1.本发明属于光电材料技术领域,尤其涉及一种有机室温电致磷光材料、制备方法及其有机电致发光二极管。
背景技术:
2.有机发光显示(oled)和液晶显示(lcd)相比,具有自发光、宽视角、高响应速度、超薄、耐低温等突出特点,已经成为国际竞相发展的新一代显示技术。根据发光机理,现有的有机电致发光材料可以分为荧光材料、磷光金属配合物和热活化延迟荧光材料。对于荧光材料,只能利用25%的单线态激子,75%的三线态激子发生非辐射衰减损失,因此最大理论内量子效率限制在25%。对于磷光金属配合物,由于贵重金属导致的自旋轨道耦合效应,能够同时利用单线态和三线态激子,理论内量子效率可以达到100%。但是,贵重金属的使用,存在地球储量有限、价格昂贵等问题。针对这一问题,兼具荧光材料低成本和磷光金属配合物高效率的热活化延迟荧光材料随后被开发出来。热活化延迟荧光材料中三线态激子能够利用环境热,反向系间窜越到单线态,在瞬时荧光之外产生额外的延迟荧光发射,从而实现100%理论内量子效率,但热活化延迟荧光材料加工性能差。
3.近年来引起了人们的广泛关注为纯有机室温磷光材料,纯有机室温磷光材料中三线态激子可以直接辐射跃迁到基态,达到提高激子利用率的目的。这类纯有机室温磷光材料具有成本低、功能可调、细胞毒性低、加工性好等特点。
4.但是,由于三线态激子的非辐射衰减和/或周围环境淬灭,这类纯有机室温磷光材料在无定型状态下不发光,而仅在晶体状态下才能观察到强的室温磷光,严重制约了其在oled器件中的应用。
技术实现要素:
5.有鉴于此,本发明提供了一种有机室温电致磷光材料、制备方法及其有机电致发光二极管,本发明提供的有机室温电致磷光材料能够在无定形态下获得有效的室温磷光。
6.本发明提供了一种有机室温电致磷光材料,具有式i所示结构:
7.d-x-a式i;
8.其中,所述x独立地选自o或s;
9.所述a为电子受体,独立地选自含有n、p、b、-c=o、-p=o、-p=s、-cn、-cho、-s(=o)2中的一种或多种的c6~c30的芳基、含有n、p、b、-c=o、-p=o、-p=s、-cn、-cho或-s(=o)2中的一种或多种的c5~c30的芳杂基、或含有n、p、b、-c=o、-p=o、-p=s、-cn、-cho或-s(=o)2中的一种或多种的c6~c30的芳基和含有n、p、b、-p=o、-p=s、-c=o、-cn、-cho或-s(=o)2中的一种或多种的c5~c30的芳杂基的任意组合;
10.所述d为稠并芳胺类电子给体,具有式ii-1或ii-2的结构:
[0011][0012]
其中,所述ar1、ar2和ar3可以相同,也可以不同,独立地选自式iii-1~iii-9所示结构中的一种:
[0013][0014]
所述y和v可以相同,也可以不同,独立地选自c、si、n、o或s;
[0015]
当所述y或v为c或si时,所述r5或r6为取代或未取代c1~c20的直链烷基、取代或未取代c1~c20的直链烷氧基、取代或未取代c3~c20的支链烷基、取代或未取代c3~c20的支链烷氧基、苯基和氢中的任意两种;
[0016]
当所述y或v为n时,所述r5或r6为取代或未取代c1~c20的直链烷基、取代或未取代c1~c20的直链烷氧基、取代或未取代c3~c20的支链烷基、取代或未取代c3~c20的支链烷氧基和苯基中的任意一种;
[0017]
当所述y或v为o或s时,所述r5或r6为无;
[0018]
所述
①
独立地为五元环或六元环;
[0019]
当所述
①
独立地为五元环时,所述z1独立地为单键;
[0020]
当所述
①
独立地为六元环时,所述z1独立地为c、si、n、o或s;
[0021]
所述
②
或
③
独立地为不成环、五元环或六元环;
[0022]
当所述
②
或
③
为不成环时,所述z2或z3为无;
[0023]
当所述
②
或
③
独立地为五元环时,所述z2或z3独立地为单键;
[0024]
当所述
②
或
③
独立地为六元环时,所述z2或z3独立地为c、si、n、o或s;
[0025]
所述r1~r4独立地为取代或未取代c1~c20的直链烷基、取代或未取代c1~c20的直链烷氧基、取代或未取代c3~c20的支链烷基、取代或未取代c3~c20的支链烷氧基或氢;
[0026]
所述n1~n3独立地为为1~5的整数。
[0027]
优选的,所述d为式
ⅳ‑
1~式
ⅳ‑
52所示结构中的一种:
[0028][0029]
[0030][0031]
优选的,所述a为式v-1~v-34所示结构中的一种:
[0032][0033]
本发明提供了上述技术方案所述的有机室温电致磷光材料的制备方法,在保护气氛中,将中间体d-xh、中间体f-a、有机溶剂、脱水剂和碱性催化剂进行亲核取代反应,得到具有式i结构的所述有机室温电致磷光材料。
[0034]
优选的,所述亲核取代反应包括以下步骤:
[0035]
在保护气氛中,将中间体d-xh、极性有机溶剂、脱水剂和碱性催化剂进行预脱水反应,得到中间反应液;所述反应温度为130-160℃,反应时间为1~3h后;
[0036]
将中间反应液和中间体f-a混合进行亲核取代反应,得到所述有机室温电致磷光材料;所述亲核取代反应的温度为140~220℃,所述亲核取代反应的反应时间为8~20h,所述中间体d-xh和所述中间体f-a的摩尔比为(1~1.3):1。
[0037]
本发明提高了一种有机电致发光二极管,发光层中包括至少一种上述技术方案所
述的有机室温电致磷光材料。
[0038]
优选的,上述技术方案所述的有机室温电致磷光材料本身作为发光层或作为客体掺杂在主体材料中构成发光层。
[0039]
优选的,上述技术方案所述的有机室温电致磷光材料作为主体,掺杂传统荧光材料、磷光金属配合物、热活化延迟荧光材料作为客体,构成发光层。
[0040]
