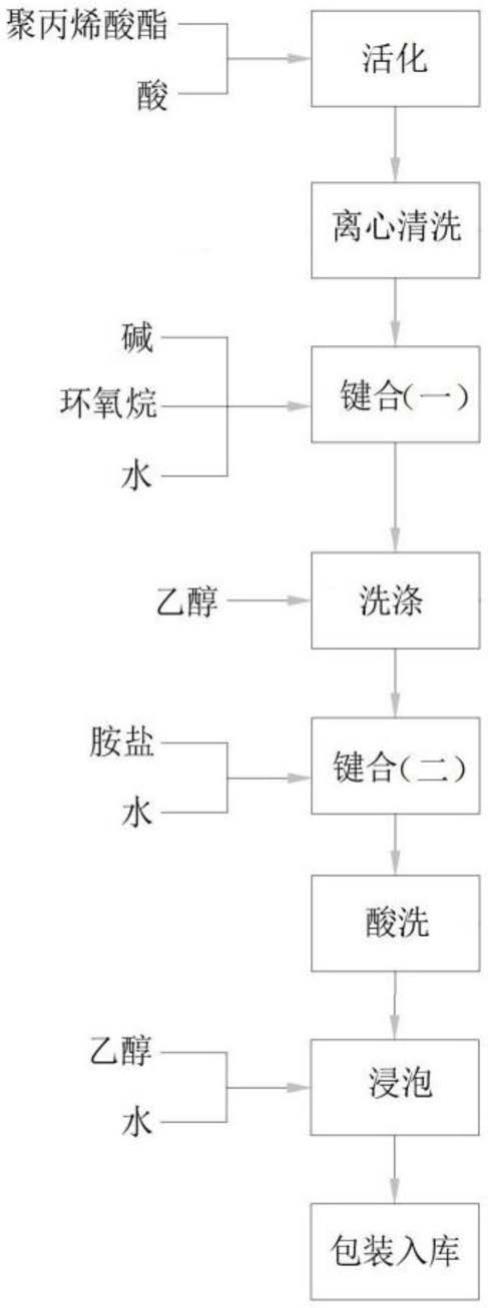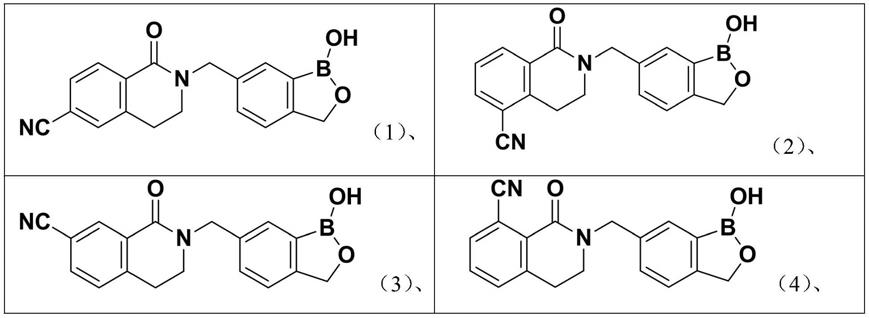
一种苯并[c][1,2]氧杂硼戊环-1(3h)-醇类合物及其用途
技术领域:
[0001]
本发明属于生物医药技术领域,具体涉及具有pde4抑制作用的苯并[c][1,2]氧杂硼戊环-1(3h)-醇类衍生物、制备方法、组合物和用途。
背景技术:
[0002]
环腺苷酸(camp)是介导细胞对大范围胞外刺激的生物相应的第二信使,当适当的激动剂与特定细胞表面受体结合时,腺苷酸环化酶被激活,将三磷酸腺苷(atp)转化为camp。研究表明,激动剂诱导的camp在细胞内的作用主要有camp依赖性蛋白激酶的作用介导,通过核苷酸转运至细胞外或通过由核苷磷酸二酯酶(pde)酶促裂解来终止camp的细胞内作用,磷酸二酯酶水解成无活性的代谢物5
’‑
腺苷磷酸(5
’‑
amp)。在人类骨髓和淋巴谱系细胞中,camp水平的升高与细胞活化的抑制有关。因此,胞内pde酶家族调节细胞中camp的水平,且为camp降解的主要贡献者。因此,pde功能的抑制便会阻止camp向无活性的代谢物5-amp转化,因而维持较高的camp水平,并因此抑制细胞活化。
[0003]
pde4对camp具有高度特异性,广泛分布于人体各种组织和细胞中,如大脑、肾脏、心肌细胞、内皮细胞和免疫细胞中。pde4参与了促进单核细胞与巨噬细胞活化、中性粒细胞浸润、血管平滑肌的增殖、血管扩张以及心肌收缩等相关生理病理过程,对中枢神经系统功能、心血管功能、炎症/免疫系统、细胞黏附等都有影响。研究人员已经对使用pde4抑制剂作为抗炎剂显示相当的兴趣。早期证据表明pde4抑制剂对多种炎症细胞(如单核细胞、巨噬细胞以及粒细胞)具有有益的作用。在这些细胞中,许多促炎介质、脂质介质、超氧化物和生物胺类的合成和/或释放被pde4抑制剂的作用所减弱。所述pde4抑制剂还影响其他细胞功能,包括t细胞增殖、响应化学毒性物质的粒细胞移行和脉管系统中内皮细胞连接的完整性。
[0004]
由于茶碱、咯利普兰(rolipram)较低的药理活性和严重的恶心、呕吐等副作用,在临床上的应用受到了限制。研究人员继续寻找选择性更高、药理活性更高、不良反应更低的pde4抑制剂。此外,一些公司现在正进行其他pde4抑制剂的临床试验。然而,关于效能和不良副作用的问题,如呕吐和中枢神经系统紊乱,仍然没有解决。
技术实现要素:
[0005]
为了解决上述问题,本发明提供了一种如式i所示苯并[c][1,2]氧杂硼戊环-1(3h)-醇类合物、其立体异构体、其溶剂合物、其水合物或其药学上可接受的盐,
[0006][0007]
其中,
[0008]
x独立地为ch或n;
[0009]
a、d独立地为c或n,a、d不同时为n;
[0010]
r1独立地为h、氰基、氨基羰基、卤素、c1-4烷基氧基、乙酰氨基、环氧丙烷甲酰氨基、甲基磺酸氨基、c1-4羟基烷基。
[0011]
根据本发明示例性的实施方案,所述x为n,所述a、d均为c;
[0012]
所述r1独立地为h、氰基、氨基羰基、氟、乙酰氨基、环氧丙烷甲酰氨基、甲基磺酸氨基、2-羟基丙烷-2-基、羟基乙烷基、羟基甲烷基、甲氧基、乙氧基。
[0013]
根据本发明示例性的实施方案,所述x为n;所述a、d独立地为c或n,且a与d不相同;所述r1独立地为h、氰基、氨基羰基、甲氧基、乙氧基。
[0014]
根据本发明示例性的实施方案,所述x为c,所述a独立地为c或n,所述d为c,r1独立地为氰基、氨基羰基。
[0015]
作为实例,所述式i所示的化合物可为如下任一化合物:
[0016]
[0017]
[0018][0019]
本发明还提供了一种药物组合物,其包含上述的化合物i、其立体异构体、其溶剂合物、其水合物或其药学上可接受的盐和药用辅料;所述的化合物i、其立体异构体、其溶剂合物、其水合物或其药学上可接受的盐的用量可为治疗有效量。
[0020]
本发明的药物组合物可根据公开的内容使用本领域技术人员已知的任何方法来制备。例如,常规混合、溶解、整粒、压片、填充、包封、包衣等。本发明所述的药物组合物可以任何形式给药,包括注射、口服给药。口服给药包括但不限于胶囊、囊片、软胶囊剂、片剂、溶液、悬浮液、乳液、酏剂;注射给药包括但不限于注射用溶液、可以溶解或悬浮在药学上可接受载体中的干制剂、注射用悬浮液和注射用乳剂。
[0021]
根据本发明,药物组合物所述的药用辅料可以包括下列辅料中的一种或多种:崩解剂、润滑剂、稀释剂、润湿剂、粘合剂、着色剂、助流剂、ph调节剂、抗氧剂、包衣料、矫味剂、掩味剂、助悬剂、助溶剂。所述的药用辅料可为药物生产领域中广泛采用的辅料。辅料主要用于提供一个安全、稳定和功能性的药物组合物,还可以提供方法,使受试者接受给药后活性成分以所期望速率溶出,或促进受试者接受组合物给药后活性成分得到有效吸收。
[0022]
根据本发明,药物组合物可以进一步地包含其他pde4抑制剂药物,所述的其他pde4抑制剂药物为罗氟司特、西洛司特或阿普斯特。
[0023]
