d,j=10.8hz,1h),4.42(d,j=10.7hz,1h),3.64(s,2h),1.08(s,9h).hrms(esi)calcd forc
27h28
n8o3[m h
]:513.2357,found:513.2538.
[0167]
(2)2-(3-(4-((7h-吡咯并[2,3-d]嘧啶-4-基)氨基)-1h-吡唑-1-基)-1-苯甲酰基氮杂环丁烷-3-基)乙腈的制备,具体制备方法如下:
[0168][0169]
将化合物(4-((1-(1-苯甲酰基-3-(氰甲基)氮杂环丁烷-3-基)-1h-吡唑-4-基)氨基)-7h-吡咯并[2,3-d]嘧啶-7-基)新戊酸甲酯(500.00mg,975.49μmol,1.0eq)用甲醇(20ml)溶解,室温下加入1m氢氧化钠水溶液(58.53mg,1.46mmol,1.5eq)室温搅拌2h。tlc监测原料反应完毕后,结束反应。加水淬灭反应,用1m盐酸调节体系ph值至7左右,淡黄色固体析出,旋掉大部分溶剂,抽滤,滤饼烘干,得到淡黄色固体产物2-(3-(4-((7h-吡咯并[2,3-d]嘧啶-4-基)氨基)-1h-吡唑-1-基)-1-苯甲酰基氮杂环丁烷-3-基)乙腈(91.49mg,收率:23.54%)。
[0170]
化合物ii-10的1h nmr(400mhz,dmso-d6)δ11.69(s,1h),9.54(s,1h),8.45(s,1h),8.28(s,1h),7.83(s,1h),7.70(d,j=7.5hz,2h),7.60
–
7.51(m,1h),7.49(t,j=7.4hz,2h),7.19(s,1h),6.66(s,1h),4.90(d,j=9.5hz,1h),4.63(d,j=9.4hz,1h),4.57(d,j=10.6hz,1h),4.42(d,j=10.7hz,1h),3.64(s,2h).hrms(esi)calcd for c
21h18
n8o[m h
]:399.1676,found:399.2090.
[0171]
除上面描述的化合物外,表1中其它化合物可参照合成实施例1-9中相似的方法制备,下文表2中给出了参照合成实施例1-9合成的部分化合物核磁和高分辨质谱数据。
[0172]
表2
[0173]
[0174]
[0175][0176]
本发明的其他通式i或通式ii化合物可参照上述方法合成。
[0177]
药理活性试验实施例
[0178]
实施例1.化合物物理化学参数如下表3
[0179]
表3化合物物理化学性质相关参数
[0180]
[0181][0182]
注:化合物的物理化学属性(logp、clogp和tpsa值)为chemoffice软件包中的chemdraw软件预测数值。
[0183]
结果显示,该类化合物的物理化学属性(logp、clogp和tpsa等)与阳性药巴瑞替尼(baricitinib)相当,具有良好的成药性。
[0184]
实施例2:化合物对jak激酶的体外抑制活性
[0185]
实验材料:jaktide peptide底物、irstide peptide底物,激酶jak1/jak2/jak3/tyk2,三磷酸腺苷(atp),阳性对照baricitinib等
[0186]
实验仪器:perkinelmer的设备caliper
[0187]
实验方法:荧光检测法
[0188]
实验原理:本实验采用迁移率改变法(mobility shift assay)来检测化合物的半抑制浓度ic
50
(把酶活性抑制至50%时化合物的浓度)。perkinelmer公司的ez reader可用来检测被激酶催化多肽底物的磷酸化水平,该设备基于微控流体分离技术,可以直接检测荧光标记的底物和产物,分离步骤在微流体芯片内通过控制压力和电场强度来实现。使用真空压力,将反应引入通过熔融硅小孔的芯片底部,由于芯片中分离通道上被施加了电位差,带有荧光标记的多肽底物和反应产物由于电荷的不同被分离,然后在检测窗口进行信号的激发和检测,在检测每一个样品时,都可以同时看到底物和产物的信号,通过多肽产物以及多肽底物的峰高比较计算,就可以获知实际转化率。实验结果如下表4:
[0189]
表4化合物对jak激酶抑制作用研究
[0190]
[0191][0192]
阳性化合物结构如下:
[0193][0194]
实验结论:在生物活性评价中,我们选用baricitinib作为阳性对照药,测试结果表明合成的部分化合物对jak1和jak2具有较好的抑制活性,ic
50
值在纳摩尔水平,其中活性最好的化合物i-7与阳性对照药活性相当,具有潜在治疗该靶点相关疾病的功效。
[0195]
本发明通过上述讲授内容来说明本发明的吡咯并嘧啶五元氮杂环衍生物及其制
备方法和用途,但本发明并不局限于上述所述内容。所属技术领域的技术人员应该明了,对本发明作各种改动或修改,这些等价形式同样落于本技术所附权利要求书所限定的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。