本发明提供的有机室温电致磷光材料具有稠并芳胺电子给体d和a电子受体特征结构,其中稠并芳胺电子给体d具有式ii-1或ii-2所述结构,稠并芳胺电子给体提供分子刚性,抑制三线态激子非辐射衰减;x调控分子内电荷转移,同时增强自旋轨道耦合效应,a作为电子受体接收d提供的电子,最终在无定型状态下获得有效的室温磷光。
附图说明
[0041]
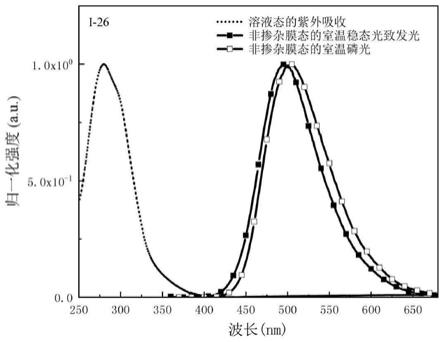
图1为本发明实施例1的分子i-26在二氯甲烷溶液中的吸收光谱、非掺杂薄膜稳态光致发光光谱和非掺杂薄膜室温磷光光谱;
[0042]
图2为本发明实施例1的分子i-26非掺杂薄膜稳态光致发光温度依赖性光谱;
[0043]
图3为本发明实施例1的分子i-26非掺杂薄膜瞬态光致发光温度依赖性光谱;
[0044]
图4为本发明实施例8和对比例2制备的oled器件外量子效率-亮度曲线;
[0045]
图5为本发明实施例8和对比例2制备的oled器件电致发光光谱;
[0046]
图6为本发明实施例9和对比例3制备的有机发光二极管器件的外量子效率-亮度曲线对比图;
[0047]
图7为本发明器件实施例9和对比例3制备的有机发光二极管器件的电致发光光谱。
具体实施方式
[0048]
本发明提供了一种有机室温电致磷光材料,具有式i所示结构:
[0049]
d-x-a式i;
[0050]
其中,所述x独立地选自o或s;
[0051]
所述a为电子受体,独立地选自含有n、p、b、-c=o、-p=o、-p=s、-cn、-cho、-s(=o)2中的一种或多种的c6~c30的芳基、含有n、p、b、-c=o、-p=o、-p=s、-cn、-cho或-s(=o)2中的一种或多种的c5~c30的芳杂基、或含有n、p、b、-c=o、-p=o、-p=s、-cn、-cho或-s(=o)2中的一种或多种的c6~c30的芳基和含有n、p、b、-p=o、-p=s、-c=o、-cn、-cho或-s(=o)2中的一种或多种的c5~c30的芳杂基的任意组合;
[0052]
所述d为稠并芳胺类电子给体,具有式ii-1或ii-2的结构:
[0053][0054]
其中,所述ar1、ar2和ar3可以相同,也可以不同,独立地选自式iii-1~iii-9所示结构中的一种:
[0055][0056]
所述y和v可以相同,也可以不同,独立地选自c、si、n、o或s;
[0057]
当所述y或v为c或si时,所述r5或r6为取代或未取代c1~c20的直链烷基、取代或未取代c1~c20的直链烷氧基、取代或未取代c3~c20的支链烷基、取代或未取代c3~c20的支链烷氧基、苯基中的任意两种;
[0058]
当所述y或v为n时,所述r5或r6为取代或未取代c1~c20的直链烷基、取代或未取代c1~c20的直链烷氧基、取代或未取代c3~c20的支链烷基、取代或未取代c3~c20的支链烷氧基、苯基中的任意一种;
[0059]
当所述y或v为o或s时,所述r5或r6为无;
[0060]
所述
①
独立地为五元环或六元环;
[0061]
当所述
①
独立地为五元环时,所述z1独立地为单键;
[0062]
当所述
①
独立地为六元环时,所述z1独立地为c、si、n、o或s;
[0063]
所述
②
或
③
独立地为不成环、五元环或六元环;
[0064]
当所述
②
或
③
为不成环时,所述z2或z3为无;
[0065]
当所述
②
或
③
独立地为五元环时,所述z2或z3独立地为单键;
[0066]
当所述
②
或
③
独立地为六元环时,所述z2或z3独立地为c、si、n、o或s;
[0067]
所述r1~r4独立地为取代或未取代c1~c20的直链烷基、取代或未取代c1~c20的直链烷氧基、取代或未取代c3~c20的支链烷基、取代或未取代c3~c20的支链烷氧基或氢;
[0068]
所述n1~n3独立地为为1~5的整数。
[0069]
本发明提供的有机室温电致磷光材料的式i所述结构中,所述d作为电子给体,所述d优选为式ii-1或ii-2所示结构中的一种。
[0070]
在本发明中,所述d优选为式
ⅳ‑
1~
ⅳ‑
52所示结构中的一种:
[0071]
[0072]
[0073][0074]
本发明提供的有机室温电致磷光材料的式i所述结构中,所述x为o或s,所述x调控分子内电荷转移,同时增强自旋轨道耦合效应。
[0075]
本发明提供的有机室温电致磷光材料的式i所述结构中,所述a作为电子受体接收d提供的电子。在本发明中,所述a优选为含有n、p、-c=o、-p=o、-p=s、或-s(=o)2中的一
种或多种的c6~c30的芳基、含有n、p、-c=o、-p=o、-p=s或-s(=o)2中的一种或多种的c5~c30的芳杂基、或含有n、p、-c=o、-p=o、-p=s或-s(=o)2中的一种或多种的c6~c30的芳基和含有n、p、-c=o、-p=o、-p=s或-s(=o)2中的一种或多种的c5~c30的芳杂基的任意组合。
[0076]
在本发明中,所述a优选为式v-1~v-34所示结构中的一种:34所示结构中的一种:34所示结构中的一种:
[0077]
在本发明中,所述有机室温电致磷光材料优选具有式i-1~i-208所示结构中的一种:
[0078]
[0079]
[0080]
[0081]
[0082]
[0083][0084]
本发明提供了上述技术方案所述的有机室温电致磷光材料的制备方法,包括以下步骤:
[0085]
在保护气氛中,将中间体d-xh、中间体f-a、有机溶剂、脱水剂和碱性催化剂进行亲核取代反应,得到具有式i结构的所述有机室温电致磷光材料。
[0086]
在本发明中,-xh与所述d结构中的苯环相连。
[0087]
在本本发明中,所述d-xh具体为中间体d-oh或中间体d-sh。
[0088]
本发明对所述中间体d-xh的来源没有特殊要求,采用市售产品或自制产品均可。