本发明还提供了一种上述的化合物i、其立体异构体、其溶剂合物、其水合物或其药学上可接受的盐在制备pde4抑制剂中的应用,优选地,所述pde4抑制剂用于制备炎症或自身免疫性疾病的药物中的应用。化合物i、其立体异构体、其溶剂合物、其水合物或其药学上可接受的盐可以单独使用用于治疗炎症或自身免疫性疾病,也可以多种联合使用治疗炎症或自身免疫性疾病。包含化合物i、其立体异构体、其溶剂合物、其水合物或其药学上可接受的盐的组合物为pde4抑制剂。
[0024]
在一些实施例中,所述炎症或自身免疫性疾病是指类风湿性关节炎、骨关节炎、痛风性关节炎、脊椎炎、脓毒症、脓毒性休克、内毒素性休克、革兰氏阴性脓毒症、革兰氏阳性脓毒症、中毒性休克综合征、哮喘、慢性支气管炎、成人型呼吸窘迫综合征、慢性阻塞性肺疾病、硅肺病、肺结节病、囊性纤维化病、慢性肾小球肾炎、狼疮、炎性肠病和炎性皮肤病,例如特应性皮炎、银屑病和荨麻疹。
[0025]
术语定义和说明
[0026]
在本发明的各部分,描述了连接取代基。当该结构清楚地需要连接基团时,针对该基团所列举的马库什变量应理解为连接基团。例如,如果该结构需要连接基团并且针对该变量的马库什基团定义列举了“烷基”,则应该理解,该“烷基”代表连接的亚烷基基团,如-ch3。
[0027]
术语“取代的”表示所给结构中的一个或多个氢原子被具体取代基所取代。当所给出的结构式中不止一个位置能被选自具体基团的一个或多个取代基所取代,那么取代基可以相同或不同地在各个位置取代。
[0028]
另外,需要说明的是,除非以其他方式明确指出,在本发明中
“…
独立地为”均应做广义理解,其既可以是指在不同基团中,相同符号之间所表达的具体选项之间互相不影响,也可以表示在相同的基团中,相同符号之间所表达的具体选项之间互相不影响。
[0029]
术语“立体异构体”是指具有相同化学构造,但原子或基团在空间上排列方式不同的化合物。立体异构体包括对映异构体、非对映异构体、构象异构体(旋转异构体)、几何异构体、阻转异构体,等等。可以用已知的方法将任何所得终产物或中间体的外消旋体通过本领域技术人员熟悉的方法拆分成光学对映体,如,通过对获得的其非对映异构进行分离。外消旋的产物也可以通过手性色谱来分离,如,使用手性吸附剂的高效液相色谱(hplc)。特别
地,对映异构体可以通过不对称合成制备。
[0030]
术语“烷基”或“烷基基团”,表示饱和的直链或支链一价烃基基团。
[0031]
术语“杂原子”是指o、n、s,包括n任何氧化态的形式。
[0032]
术语“溶剂化物”是指一个或多个溶剂分子与本发明的化合物所形成的缔合物。形成溶剂化物的溶剂包括,但并不限于,水,乙醇,甲醇和乙酸。术语“水合物”是指溶剂分子是水所形成的缔合物。
[0033]
术语“药学上可接受的盐”是指本发明化合物的盐,由本发明发现的具有特定取代基的化合物与相对无毒的酸制备。药学上可接受的盐可以是例如在链或环中具有氮原子的具有足够碱性的本发明的化合物的酸加成盐,例如与如下无机酸形成的酸加成盐:例如盐酸、氢溴酸、硫酸或磷酸,或者与如下有机酸形成的酸加成盐:例如甲酸、乙酸、月桂酸、苯甲酸、水杨酸、苯磺酸、对甲苯磺酸、甲磺酸、柠檬酸或酒石酸。
[0034]
有机化学一般原理可参考"organic chemistry",thomas sorrell,university science books,sausalito:1999,和"march's advanced organic chemistry”by michael b.smith and jerry march,johnwiley&sons,newyork:2007中的描述,其全部内容通过引用并入本文。如可以按照本领域常规的化学合成方法制备得到,其步骤和条件可参考本领域类似反应的步骤和条件,例如通过萃取、过滤或柱层析来分离目标化合物。
[0035]
术语“有效量”或者“治疗有效量”是指足以实现预期应用(包括但不限于如下定义的疾病治疗)的本发明所述化合物的量。治疗有效量可以取决于以下因素而改变:预期应用(体外或者体内),或者所治疗的受试者和疾病病症如受试者的重量和年龄、疾病病症的严重性和给药方式等,其可以由本领域普通技术人员容易地确定。具体剂量将取决于以下因素而改变:所选择的特定化合物、所依据的给药方案、是否与其它化合物组合给药、给药的时间安排、所给药的组织和所承载的物理递送系统。
[0036]
术语“组合物”是指包含规定量的活性成分的产物。本发明药物组合物包括通过将本发明化合物与药用辅料混合而制备的任何组合物。术语“药用辅料”是指可药用惰性成分,分为稀释剂、填充剂、粘合剂、崩解剂、润滑剂、助流剂、造粒剂、包衣剂、润湿剂、溶剂、共溶剂、助悬剂、乳化剂、甜味剂、矫味剂、掩味剂、着色剂、防结块剂、保湿剂、螯合剂、塑化剂、增粘剂、抗氧化剂、防腐剂、稳定剂、表面活性剂和缓冲剂等。合适的药学上可接受的药用辅料会依所选具体剂型而不同。此外,可根据它们在组合物中的特定功能来选择药学上可接受的药用辅料。技术人员可认识到,某些药学上可接受的药用辅料可提供不止一种功能,并提供可供选择的功能,这取决于制剂中存在多少该药用辅料和制剂中存在哪些其他药用辅料。