在本发明中,当所述中间体d-oh或中间体d-sh优选为自制产品时,本发明对所述中间体d-oh或中间体d-sh的制备方法没有特殊要求,采用本领域技术人员熟知的制备方法即可。
[0089]
在本发明的具体实施例中,当所述中间体d-oh的结构式具体为m1时,所述m1的制备方法具体为:
[0090][0091]
将9,9-二氢吖啶(21g,100mmol)、n-溴代琥珀酰亚胺(nbs)(19g,105mmol)在n,n-二甲基甲酰胺(dmf)中混合在0℃进行溴化反应,得到溴化9,9-二氢吖啶;
[0092]
将溴化9,9-二氢吖啶(19g,65mmol)、新制甲醇钠(40.7g,750mmol)、碘化亚铜(37g,195mmol)和dmf混合在85℃进行甲基化反应,得到甲氧基9,9-二氢吖啶;
[0093]
将甲氧基9,9-二氢吖啶(8g,33.4mmol)、溴苯(6.3g,40.1mmol)、三二亚苄基丙酮二钯(pd2(dba)3)(1.23g,1.336mmol)、叔丁基醇钠(t-buona)(8g,83.5mmol)、四氟硼酸三正丁基磷([hp(t-bu)3]bf4)(1.93g,6.68mmol)在甲苯中混合在120℃进行buchwald-hlartwig偶联反应,得到苯环取代甲氧基9,9-二氢吖啶;
[0094]
将苯环取代甲氧基9,9-二氢吖啶(10.1g,32mmol)、bbr3(16.1g,64mmol)和二氯甲烷混合在0℃进行脱甲氧基得到m1(9.04g)。
[0095]
本发明实施例中,m1的核磁数据为:1hnmr(500mhz,dmso-d6,δppm):7.67(t,j=7.7hz,2h),7.54(t,j=7.4hz,1h),7.43(d,j=7.7hz,1h),7.32(d,j=7.7hz,2h),6.93(t,j=7.4hz,1h),6.89(d,j=2.6hz,1h),6.84(t,j=7.4hz,1h),6.42(dd,j=2.5,8.8hz,1h),6.10(d,j=8.1hz,1h),5.98(d,j=8.8hz,1h),1.58(s,6h)。
[0096]
在本发明的具体实施例中,所述m1的合成路线为:
[0097][0098]
在本发明的具体实施例中,当所述中间体d-oh的结构式具体为m3时,所述m3的制备方法具体为:
[0099][0100]
将9,9-二氢吖啶(21g,100mmol)、n-溴代琥珀酰亚胺(nbs)(19g,105mmol)在n,n-二甲基甲酰胺(dmf)中混合在0℃进行溴化反应,得到溴化9,9-二氢吖啶;
[0101]
将溴化9,9-二氢吖啶(19g,65mmol)、甲醇钠(40.7g,750mmol)、碘化亚铜(37g,
195mmol)和dmf混合在85℃进行甲基化反应,得到甲氧基9,9-二氢吖啶;
[0102]
将甲氧基9,9-二氢吖啶(5g,20.1mmol)、ich3(3.6g,25.36mmol)、nah(0.8g,34.12mmol)和四氢呋喃(thf)在室温进行甲基取代,得到甲基取代甲氧基9,9-二氢吖啶;
[0103]
将甲基取代甲氧基9,9-二氢吖啶(5g,19.75mmol)、bbr3(9.9g,39.50mmol)和二氯甲烷混合在0℃进行脱甲氧基得到m3。
[0104]
本发明实施例中,m3的核磁数据为:1hnmr(500mhz,dmso-d6,δppm):7.36(dd,j=7.6,1.6hz,1h),7.19(td,j=7.6,1.4hz,1h),6.91-6.99(m,4h),6.80(dd,j=8.8,2.8hz,1h),3.73(s,3h),3.34(s,3h),1.44(s,6h)。
[0105]
在本发明的具体实施例中,所述m3的合成路线为:
[0106][0107]
在本发明的具体实施例中,当所述中间体d-oh的结构式具体为m4时,所述m4的制备方法具体为:
[0108][0109]
将咔唑(16.7g,100mmol)、n-溴代琥珀酰亚胺(nbs)(19g,105mmol)和n,n-二甲基甲酰胺(dmf)混合在0℃进行溴化反应,得到溴化咔唑;
[0110]
将溴化咔唑(18g,73mmol)、甲醇钠(40.7g,750mmol)、碘化亚铜(41.7g,219mmol)和dmf混合在85℃进行甲基化反应,得到甲氧基咔唑;
[0111]
将甲氧基咔唑(9.6g,48.7mmol)、溴苯(9.2g,58.44mmol)、三二亚苄基丙酮二钯(pd2(dba)3)(1.78g,1.948mmol)、叔丁基醇钠(t-buona)(11.7g,121.75mmol)、四氟硼酸三正丁基磷([hp(t-bu)3]bf4)(2.83g,9.74mmol)和甲苯混合在120℃进行buchwald-hlartwig偶联反应,得到苯环取代甲氧基咔唑;
[0112]
将苯环取代甲氧基咔唑(12.3g,45mmol)、bbr3(22.5g,90mmol)和二氯甲烷混合在0℃进行脱甲氧基得到m4。
[0113]
在本发明的实施例中,m4的核磁数据为:1h nmr(500mhz,dmso-d6,δppm):9.18(s,1h),8.13(d,j=7.7hz,1h),7.66(m,2h),7.60(m,2h),7.55(d,j=2.34hz,1h),7.50(td,j=7.7,1.34hz,1h),7.37(m,2h),7.21(m,2h),6.93(dd,j=8.9,2.3hz,1h)。
[0114]
在本发明的具体实施例中,所述m4的合成路线为:
[0115][0116]
在本发明的具体实施例中,当所述中间体d-oh的结构式具体为m5时,所述m5的制备方法具体为:
[0117][0118]
将吩噁嗪(12.8g,75mmol)、n-溴代琥珀酰亚胺(nbs)(14.5g,80mmol)和n,n-二甲基甲酰胺(dmf)混合在0℃进行溴化反应,得到溴化吩噁嗪;
[0119]
将溴化吩噁嗪(13.05g,50mmol)、甲醇钠(32.6g,750mmol)、碘化亚铜(38.1g,219mmol)和dmf混合在85℃进行甲基化反应,得到甲氧基吩噁嗪;
[0120]
将甲氧基吩噁嗪(6.4g,30mmol)、溴苯(5.5g,58.44mmol)、三二亚苄基丙酮二钯(pd2(dba)3)(1.09g,1.2mmol)、叔丁基醇钠(t-buona)(7.2g,75mmol)、四氟硼酸三正丁基磷([hp(t-bu)3]bf4)(2.74g,9.74mmol)和甲苯混合在120℃进行buchwald-hlartwig偶联反应,得到苯环取代甲氧基吩噁嗪;
[0121]
将苯环取代甲氧基吩噁嗪(7.5g,26mmol)、bbr3(13g,52mmol)和二氯甲烷混合在0℃进行脱甲氧基得到m5。
[0122]
本发明实施例中,m5的核磁数据为:1h nmr(500mhz,dmso-d6,δppm):9.89(s,1h),7.76(d,j=7.8hz,2h),7.36(d,j=7.8hz,2h),7.23(d,j=8.3hz,1h),7.01(m,3h),6.94(m,1h),6.54(m,2h)。
[0123]
在本发明的具体实施例中,所述m5的合成路线为:
[0124][0125]
在本发明的具体实施例中,当所述中间体d-oh的结构式具体为m6时,所述m6的制备方法具体为:
[0126][0127]
将4,4,8,8,12,12-六甲基-8,12-二氢-4h-苯并[9,1]喹喔啉[3,4,5,6,7-defg]吖啶(18.3g,50mmol)、n-溴代琥珀酰亚胺(nbs)(10g,55mmol)和n,n-二甲基甲酰胺(dmf)混合在0℃进行溴化反应,得到溴化4,4,8,8,12,12-六甲基-8,12-二氢-4h-苯并[9,1]喹喔啉[3,4,5,6,7-defg]吖啶;
[0128]
将溴化4,4,8,8,12,12-六甲基-8,12-二氢-4h-苯并[9,1]喹喔啉[3,4,5,6,7-defg]吖啶(19.05g,43mmol)、甲醇钠(21.7g,500mmol)、碘化亚铜(26.1g,150mmol)混合在85℃进行甲基化反应,得到甲氧基4,4,8,8,12,12-六甲基-8,12-二氢-4h-苯并[9,1]喹喔啉[3,4,5,6,7-defg]吖啶;
[0129]
将甲氧基4,4,8,8,12,12-六甲基-8,12-二氢-4h-苯并[9,1]喹喔啉[3,4,5,6,7-defg]吖啶(11.1g,28mmol)、bbr3(14g,56mmol)和二氯甲烷混合在0℃进行脱甲氧基得到m6。
[0130]
本发明实施例中,m6的核磁数据为:1hnmr(500mhz,dmso-d6,δppm):7.77(t,j=7.7hz,2h),7.64(t,j=7.4hz,1h),7.45(d,j=7.7hz,1h),7.32(d,j=7.7hz,2h),6.93(t,j=7.4hz,1h),6.89(d,j=2.6hz,1h),6.84(t,j=7.4hz,1h),6.42(dd,j=2.5,8.8hz,1h),6.10(d,j=8.1hz,1h),5.98(d,j=8.8hz,1h),1.60(s,18h)。
[0131]
在本发明的具体实施例中,所述m6的合成路线为:
[0132][0133]
本发明对所述中间体f-a的来源没有特殊要求,采用市售产品即可。
[0134]
在本发明中,-f与所述a的结构中的苯环相连。
[0135]
在本发明的具体实施例中,所述f-a具体为m2或m7
[0136][0137]
在本发明中,所述中间体d-xh和所述中间体f-a的摩尔比优选为(1~1.3):1,更优选为(1.15~1.28):1。
[0138]
在本发明中,所述极性有机溶剂优选为n-甲基吡咯烷酮和/或n,n-二甲基甲酰胺。
[0139]
在本发明中,所述中间体d-xh的物质的量和所述极性有机溶剂的体积比优选为(0.25~0.5)mmol:1ml。
[0140]
在本发明中,所述脱水剂优选为甲苯和/或苯,更优选为甲苯。
[0141]
在本发明中,所述极性有机溶剂和脱水剂的体积比优选为1:1。
[0142]
在本发明中,所述碱性催化剂优选包括碱金属碳酸盐,更优选包括碳酸钾和/或碳酸铯。
[0143]
在本发明中,所述中间体d-xh和所述碱性催化剂的摩尔比优选为1:(1.15~1.2)。
[0144]
在本发明中,所述亲核取代反应包括以下步骤:
[0145]
在保护气氛中,将中间体d-xh、极性有机溶剂、脱水剂和碱性催化剂进行脱水反应,得到中间反应液;所述反应温度为130-160℃,反应时间为1~3h;
[0146]
将所述中间反应液和中间体f-a混合进行亲核取代反应,得到所述有机室温电致磷光材料;所述亲核取代反应的温度为140~220℃,所述亲核取代反应的反应时间为8~20h,所述中间体d-xh和所述中间体f-a的摩尔比为(1~1.3):1。。
[0147]
在本发明中,所述脱水反应的温度为130~160℃,更优选为140~155℃,在本发明中,所述脱水反应的时间优选为1~3h,更优选为2h。
[0148]
在本发明中,所述脱水反应优选在保护气氛中进行,所述保护气氛优选为氮气或惰性气体,更优选为惰性气体。
[0149]
在本发明中,所述脱水反应的具体实施方式优选为加热回流,在本发明中,所述加热回流优选用连接有分水器的冷凝管进行。本发明优选通过加热回流去除所述第一混合液中的水分。在本发明中,所述脱水反应时加料的的顺序优选为将所述中间体d-xh和碱性催化剂混合,然后在保护气氛中与所述极性有机溶剂混合,最后与所述脱水剂混合加热回流进行脱水反应,得到中间反应液。
[0150]