[0037]
技术人员掌握本领域的知识和技能,以使他们能选择用于本发明的适当量的合适的药学上可接受的药用辅料。此外,存在大量技术人员可获得的资源,他们描述药学上可接受的赋形剂,并用于选择合适的药学上可接受的药用辅料(即赋形剂)。实例包括remington's pharmaceutical sciences(mack publishing company),the hand book of pharmaceutical additives(gower publishing limited),and the hand book of pharmaceutical excipients(the american pharmaceutical association and the pharmaceutical press)。
[0038]
术语“包含”为开放式表达,即包括本发明所指明的内容,但并不排除其他方面的
内容。
[0039]
术语“活性成分”、“治疗剂”、或“活性物质”是指一种化学实体,它可以有效地治疗目标疾病或病症。
[0040]
除非另有说明,本文使用的所有技术术语和科学术语具有要求保护主题所属领域的标准含义。倘若对于某术语存在多个定义,则以本文定义为准。
[0041]
除非另有说明,本发明采用质谱、nmr、hplc、生物化学或药理检测的传统方法,各步骤和条件可参照本领域常规的操作步骤和条件。
[0042]
除非另有说明,本发明采用分析化学、有机合成化学和医药化学的标准命名及标准实验室步骤和技术。在某些情况下,标准技术被用于化学合成、化学分析、药物制备。
[0043]
除非另有说明,本技术说明书和权利要求书中记载的基团和术语定义,包括其作为实例的定义、示例性的定义、优选的定义、实施例中具体化合物的定义等,可以彼此之间任意组合和结合。这样的组合和结合后的基团定义及化合物结构,应当属于本技术说明书记载的范围内。
[0044]
有益效果:
[0045]
本发明化合物可作为作pde4抑制剂,其具有良好的pde4调节活性,呈现高活性,具有临床应用价值,化合物适合开发成治疗慢性阻塞性肺疾病、银屑病等疾病的药物。此外,本发明化合物合成步骤简单,因此具备良好的经济利用价值。
具体实施方式
[0046]
通过下面的实施例可以对本发明进行进一步的描述,然而,本发明的范围并不限于下述实施例。
[0047]
实施例1
[0048]
2-((1-羟基-1,3-二氢苯并[c][1,2]氧杂硼杂烷-6-基)甲基)-1-氧代-1,2,3,4-四氢异喹啉-6-甲腈
[0049]
第一步:
[0050]
将化合物1a(31.4g,200.0mmol)和甲基磺酸(50ml)溶于二氯甲烷(200ml)中,冰浴下加入nan3(26g,400.0mmol),室温搅拌反应12小时,tlc检测反应,反应完毕后将反应液倒入10%氢氧化钠水溶液(500ml)中,二氯甲烷(200mlx2)萃取,合并有机层,干燥,柱层析分离得到类白色固体25.7g,收率74.7%。
[0051]
第二步:
[0052]
将化合物1b(25.0g,145.3mmol)溶于dmf(200ml),室温下加入氢化钠(4.2g,174.4mmol),搅拌反应1小时,然后加入化合物1c(44.4g,145.3mmol),于40℃搅拌反应4小时,tlc检测反应,反应完毕后用水(200ml)淬灭反应,用乙酸乙酯(300mlx2)萃取,合并有机层,干燥柱层析分离得到淡黄色固体35.6g,收率61.6%。
[0053]
第三步
[0054]
将化合物1d(35.0g,87.9mmol)、化合物1e(26.8g,105.5mmol)、pd(dppf)cl2(3.7g,5.0mmol)、koac(9.8g,100.0mmol)溶于dmf(150ml)中,升温至100℃反应6小时,tlc检测反应,反应完毕后加入乙酸乙酯(400ml)和水(200ml)淬灭反应,有机层干燥,柱层析分离得到类白色固体28.3g,收率72.2%。
[0055]
第四步:
[0056]
将中间体1f(4.46g,10.0mmol)溶于甲醇(100ml)中,室温下加入硼氢化钠(1.1g,30.0mmol),搅拌反应6分钟,tlc检测反应,反应完毕后加水(50ml)淬灭反应,乙酸乙酯(50mlx2)提取,合并有机层,然后加入1m盐酸10ml搅拌2小时,有机层干燥,柱层析分离得到类白色固体2.3g,收率72.3%。esi( )m/z=319.1。
[0057]
实施例2
[0058]
2-((1-羟基-1,3-二氢苯并[c][1,2]氧杂硼杂烷-6-基)甲基)-1-氧代-1,2,3,4-四氢异喹啉-5-甲腈
[0059]
第一步:
[0060]
将化合物2a(15.7g,100.0mmol)和甲基磺酸(30ml)溶于二氯甲烷(200ml)中,冰浴下加入nan3(13g,200.0mmol),室温搅拌反应12小时,tlc检测反应,反应完毕后将反应液倒入10%氢氧化钠水溶液(500ml)中,二氯甲烷(200mlx2)萃取,合并有机层,干燥,柱层析分离得到类白色固体12.1g,收率70.3%。