得到中间反应液后,本发明将所述中间反应液和所述中间体f-a进行亲核取代反应,得到所述有机室温电致磷光材料;所述亲核取代反应的温度为140~220℃,所述亲核取代反应的反应时间为8~20h,所述中间体d-xh和所述中间体f-a的摩尔比为(1~1.3):1。
[0151]
在本发明中,所述中间体f-a优选以中间体f-a溶液的形式使用,在本发明中,所述中间体f-a溶液中的溶剂优选为n-甲基吡咯烷酮和/或n,n-二甲基甲酰胺。本发明对所述中间体f-a溶液的摩尔浓度没有特殊要求,能够将所述中间体f-a完全溶解即可。
[0152]
在本发明中,所述亲核取代反应的温度优选为140~220℃,更优选为190~210℃。在本发明中,所述亲核取代反应的时间优选为8~20h,更优选为8.5~18h。本发明优选通过薄膜色谱监测所述亲核取代反应的反应进程。
[0153]
所述亲核取代反应后,本发明优选对所述亲核取代反应得到的亲核取代反应液进行后处理,得到所述有机室温电致磷光材料。在本发明中,所述后处理优选包括:依次进行水洗、有机溶剂萃取、有机相干燥、浓缩、硅胶柱层析、洗脱液干燥。
[0154]
在本发明中,所述水洗优选为将所述亲核取代反应液和水混合,所述水优选为去离子水,所述水洗的次数优选为1~3次,每次水洗时,所述亲核取代反应液和水的体积优选为2:1。在本发明中,所述水洗优选在室温进行进行。本发明提供过水洗除去反应液中的高沸点溶剂。
[0155]
本发明优选对所述水洗后的反应液进行有机溶剂萃取,所述有机溶剂萃取用有机溶剂优选为二氯甲烷,所述水洗后的反应液和所述有机溶剂的体积比优选为2:1。在本发明中,所述有机溶剂萃取的次数优选为1次。在本发明中,所述有机溶剂萃取后得到有机相和萃余相。
[0156]
本发明优选对所述有机相干燥,在本发明中,所述干燥用干燥剂优选为无水硫酸钠。本发明对所述有机相的体积和干燥剂的质量比没有特殊要求。
[0157]
本发明优选对所述干燥后的有机相进行浓缩,所述浓缩优选为加热浓缩,所述浓缩后有机相的体积优选为浓缩前有机相体积的0.3~0.5。
[0158]
本发明优选对浓缩后的有机相进行硅胶柱层析,所述硅胶柱层析的洗脱溶剂优选为石油醚和二氯甲烷的混合溶剂,所述石油醚和二氯甲烷的体积比优选为5:1。在本发明的具体实施例中,所述洗脱时溶解样品的溶剂优选与洗脱溶剂相同。
[0159]
本发明优选对所述硅胶柱层析得到的产品干燥得到白色固体即为式i所示有机室温电致磷光材料。本发明对所述干燥的具体实施过程没有特殊要求。
[0160]
本发明提供了一种有机电致发光二极管,发光层中包括至少一种如权利要求1所述的有机室温电致磷光材料。
[0161]
在本发明中,上述技术方案所述的有机室温电致磷光材料本身作为发光层或优选作为客体掺杂在主体材料中构成发光层。
[0162]
在本发明中,上述技术方案所述的有机室温电致磷光材料优选作为主体,掺杂传统荧光材料、磷光金属配合物和热活化延迟荧光材料中的一种或多种作为客体,构成发光层。
[0163]
在本发明中的具体实施例中,当所述所述有机室温电致磷光材料优选与传统荧光材料、磷光金属配合物或热活化延迟荧光材料的中一种或多种混合作为发光材料时,所述发光材料中有机室温电致磷光材料的质量百分比优选为90~95%。
[0164]
在本发明的具体实施例中,所述有机发光二极管包括衬底和依次设置于所述衬底表面的阳极层、空穴注入层(hat-cn)、第一空穴传输层(npb)、第二空穴传输层(tcta)、发光层、插入层(tmpypb)、电子传输层(etl)和阴极。本发明对所述衬底的材质没有特殊要求。在本发明中,所述阳极层优选为氧化铟锡(ito),本发明对所述阳极层的厚度没有特殊要求。在本发明中,所述空穴注入层优选为2,3,6,7,10,11-六氰基-1,4,5,8,9,12-六氮杂苯并菲,所述空穴注入层的厚度优选为3nm。在本发明中,所述第一空穴传输层优选为n,n'-二苯基-n,n'-(1-萘基)-1,1'-联苯-4,4'-二胺,所述第一空穴传输层的厚度优选为40nm。在本发明中,所述第二空穴传输层优选为4,4',4'-三(咔唑-9-基)三苯胺,所述第二空穴传输层的厚度优选为10nm。
[0165]
在本发明中,所述发光层优选包括上述技术方案所述的有机室温电致磷光材料或上述技术方案所述制备方法制备的有机室温电致磷光材料。
[0166]
在本发明中,所述发光层优选包括所述有机室温电致磷光材料与传统荧光材料、磷光金属配合物或热活化延迟荧光材料的中一种或多种混合发光材料。所述混合发光材料中有机室温电致磷光材料的质量百分比优选为90~95%。
[0167]
在本发明中,所述发光层的厚度优选为15nm。
[0168]
在本发明中,所述插入层优选为1,3,5-三[(3-吡啶基)-3-苯基]苯,所述插入层的
厚度优选为50nm。在本发明中,所述电子传输层优选为8-羟基喹啉-锂,所述电子传输层的厚度优选为1nm。在本发明中,所述阴极优选为铝,所述阴极的厚度优选为100nm。
[0169]
本发明对所述有机发光二级管的制备方法没有特殊要求。采用本领域技术人员数值的方法进行即可,例如化学沉积法。
[0170]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0171]
实施例1
[0172]
将m1(1g,3.32mmol)、碳酸钾(0.55g,3.98mmol)加入三口反应瓶中,氩气保护下,加入5mln-甲基吡咯烷酮,5ml甲苯。140℃进行,用连有分水器的冷凝管回流。2h后,将反应冷却至室温。加入5mln-甲基吡咯烷酮溶解的m2(0.86g,2.59mmol),升温至190℃进行亲和取代反应18h,薄层色谱检测反应。反应结束后,去离子水洗涤3次,二氯甲烷萃取有机相,无水硫酸钠干燥,浓缩。硅胶柱层析,石油醚:二氯甲烷体积比5:1的混合溶剂洗脱,干燥得到白色固体有机室温电致磷光材料,结构式为i-26,1.554g,产率60%。产品的核磁图谱1h nmr(500mhz,dmso-d6,δppm):8.71(d,j=8.3hz,6h),7.70(m,4h),7.64(t,j=7.8hz,4h),7.59(t,j=7.5hz,1h),7.49(dd,j=7.6,1.6hz,1h),7.42(d,j=7.4hz,2h),7.35(d,j=2.8hz,1h),7.13(d,j=8.9hz,2h),6.99(td,j=7.5,1.5hz,1h),6.91(td,j=7.5,1.2hz,1h),6.86(dd,j=8.9,2.7hz,1h),6.23(d,j=8.9hz,1h),6.17(dd,j=8.3,1.2hz,1h),1.64(s,6h)。分子式c
42h32
n4o;m/z=609.3理论值:m/z:608.26(100.0%),609.26(45.4%),610.26(10.1%),609.25(1.5%)。元素分析:c,82.58;h,5.24;n,8.96。结构为式i-26有机室温电致磷光材料的的合成路线为:
[0173][0174]
图1为本发明实施例1的分子i-26在二氯甲烷溶液中的吸收光谱、非掺杂薄膜稳态光致发光光谱和非掺杂薄膜室温磷光光谱。由图1可知,实施例1的分子i-26的吸收峰位于280nm、298nm;非掺杂薄膜稳态光致发光峰位于497nm,非掺杂薄膜室温磷光峰位于502nm。
[0175]
图2为实施例1的分子i-26非掺杂薄膜稳态光致发光温度依赖性光谱。由图2可知,随着温度上升,非掺杂薄膜稳态光致发光强度减弱。
[0176]
图3为实施例1的分子i-26非掺杂薄膜瞬态光致发光温度依赖性光谱。由图3可知,非掺杂薄膜随着温度升高,寿命变短。
[0177]
实施例2
[0178]
将m3(0.36g,1.53mmol)、碳酸钾(0.25g,1.83mmol)加入三口反应瓶中,氩气保护下,加入5mln-甲基吡咯烷酮,5ml甲苯。140℃进行,用连有分水器的冷凝管回流。2h后,将反应冷却至室温。加入5mln-甲基吡咯烷酮溶解的m2(0.396g,1.19mmol),升温至190℃,薄层色谱检测反应。反应结束后,去离子水洗滤液,二氯甲烷萃取有机相,无水硫酸钠干燥,浓
缩。硅胶柱层析,石油醚:二氯甲烷8:1的混合溶剂洗脱,得到白色固体有机室温电致磷光材料,结构式为i-25,0.5g,产率76.9%。1h nmr(500mhz,cdcl3,δppm):8.75(m,6h),7.59(m,6h),7.43(dd,j=7.6,1.5hz,1h),7.29(m,1h),7.24(d,j=2.6hz,1h),7.10(m,2h),6.97-7.05(m,4h),3.50(s,3h),1.56(s,6h)。分子式c
37h30
n4o;m/z=546.2理论值:m/z:546.24(100.0%),547.25(40.0%),548.25(7.8%),547.24(1.5%)。元素分析:c,81.58;h,5.23;n,10.25。结构为式i-25有机室温电致磷光材料的的合成路线为:
[0179][0180]
对实施例2制备的材料进行光学性能检测,与实施例1制备的产品性能相似。
[0181]
实施例3
[0182]
将m4(1.20g,4.6mmol)、碳酸钾(0.83g,6mmol)加入三口反应瓶中,氩气保护下,加入15mln-甲基吡咯烷酮,15ml甲苯。140℃进行,用连有分水器的冷凝管回流。2h后,将反应冷却至室温。加入15mln-甲基吡咯烷酮溶解的m2(1.31g,4mmol),升温至190℃,薄层色谱检测反应。反应结束后,去离子水洗滤液,二氯甲烷萃取有机相,无水硫酸钠干燥,浓缩。硅胶柱层析,石油醚:二氯甲烷10:1的混合溶剂洗脱,得到白色固体有机室温电致磷光材料,结构式为i-24,1.6g,产率70%。1h nmr(500mhz,cdcl3,δppm):8.77(m,6h),8.10(td,j=7.8,1.0hz,1h),7.92(d,j=2.4,1h),7.61(m,10h),7.51(m,1h),7.45(m,3h),7.30(m,1h),7.24(dd,j=8.7,2.4hz,1h),7.18(m,2h).分子式c
39h26
n4o;m/z=566.2理论值:m/z:566.21(100.0%),567.21(42.2%),568.22(8.7%),567.21(1.5%)。元素分析:c,82.68;h,4.53;n,9.89。结构为式i-24有机室温电致磷光材料的的合成路线为:
[0183][0184]
对实施例3制备的材料进行光学性能检测,与实施例1制备的产品性能相似。
[0185]
实施例4
[0186]
将m5(0.58g,2.1mmol)、碳酸钾(0.34g,2.51mmol)加入三口反应瓶中,氩气保护下,加入5mln-甲基吡咯烷酮,5ml甲苯。140℃下,用连有分水器的冷凝管回流。2h后,将反应冷却至室温。加入5mln-甲基吡咯烷酮溶解的m-5(0.55g,1.64mmol),升温至190℃,薄层色谱检测反应。反应结束后,去离子水洗滤液,二氯甲烷萃取有机相,无水硫酸钠干燥,浓缩。硅胶柱层析,石油醚:二氯甲烷12:1的混合溶剂洗脱,得到白色固体有机室温电致磷光材料,结构式为i-27,0.70g,产率73.2%。1h nmr(500mhz,cdcl3,δppm):8.47(m,4h),8.31(d,j=8.3hz,2h),7.65(m,6h),7.24(m,3h),7.13(m,3h),7.05(m,3h),6.98(m,3h),6.88(m,2h)分子式c
39h26