[0061]
第二步:
[0062]
将化合物2b(12.0g,69.8mmol)溶于dmf(100ml),室温下加入氢化钠(3.3g,137.5mmol),搅拌反应1小时,然后加入化合物1c(21.4g,69.8mmol),于40℃搅拌反应4小时,tlc检测反应,反应完毕后用水(100ml)淬灭反应,用乙酸乙酯(100mlx2)提取,合并有机层,柱层析分离得到淡黄色固体17.2g,收率61.2%。
[0063]
第三步
[0064]
将化合物2c(17.0g,42.7mmol)、化合物1e(21.7g,85.4mmol)、pd(dppf)cl2(2.2g,
3.0mmol)、koac(4.9g,50.0mmol)溶于dmf(100ml)中,升温至100℃反应6小时,tlc检测反应,反应完毕后加入乙酸乙酯(200ml)和水(100ml)淬灭反应,有机层干燥,柱层析分离得到类白色固体13.1g,收率68.9%。
[0065]
第四步:
[0066]
将中间体2d(4.46g,10.0mmol)溶于甲醇(100ml)中,室温下加入硼氢化钠(1.1g,30.0mmol),搅拌反应6分钟,tlc检测反应,反应完毕后加水(50ml)淬灭反应,乙酸乙酯(50mlx2)提取,合并有机层,然后加入1m盐酸10ml搅拌2小时,有机层干燥,柱层析分离得到类白色固体1.9g,收率59.7z%,si( )m/z=319.1。
[0067]
实施例3
[0068]
2-((1-羟基-1,3-二氢苯并[c][1,2]氧杂硼杂烷-6-基)甲基)-1-氧代-1,2,3,4-四氢异喹啉-7-甲腈
[0069]
第一步:
[0070]
将化合物3a(15.7g,100.0mmol)和甲基磺酸(30ml)溶于二氯甲烷(200ml)中,冰浴下加入nan3(13.0g,200.0mmol),室温搅拌反应12小时,tlc检测反应,反应完毕后将反应液倒入10%氢氧化钠水溶液(500ml)中,二氯甲烷(200mlx2)萃取,合并有机层,干燥,柱层析分离得到类白色固体12.7g,收率73.8%。
[0071]
第二步至第四步参照实施例1,得到标题化合物(类白色固体1.5g,收率47.2%),esi( )m/z=319.1。
[0072]
实施例4
[0073]
2-((1-羟基-1,3-二氢苯并[c][1,2]氧杂硼杂烷-6-基)甲基)-1-氧代-1,2,3,4-四氢异喹啉-8-甲腈
四氢异喹啉-6-甲酰胺
[0084]
第一步:
[0085]
将化合物1d(8.0g,20.0mmol)、碳酸钾(5.52g,40.0mmol)溶于meoh(100ml)中,室温下加入双氧水(10ml),搅拌反应6小时,tlc检测反应,反应完毕后减压除去甲醇,然后用稀盐酸调ph至5-6,析出白色固体,过滤,干燥得到类白色固体5.3g,收率63.7%。化合物1d参照实施例1的第一步和第二步。
[0086]
第二步:
[0087]
将化合物6a(4.16g,10.0mmol)、化合物1e(5.1g,20.0mmol)、pd(dppf)cl2(730mg,1.0mmol)、koac(1.47g,15.0mmol)溶于dmf(20ml)中,升温至100℃反应6小时,tlc检测反应,反应完毕后加入乙酸乙酯(30ml)和水(10ml)淬灭反应,有机层干燥,柱层析分离得到类白色固体2.6g,收率56.3%。
[0088]
第三步:
[0089]
将中间体6b(2.32g,5.0mmol)溶于甲醇(50ml)中,室温下加入硼氢化钠(0.56g,15.0mmol),搅拌反应60分钟,tlc检测反应,反应完毕后加水(20ml)淬灭反应,乙酸乙酯(50mlx2)提取,合并有机层,然后加入1m盐酸10ml搅拌2小时,有机层干燥,柱层析分离得到类白色固体960mg,收率57.1%,esi( )m/z=337.1。
[0090]
实施例7
[0091]
2-((1-羟基-1,3-二氢苯并[c][1,2]氧杂硼杂烷-6-基)甲基)-1-氧代-1,2,3,4-四氢异喹啉-5-甲酰胺
[0092]
标题化合物参照实施例6,起始物料用2c替换1d。起始物料化合物2c参照实施例2的第一步和第二步。得到标题化合物(类白色固体1.1g,收率65.5%),esi( )m/z=337.1。2c化合物的结构式为:
[0093][0094]
实施例8
[0095]
2-((1-羟基-1,3-二氢苯并[c][1,2]氧杂硼杂烷-6-基)甲基)-1-氧代-1,2,3,4-四氢异喹啉-7-甲酰胺
[0096]
第一步:
[0097]
将化合物3c(8.0g,20.0mmol)、碳酸钾(5.52g,40.0mmol)溶于meoh(100ml)中,室温下加入双氧水(10ml),搅拌反应6小时,tlc检测反应,反应完毕后减压除去甲醇,然后用稀盐酸调ph至5-6,析出白色固体,过滤,干燥得到类白色固体5.5g,收率66.1%。化合物3c参照实施例3的第一步和第二步。
[0098]