n4o2;m/z=582.2理论值:m/z:582.21(100.0%),583.21(42.2%),584.21(8.7%),583.20(1.5%)。元素分析:c,80.41;h,4.70;n,9.59。结构为式i-27有机室温电致磷光材料的的合成路线为:
[0187][0188]
对实施例4制备的材料进行光学性能检测,与实施例1制备的产品性能相似。
[0189]
实施例5
[0190]
将m6(1.46g,3.8mmol)、碳酸钾(0.63g,4.58mmol)加入三口反应瓶中,氩气保护下,加入15mln-甲基吡咯烷酮,15ml甲苯。140℃进行下,用连有分水器的冷凝管回流。2h后,将反应冷却至室温。加入15mln-甲基吡咯烷酮溶解的m2(0.99g,2.98mmol),升温至190℃,薄层色谱检测反应。反应结束后,去离子水洗滤液,二氯甲烷萃取有机相,无水硫酸钠干燥,浓缩。硅胶柱层析,石油醚:二氯甲烷15:1的混合溶剂洗脱,得到白色固体有机室温电致磷光材料,结构式为i-129,1.54g,产率75.1%。1h nmr(500mhz,dmso-d6,δppm):8.71(m,6h),7.70(m,6h),7.32(dd,j=8.4,2.5hz,2h),7.24(d,j=7.6hz,4h),6.98(d,j=7.3hz,2h),6.87(s,2h),1.66(s,18h)。分子式c
48h40
n4o;m/z=688.3理论值:m/z:688.32(100.0%),689.32(51.9%),690.33(13.2%),689.32(1.5%),691.33(1.4%)。元素分析:c,83.81;h,5.85;n,8.16。结构为式i-129有机室温电致磷光材料的的合成路线为:
[0191][0192]
对实施例5制备的材料进行光学性能检测,与实施例1制备的产品性能相似。
[0193]
实施例6
[0194]
将m1(0.72g,3.12mmol)、碳酸钾(0.5g,3.66mmol)加入三口反应瓶中,氩气保护下,加入10mln-甲基吡咯烷酮,10ml甲苯。140℃进行,用连有分水器的冷凝管回流。2h后,将反应冷却至室温。加入10mln-甲基吡咯烷酮溶解的m7(0.49g,2.44mmol),升温至190℃,薄层色谱检测反应。反应结束后,去离子水洗滤液,二氯甲烷萃取有机相,无水硫酸钠干燥,浓缩。硅胶柱层析,石油醚:二氯甲烷3:1的混合溶剂洗脱,得到白色固体有机室温电致磷光材料,结构式为i-46,0.94g,产率80.5%。1h nmr(500mhz,cdcl
3,
δppm):7.75(dd,j=7.8,1.8hz,6h),7.19(d,j=8.1hz,4h),7.28(s,1h),7.19(s,2h),6.96(d,j=8.1hz,4h),6.72(d,j=7.8hz,2h),6.33(m,2h),1.64(s,6h)。分子式c
34h27
no2;m/z=481.2理论值:m/z:481.20(100.0%),482.21(36.8%),483.21(3.9%),483.21(2.7%)。元素分析:c,85.01;h,5.85;n,2.91。结构为式i-46有机室温电致磷光材料的的合成路线为:
[0195][0196]
对实施例6制备的材料进行光学性能检测,与实施例1制备的产品性能相似。
[0197]
实施例7
[0198]
将m1(1.08g,4.59mmol)、碳酸钾(0.75g,5.49mmol)加入三口反应瓶中,氩气保护下,加入5mln-甲基吡咯烷酮,5ml甲苯。140℃进行,用连有分水器的冷凝管回流。2h后,将反应冷却至室温。加入5mln-甲基吡咯烷酮溶解的m8(0.43g,3.57mmol),升温至190℃,薄层色谱检测反应。反应结束后,去离子水洗滤液,二氯甲烷萃取有机相,无水硫酸钠干燥,浓缩。硅胶柱层析,石油醚:二氯甲烷5:1的混合溶剂洗脱,得到白色固体有机室温电致磷光材料,结构式为i-2,0.86g,产率60%。1h nmr(500mhz,cdcl3,δppm):7.95(d,j=7.5hz,2h),7.24(dd,j=7.5,1.5hz,2h)7.17(m,6h),7.08(d,j=7.6hz,2h),6.99(dd,j=7.5,1.5hz,2h),6.95(d,j=7.5hz,1h),6.87(s,1h),1.65(s,6h)。分子式c
28h22
n2o;m/z=407.2理论值:m/z:m/z:402.17(100.0%),403.18(30.3%),404.18(2.7%),404.18(1.7%)。元素分析:c,83.01;h,5.56;n,7.01。结构为式i-2有机室温电致磷光材料的的合成路线为:
[0199][0200]
对实施例7制备的材料进行光学性能检测,与实施例1制备的产品性能相似。
[0201]
对比例1
[0202]
将二苯胺(08.45g,50mmol)、4-甲氧基碘苯(11.7g,50mmol)、三二亚苄基丙酮二钯(pd2(dba)3)(1.84g,2mmol)、叔丁基醇钠(t-buona)(2.96g,10mmol)、四氟硼酸三正丁基磷([hp(t-bu)3]bf4)(12.g,125mmol)和甲苯混合在120℃进行buchwald-hlartwig偶联反应,得到甲氧基三苯胺;将甲氧基三苯胺、bbr3和二氯甲烷混合在0℃进行脱甲氧基得到m9。
[0203]
m9的核磁数据为:1h nmr(500mhz,cdcl
3,
δppm):7.21(s,4h),7.01(s,7h),6.76(s,3h)。m9的合成路线为:
[0204][0205]