第二步:
[0099]
将化合物8a(4.16g,10.0mmol)、化合物1e(5.1g,20.0mmol)、pd(dppf)cl2(730mg,1.0mmol)、koac(1.47g,15.0mmol)溶于dmf(20ml)中,升温至100℃反应6小时,tlc检测反应,反应完毕后加入乙酸乙酯(30ml)和水(10ml)淬灭反应,有机层干燥,柱层析分离得到类白色固体2.8g,收率60.3%。
[0100]
第三步:
[0101]
将中间体8b(2.32g,5.0mmol)溶于甲醇(50ml)中,室温下加入硼氢化钠(0.56g,15.0mmol),搅拌反应60分钟,tlc检测反应,反应完毕后加水(20ml)淬灭反应,乙酸乙酯(50mlx2)提取,合并有机层,然后加入1m盐酸10ml搅拌2小时,有机层干燥,柱层析分离得到类白色固体970mg,收率57.7%,esi( )m/z=337.1。
[0102]
实施例9
[0103]
2-((1-羟基-1,3-二氢苯并[c][1,2]氧杂硼杂烷-6-基)甲基)-1-氧代-1,2,3,4-四氢异喹啉-8-甲酰胺
[0104]
标题化合物参照实施例6,起始物料用4c替换1d。起始物料化合物4c参照实施例4的第一步和第二步。得到标题化合物(类白色固体1.0g,收率59.5%),esi( )m/z=337.1。4c化合物的结构式为:
[0105][0106]
实施例10
[0107]
7-((1-羟基-1,3-二氢苯并[c][1,2]氧杂硼杂烷-6-基)甲基)-8-氧代-5,6,7,8-四氢-2,7-萘啶-3-甲酰胺
(2.2g,3.0mmol)、koac(4.9g,50.0mmol)溶于dmf(100ml)中,升温至100℃反应6小时,tlc检测反应,反应完毕后加入乙酸乙酯(200ml)和水(100ml)淬灭反应,有机层干燥,浓缩后出层析分离得到类白色固体12.7g,收率80.5%。
[0120]
第四步:
[0121]
将中间体11d(4.22g,10.0mmol)溶于甲醇(100ml)中,室温下加入硼氢化钠(1.1g,30.0mmol),搅拌反应6分钟,tlc检测反应,反应完毕后加水(50ml)淬灭反应,乙酸乙酯(50mlx2)提取,合并有机层,然后加入1m盐酸10ml搅拌2小时,有机层干燥,柱层析分离得到类白色固体1.5g,收率51.0%,esi( )m/z=295.1。
[0122]
实施例12
[0123]
2-((1-羟基-1,3-二氢苯并[c][1,2]氧杂硼杂烷-6-基)甲基)-7-甲氧基-3,4-二氢-2,6-萘啶-1(2h)-酮
[0124][0125]
第一步:
[0126]
将化合物12a(16.3g,100.0mmol)和甲基磺酸(30ml)溶于二氯甲烷(200ml)中,冰浴下加入nan3(13g,200.0mmol),室温搅拌反应12小时,tlc检测反应,反应完毕后将反应液倒入10%氢氧化钠水溶液(500ml)中,二氯甲烷(200mlx2)萃取,合并有机层,干燥,柱层析分离得到类白色固体12.7g,收率71.3%。
[0127]
第二步至第四步参照实施例11,得到标题化合物(类白色固体1.8g,收率55.6%),esi( )m/z=325.1。
[0128]
实施例13
[0129]
2-((1-羟基-1,3-二氢苯并[c][1,2]氧杂硼杂烷-6-基)甲基)-6-甲氧基-3,4-二氢-2,7-萘啶-1(2h)-酮
[0130][0131]
第一步:
[0132]
将化合物13a(16.3g,100.0mmol)和甲基磺酸(30ml)溶于二氯甲烷(200ml)中,冰浴下加入nan3(13g,200.0mmol),室温搅拌反应12小时,tlc检测反应,反应完毕后将反应液倒入10%氢氧化钠水溶液(500ml)中,二氯甲烷(200mlx2)萃取,合并有机层,干燥,柱层析分离得到类白色固体11.1g,收率62.4%。
[0133]
第二步至第四步参照实施例11,得到标题化合物(类白色固体1.6g,收率49.4%),esi( )m/z=325.1。
[0134]
实施例14
[0135]
2-((1-羟基-1,3-二氢苯并[c][1,2]氧杂硼杂烷-6-基)甲基)-1-氧代-1,2,3,4-四氢异喹啉-6-磺酰胺
[0136]
第一步:
[0137]
将化合物14a(22.6g,100.0mmol)溶于dmf(100ml),室温下加入氢化钠(4.8g,200.0mmol),搅拌反应1小时,然后加入化合物1c(30.6g,100.0mmol),于40℃搅拌反应4小时,tlc检测反应,反应完毕后用水(100ml)淬灭反应,用乙酸乙酯(100mlx2)提取,合并有机层,干燥柱层析分离得到淡黄色固体12.5g,收率27.8%。
[0138]
第二步
[0139]
将化合物14b(12.0g,26.5mmol)、化合物1e(6.73g,26.5mmol)、pd(dppf)cl2(1.46g,2.0mmol)、koac(2.94g,30mmol)溶于dmf(100ml)中,升温至100℃反应8小时,tlc检测反应,反应完毕后加入乙酸乙酯(200ml)和水(100ml)淬灭反应,有机层干燥,浓缩后出层析分离得到类白色固体9.8g,收率74.2%。
[0140]
第三步:
[0141]
将中间体14c(5.0g,10.0mmol)溶于甲醇(100ml)中,室温下加入硼氢化钠(1.1g,30.0mmol),搅拌反应6小时,tlc检测反应,反应完毕后加水(50ml)淬灭反应,乙酸乙酯(50mlx2)提取,合并有机层,然后加入1m盐酸10ml搅拌2小时,有机层干燥,柱层析分离得到类白色固体1.4g,收率37.6%,esi( )m/z=373.1。
[0142]
实施例15
[0143]
n-(2-((1-羟基-1,3-二氢苯并[c][1,2]氧杂硼杂烷-6-基)甲基)-1-氧代-1,2,3,4-四氢异喹啉-6-基)乙酰胺
[0144][0145]
第一步:
[0146]
将化合物15a(20.4g,100.0mmol)溶于dmf(100ml),室温下加入氢化钠(4.8g,200.0mmol),搅拌反应1小时,然后加入化合物1c(30.5g,100.0mmol),于40℃搅拌反应4小时,tlc检测反应,反应完毕后用水(100ml)淬灭反应,用乙酸乙酯(100mlx2)提取,合并有机层,干燥柱层析分离得到淡黄色固体11.7g,收率27.2%。
[0147]
第二步
[0148]
将化合物15b(11.0g,25.6mmol)、化合物1e(6.5g,25.6mmol)、pd(dppf)cl2(1.46g,2.0mmol)、koac(2.94g,30mmol)溶于dmf(100ml)中,升温至100℃反应8小时,tlc检测反应,反应完毕后加入乙酸乙酯(200ml)和水(100ml)淬灭反应,有机层干燥,浓缩干燥后出层析分离得到类白色固体8.7g,收率71.1%。
[0149]
第三步:
[0150]
将中间体15c(4.78g,10.0mmol)溶于甲醇(100ml)中,室温下加入硼氢化钠(1.1g,30.0mmol),搅拌反应6小时,tlc检测反应,反应完毕后加水(50ml)淬灭反应,乙酸乙酯(50mlx2)提取,合并有机层,然后加入1m盐酸10ml搅拌2小时,有机层干燥,柱层析分离得到类白色固体1.9g,收率54.3%,esi( )m/z=351.1。
[0151]
实施例16
[0152]
n-(2-((1-羟基-1,3-二氢苯并[c][1,2]氧杂硼杂烷-6-基)甲基)-1-氧代-1,2,3,4-四氢异喹啉-5-基)乙酰胺
[0153][0154]
第一步:
[0155]
将化合物16a(20.4g,100.0mmol)溶于dmf(100ml),室温下加入氢化钠(4.8g,200.0mmol),搅拌反应1小时,然后加入化合物1c(30.5g,100.0mmol),于40℃搅拌反应4小时,tlc检测反应,反应完毕后用水(100ml)淬灭反应,用乙酸乙酯(100mlx2)提取,合并有机层,干燥,柱层析分离得到淡黄色固体13.2g,收率30.7%。
[0156]
第二步至第三步参照实施例15,得到标题化合物(类白色固体2.3g,收率65.7%),esi( )m/z=351.1。
[0157]
实施例17
[0158]
n-(2-((1-羟基-1,3-二氢苯并[c][1,2]氧杂硼杂烷-6-基)甲基)-1-氧代-1,2,3,4-四氢异喹啉-7-基)环丙基甲酰胺
[0159][0160]
第一步:
[0161]
将化合物17a(23.0g,100.0mmol)溶于dmf(100ml),室温下加入氢化钠(4.8g,200.0mmol),搅拌反应1小时,然后加入化合物1c(30.6g,100.0mmol),于40℃搅拌反应4小时,tlc检测反应,反应完毕后用水(100ml)淬灭反应,用乙酸乙酯(100mlx2)提取,合并有机层,干燥,柱层析分离得到淡黄色固体13.1g,收率28.7%。
[0162]
第二步至第三步参照实施例15,得到标题化合物(类白色固体2.1g,收率55.9%),esi( )m/z=377.1。
[0163]
实施例18
[0164]
n-(2-((1-羟基-1,3-二氢苯并[c][1,2]氧杂硼杂烷-6-基)甲基)-1-氧代-1,2,3,4-四氢异喹啉-8-基)乙酰胺
[0165][0166]
第一步:
[0167]
将化合物18a(20.4g,100.0mmol)溶于dmf(100ml),室温下加入氢化钠(4.8g,200.0mmol),搅拌反应1小时,然后加入化合物1c(30.5g,100.0mmol),于40℃搅拌反应4小时,tlc检测反应,反应完毕后用水(100ml)淬灭反应,用乙酸乙酯(100mlx2)提取,合并有机层,干燥,柱层析分离得到淡黄色固体12.1g,收率28.1%。
[0168]
第二步至第三步参照实施例15,得到标题化合物(类白色固体1.6g,收率45.7%),esi( )m/z=351.1。
[0169]
实施例19
[0170]
n-(2-((1-羟基-1,3-二氢苯并[c][1,2]氧杂硼杂烷-6-基)甲基)-1-氧代-1,2,3,4-四氢异喹啉-6-基)甲基磺酰胺