将m9(0.9g,3.45mmol)、碳酸钾(0.32g,4.5mmol)加入三口反应瓶中,氩气保护下,加入5mln-甲基吡咯烷酮,5ml甲苯。140℃下,用连有分水器的冷凝管回流。2h后,将反应冷却至室温。加入5mln-甲基吡咯烷酮溶解的m2(0.98g,3mmol),升温至190℃,薄层色谱检测反应。反应结束后,去离子水洗滤液,二氯甲烷萃取有机相,无水硫酸钠干燥,浓缩。硅胶柱层析,石油醚:二氯甲烷10:1的混合溶剂洗脱,得到白色固体0.5g,产率76.9%。1h nmr(500mhz,cdcl3,δppm):8.78(m,6h),7.60(m,6h),7.29(m,4h),7.12-7.19(m,8h),7.04(m,4h)。分子式c
39h28
n4o;m/z=568.2理论值:m/z:m/z:568.23(100.0%),569.23(42.2%),570.23(8.7%),569.22(1.5%)。元素分析:c,82.01;h,5.05;n,9.75。对比例1的产物的合成路线为:
[0206][0207]
实施例8
[0208]
将ito透明导电玻璃在清洗剂中超声处理,在洁净的环境下烘烤至完全除去水分,用紫外光和臭氧清洗,并用低能阳离子轰击;按照有机发光二极管器件结构:氧化铟锡(ito)/2,3,6,7,10,11-六氰基-1,4,5,8,9,12-六氮杂苯并菲(hat-cn)(3nm)/n,n'-二苯基-n,n'-(1-萘基)-1,1'-联苯-4,4'-二胺(npb)(40nm)/4,4',4'-三(咔唑-9-基)三苯胺(tcta)(10nm)/实施例1制备得到的分子i-26(15nm)/1,3,5-三[(3-吡啶基)-3-苯基]苯(tmpypb)(50nm)/8-羟基喹啉-锂(liq)(1nm)/铝(al)(100nm)制备发光二极管,其中,发光层采用电子蒸镀法制备,其它层采用化学沉积法。
[0209]
对比例2
[0210]
将ito透明导电玻璃在清洗剂中超声处理,在洁净的环境下烘烤至完全除去水分,用紫外光和臭氧清洗,并用低能阳离子轰击;按照有机发光二极管器件结构:氧化铟锡(ito)/2,3,6,7,10,11-六氰基-1,4,5,8,9,12-六氮杂苯并菲(hat-cn)(3nm)/n,n'-二苯基-n,n'-(1-萘基)-1,1'-联苯-4,4'-二胺(npb)(40nm)/4,4',4'-三(咔唑-9-基)三苯胺(tcta)(10nm)/对比例1产品(15nm)/1,3,5-三[(3-吡啶基)-3-苯基]苯(tmpypb)(50nm)/8-羟基喹啉-锂(liq)(1nm)/铝(al)(100nm)制备发光二极管,其中,发光层采用电子蒸镀法制备,其它层采用化学沉积法。
[0211]
实施例9
[0212]
将ito透明导电玻璃在清洗剂中超声处理,在洁净的环境下烘烤至完全除去水分,用紫外光和臭氧清洗,并用低能阳离子轰击;按照有机发光二极管器件结构:氧化铟锡(ito)/2,3,6,7,10,11-六氰基-1,4,5,8,9,12-六氮杂苯并菲(hat-cn)(3nm)/n,n'-二苯基-n,n'-(1-萘基)-1,1'-联苯-4,4'-二胺(npb)(40nm)/4,4',4'-三(咔唑-9-基)三苯胺(tcta)(10nm)/以实施例1制备的分子i-26为主体,磷光材料po-01作为客体,掺杂量为8%,(15nm)/1,3,5-三[(3-吡啶基)-3-苯基]苯(tmpypb)(50nm)/8-羟基喹啉-锂(liq)(1nm)/铝(al)(100nm)制备发光二极管,其中,发光层采用电子蒸镀法制备,其它层采用化学沉积法。
[0213]
对比例3
[0214]
将ito透明导电玻璃在清洗剂中超声处理,在洁净的环境下烘烤至完全除去水分,用紫外光和臭氧清洗,并用低能阳离子轰击;按照有机发光二极管器件结构:氧化铟锡(ito)/2,3,6,7,10,11-六氰基-1,4,5,8,9,12-六氮杂苯并菲(hat-cn)(3nm)/n,n'-二苯基-n,n'-(1-萘基)-1,1'-联苯-4,4'-二胺(npb)(40nm)/4,4',4'-三(咔唑-9-基)三苯胺(tcta)(10nm)/以mcp为主体,磷光材料po-01作为客体,掺杂量为8%,(15nm)/1,3,5-三[(3-吡啶基)-3-苯基]苯(tmpypb)(50nm)/8-羟基喹啉-锂(liq)(1nm)/铝(al)(100nm)制备发光二极管,其中,发光层采用电子蒸镀法制备,其它层采用化学沉积法。
[0215]
测试例
[0216]
对实施例8与对比例2和实施例9与对比例3制备的oled器件进行发光性能测试。其
中,实施例8和对比例2得到的器件的发光性能测试如表1所示。图4为本发明实施例8和对比例2制备的的oled器件外量子效率-亮度曲线。由图4可以得出,实施例8和对比例2的oled器件的外量子效率最大值分别为12.5%和7.1%。图5为本发明实施例8和对比例2的oled器件电致发光光谱。本发明实施例8和对比例2的电致发光峰位分别为518nm和505nm。对比可知,当稠并芳胺作为电子给体d时,分子刚性增加,抑制了三线态激子的非辐射衰减过程,最终提升器件外量子效率。
[0217]
表1.实施例8和对比例2制备的oled器件发光性能测试结果
[0218][0219]
实施例9和对比例3得到的器件的发光性能测试如表2所示。图6为实施例9和对比例3制备的oled器件的外量子效率-亮度曲线对比图,由图6可知,实施例9和对比例3制备的oled器件最大外量子效率分别为22.1%和19.2%。图7为实施例9和对比例3制备的oled器件的电致发光光谱。由图7可知,实施例9和对比例3制备的oled器件的点值发光峰位和峰形均不发生改变。对比可知,当采用权利要求1所述的有机室温电致磷光材料作为主体来代替传统主体mcp时,器件驱动电压降低,功率效率和外量子效率增加。
[0220]
表2.实施例9和对比例3制备的oled器件发光性能测试结果
[0221][0222]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。