[0171][0172]
第一步:
[0173]
将化合物19a(24.0g,100.0mmol)溶于dmf(100ml),室温下加入氢化钠(4.8g,200.0mmol),搅拌反应1小时,然后加入化合物1c(30.6g,100.0mmol),于40℃搅拌反应4小时,tlc检测反应,反应完毕后用水(100ml)淬灭反应,用乙酸乙酯(100mlx2)提取,合并有机层,干燥,柱层析分离得到淡黄色固体13.1g,收率28.1%。
[0174]
第二步至第三步参照实施例14,得到标题化合物(2.0g,收率51.8%),esi( )m/z=387.1。
[0175]
实施例20
[0176]
6-氟-2-((1-羟基-1,3-二氢苯并[c][1,2]氧杂硼杂烷-6-基)甲基)-3,4-二氢异喹啉-1(2h)-酮
[0177]
第一步:
[0178]
将化合物20a(16.5g,100.0mmol)溶于dmf(100ml),室温下加入氢化钠(4.8g,200.0mmol),搅拌反应1小时,然后加入化合物1c(30.6g,100.0mmol),于40℃搅拌反应4小时,tlc检测反应,反应完毕后用水(100ml)淬灭反应,用乙酸乙酯(100mlx2)提取,合并有机层,干燥,柱层析分离得到淡黄色固体20.2g,收率51.7%。
[0179]
第二步至第三步参照实施例15,得到标题化合物(类白色固体1.7g,收率54.7%),esi( )m/z=312.1。
[0180]
实施例21
[0181]
2-((1-羟基-1,3-二氢苯并[c][1,2]氧杂硼杂烷-6-基)甲基)-6-(2-羟基丙烷-2-基)-3,4-二氢异喹啉-1(2h)-酮,
[0182][0183]
第一步:
[0184]
将化合物21a(20.5g,100.0mmol)溶于dmf(100ml),室温下加入氢化钠(4.8g,200.0mmol),搅拌反应1小时,然后加入化合物1c(30.6g,100.0mmol),于40℃搅拌反应4小时,tlc检测反应,反应完毕后用水(100ml)淬灭反应,用乙酸乙酯(100mlx2)提取,合并有机层,干燥,柱层析分离得到淡黄色固体13.5g,收率31.3%。
[0185]
第二步至第三步参照实施例15,得到标题化合物(类白色固体2.1g,收率59.8%),esi( )m/z=352.1。
[0186]
实施例22
[0187]
6-((1-羟基-1,3-二氢苯并[c][1,2]氧杂硼杂烷-6-基)甲基)-5-氧代-5,6,7,8-四氢萘-2-甲腈
[0188]
第一步:
[0189]
将化合物22a(17.1g,100.0mmol)溶于thf(100ml),冰浴下加入lda(200.0mmol),保温搅拌反应1小时,然后加入化合物1c(30.6g,100.0mmol),于40℃搅拌反应4小时,tlc检测反应,反应完毕后用水(100ml)淬灭反应,用乙酸乙酯(100mlx2)提取,合并有机层,干燥,柱层析分离得到淡黄色固体19.6g,收率49.4%。
[0190]
第二步
[0191]
将化合物22b(19.0g,47.9mmol)、化合物1e(12.16g,47.9mmol)、pd(dppf)cl2(1.46g,2.0mmol)、koac(4.7g,47.9mmol)溶于dmf(100ml)中,升温至100℃反应8小时,tlc检测反应,反应完毕后加入乙酸乙酯(200ml)和水(100ml)淬灭反应,有机层干燥,浓缩干燥后出层析分离得到类白色固体15.3g,收率71.8`%。
[0192]
第三步:
[0193]
将中间体22c(4.45g,10.0mmol)溶于甲醇(100ml)中,室温下加入硼氢化钠(1.1g,30.0mmol),搅拌反应6小时,tlc检测反应,反应完毕后加水(50ml)淬灭反应,乙酸乙酯(50mlx2)提取,合并有机层,然后加入1m盐酸10ml搅拌2小时,有机层干燥,柱层析分离得到类白色固体1.6g,收率50.5%,esi( )m/z=318.1。
[0194]
实施例23
[0195]
6-((1-羟基-1,3-二氢苯并[c][1,2]氧杂硼杂烷-6-基)甲基)-5-氧代-5,6,7,8-四氢萘-1-甲腈
[0196]
第一步:
[0197]
将化合物23a(17.1g,100.0mmol)溶于thf(100ml),冰浴下加入lda(200.0mmol),保温搅拌反应1小时,然后加入化合物1c(30.6g,100.0mmol),于40℃搅拌反应4小时,tlc检测反应,反应完毕后用水(100ml)淬灭反应,用乙酸乙酯(100mlx2)提取,合并有机层,干燥,
柱层析分离得到淡黄色固体18.4g,收率46.3%。
[0198]
第二步至第三步参照实施例22,得到标题化合物(类白色固体1.7g,收率53.8%),esi( )m/z=318.1。
[0199]
实施例24
[0200]
7-((1-羟基-1,3-二氢苯并[c][1,2]氧杂硼杂烷-6-基)甲基)-8-氧代-5,6,7,8-四氢萘-2-甲腈
[0201]
第一步:
[0202]
将化合物24a(17.1g,100.0mmol)溶于thf(100ml),冰浴下加入lda(200.0mmol),保温搅拌反应1小时,然后加入化合物1c(30.6g,100.0mmol),于40℃搅拌反应4小时,tlc检测反应,反应完毕后用水(100ml)淬灭反应,用乙酸乙酯(100mlx2)提取,合并有机层,干燥,柱层析分离得到淡黄色固体18.4g,收率46.3%。
[0203]
第二步至第三步参照实施例22,得到标题化合物(类白色固体1.4g,收率44.2%),esi( )m/z=318.1。
[0204]
实施例25
[0205]
7-((1-羟基-1,3-二氢苯并[c][1,2]氧杂硼杂烷-6-基)甲基)-8-氧代-5,6,7,8-四氢萘-1-甲腈
[0206]
第一步:
[0207]
将化合物25a(17.1g,100.0mmol)溶于thf(100ml),冰浴下加入lda(200.0mmol),保温搅拌反应1小时,然后加入化合物1c(30.6g,100.0mmol),于40℃搅拌反应4小时,tlc检测反应,反应完毕后用水(100ml)淬灭反应,用乙酸乙酯(100mlx2)提取,合并有机层,浓缩,柱层析分离得到淡黄色固体15.6g,收率39.3%。
[0208]
第二步至第三步参照实施例22,标题化合物(得到类白色固体1.7g,收率53.6%),
四氢萘-1-甲酰胺
[0222]
第一步:
[0223]
将化合物23b(7.94g,20.0mmol)、碳酸钾(5.52g,40.0mmol)溶于meoh(100ml)中,室温下加入双氧水(10ml),搅拌反应6小时,tlc检测反应,反应完毕后减压除去甲醇,然后用稀盐酸调ph至5-6,析出白色固体,过滤,干燥得到类白色固体4.6g,收率55.4%。
[0224]
第二步至第三步参照实施例27,得到标题化合物(类白色固体0.85g,收率50.7%),esi( )m/z=336.1。
[0225]
实施例29
[0226]
7-((1-羟基-1,3-二氢苯并[c][1,2]氧杂硼杂烷-6-基)甲基)-8-氧代-5,6,7,8-四氢萘-2-甲酰胺
[0227]
第一步:
[0228]
将化合物24b(7.94g,20.0mmol)、碳酸钾(5.52g,40.0mmol)溶于meoh(100ml)中,室温下加入双氧水(10ml),搅拌反应6小时,tlc检测反应,反应完毕后减压除去甲醇,然后用稀盐酸调ph至5-6,析出白色固体,过滤,干燥得到类白色固体5.5g,收率66.3%。
[0229]
第二步至第三步参照实施例27,得到标题化合物(类白色固体0.91g,收率54.3%),esi( )m/z=336.1。
[0230]
实施例30
[0231]
7-((1-羟基-1,3-二氢苯并[c][1,2]氧杂硼杂烷-6-基)甲基)-8-氧代-5,6,7,8-四氢萘-1-甲酰胺
[0232]
第一步:
[0233]
将化合物25b(7.94g,20.0mmol)、碳酸钾(5.52g,40.0mmol)溶于meoh(100ml)中,室温下加入双氧水(10ml),搅拌反应6小时,tlc检测反应,反应完毕后减压除去甲醇,然后用稀盐酸调ph至5-6,析出白色固体,过滤,干燥得到类白色固体4.9g,收率59.0%。
[0234]
第二步至第三步参照实施例27,得到标题化合物(类白色固体1.10g,收率65.7%),esi( )m/z=336.1。
[0235]
实施例31
[0236]
7-((1-羟基-1,3-二氢苯并[c][1,2]氧杂硼杂烷-6-基)甲基)-8-氧代-5,6,7,8-四氢异喹啉-3-甲酰胺
[0237]
第一步:
[0238]
将化合物26b(7.96g,20.0mmol)、碳酸钾(5.52g,40.0mmol)溶于meoh(100ml)中,室温下加入双氧水(10ml),搅拌反应6小时,tlc检测反应,反应完毕后减压除去甲醇,然后用稀盐酸调ph至5-6,析出白色固体,过滤,干燥得到类白色固体4.1g,收率49.4%。
[0239]
第二步至第三步参照实施例27,得到标题化合物(类白色固体0.92g,收率54.8%),esi( )m/z=337.1。
[0240]
实施例32pde4体外活性测试
[0241]
实验方法:
[0242]
将化合物稀释成不同浓度,备用;
[0243]
将缓冲液(50mm tris-hcl,ph 8.010mm mgcl2和1mm dtt)加入384孔板中,将配置好的化合物、pde4b或4d及3h-camp加至缓冲液中孵育30分钟,然后加入0.2m znso4终止反应,用0.2n氢氧化钡沉淀出3h-camp的反应产物,未反应的3h-camp留在上清液,用ls1801液闪仪测定放射性,通过h-camp水解率来计算化合物的抑制活性。
[0244]
化合物的ic50结果见下表。
[0245][0246]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。