瑞德西韦中间体(s,s)-氨基磷酸酯的不对称催化合成方法
技术领域
1.本发明涉及不对称催化合成技术领域,具体地,涉及一种瑞德西韦中间体(s,s)
-ꢀ
氨基磷酸酯的不对称催化合成方法,尤其是一种采用手性咪唑类不对称催化剂催化制备瑞德西韦(remdesivir)重要中间体(s,s)-氨基磷酸酯的方法。
背景技术:
2.(s,s)-氨基磷酸酯,分子式c
21
h
27
n2o7p,是制备治疗新冠肺炎病毒(covid-19) 药物瑞德西韦(remdesivir)的重要中间体。目前新冠病毒施虐全球,除一些有效的中药治疗方法,在研的抗病毒药物瑞德西韦对该新冠病毒也有一定治疗效果。因此,瑞德西韦及其重要中间体的高效合成受到科研人员和各公司的重视。通过二氯化磷酸苯酯与(s)-氨基丙酸-(2)-乙基丁酯不对称合成(s,s)-氨基磷酸酯则具有很大的社会价值、经济价值和研究价值。
3.经过对现有技术的检索,目前有关(s,s)-氨基磷酸酯合成的文献报道很少,所有目前报道的方法(us2013/143835;wo2013/84165;us2015/133395;wo2016/69826; journal of medicinal chemistry,2017,60,1648;wo2017/184668;us2017/71964; wo2018/204198;wo2020/2469;)都是生成的是消旋的产物,就是通过手性原料(s)
-ꢀ
氨基丙酸-(2)-乙基丁酯与二氯化磷酸苯酯反应后,进行拆分。现在为止,还没有不对称的方法来合成(s,s)-氨基磷酸酯的报道。
技术实现要素:
4.针对现有技术中的缺陷,本发明的目的是提供一种瑞德西韦中间体(s,s)-氨基磷酸酯的不对称催化合成方法。
5.本发明的目的是通过以下方案实现的:
6.本发明提供一种瑞德西韦中间体(s,s)-氨基磷酸酯的不对称催化合成方法,具体为,在惰性气体下,以(s)-氨基丙酸-(2)-乙基丁酯、二氯化磷酸苯酯和4-硝基苯酚为原料,碱和/或式ⅰ所示的手性咪唑类化合物为催化剂在溶剂中反应,得到式ⅱ所示的(s,s)-氨基磷酸酯所述(s)-氨基丙酸-(2)-乙基丁酯,纯度为99wt%,对映体的比例(s/r)是95:5~99:1。分别单独在手性咪唑类化合物催化剂的催化下或在一定碱的作用下都能优势选择性地合成(s,s)-氨基磷酸酯,得到 dr值在1:1~2.5:1,而同时在手性咪唑类化合物催化剂的催化下和碱的作用下,会有更好的dr值,最高达到
5.5:1。
7.本发明中,通过手性咪唑类催化剂催化(s)-氨基丙酸-(2)-乙基丁酯、二氯化磷酸苯酯和4-硝基苯酚合成式ⅱ所示的(s,s)-氨基磷酸酯的反应方法,反应式如下:
[0008][0009]
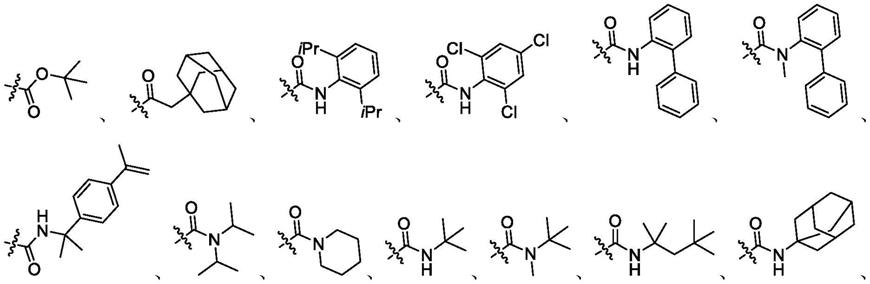
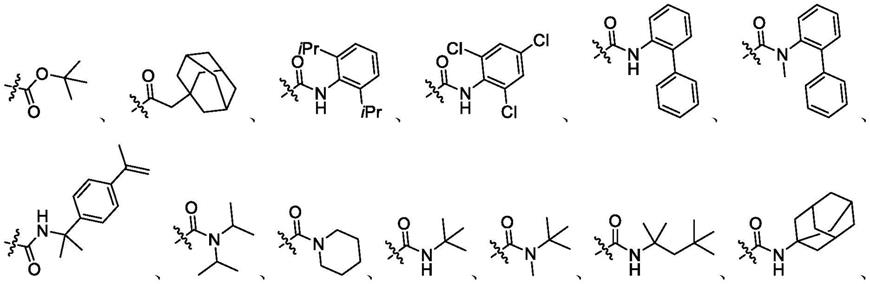
进一步地,所述手性咪唑类化合物催化剂r选自以下取代基:r选自以下取代基:r选自以下取代基:和的一种。
[0010]
20种(c1~c20)手性咪唑类化合物催化剂的结构式如下所示:
[0011][0012]
所述手性咪唑催化剂优选为c3~c5,c10~c15,c17~c18,更优选为c3,c12, c13,c14,c15,c17,进一步优选为c3,c14,c15。
[0013]
进一步地,所述碱为有机碱或者无机碱。
[0014]
进一步地,所述有机碱为具有取代基的伯、仲、叔、季胺以及其二胺,所述取代基为包含1至10个碳的脂肪取代基或者芳香烃取代基;所述无机碱为碳酸钠、碳酸钾、碳酸铯、磷酸氢二钠、碳酸氢钠和碳酸氢钾中的至少一种。
[0015]
进一步地,所述有机碱为三乙胺、乙二胺、四甲基乙二胺、三甲基乙二胺、n,n'
-ꢀ
二甲基乙二胺、n,n-二甲基乙二胺、n-甲基乙二胺、n,n'-二甲基-1,3-丙二胺和n,n'
-ꢀ
二甲基-1,4-丁二胺等中的至少一种。
[0016]
进一步地,所述溶剂为极性溶剂、非极性溶剂和离子液体溶剂中的至少一种。
[0017]
进一步地,所述非极性溶剂为甲苯、乙醚或四氢呋喃;所述极性溶剂为二氯甲烷、1,2-二氯乙烷、dmf、dmso或乙腈。所述离子液体溶剂为咪唑鎓盐离子液体、铵盐离子液体、哌啶鎓盐离子液体、硫鎓盐离子液体、吗啉盐离子液体、季磷盐离子液体、吡咯烷鎓盐离子液体或吡啶鎓盐离子液体。溶剂优选为甲苯、二氯甲烷、 1,2-二氯乙烷、乙醚、四氢呋喃、甲醇、乙醇、异丙醇和三氟乙醇中的一种或两种或多种,更优选为甲苯、二氯甲烷、四氢呋喃、甲醇、乙醇和三氟乙醇,进一步优选为甲苯、二氯甲烷和乙醇。
[0018]
进一步地,所述手性咪唑类化合物催化剂与(s)-氨基丙酸-(2)-乙基丁酯的摩尔比为1:5~1000,优选为1:50~1000,更优选为1:50~400,特别优选为1:50~200;(s)-氨基丙酸-(2)-乙基丁酯、二氯化磷酸苯酯和4-硝基苯酚的摩尔比为1:(1-1.2): (1-1.2)。
[0019]
进一步地,不对称催化反应是将(s)-氨基丙酸-(2)-乙基丁酯溶解于溶剂中进行的,所以在反应前,先将所述(s)-氨基丙酸-(2)-乙基丁酯溶解在所述溶剂中配成溶液, (s)-氨基丙酸-(2)-乙基丁酯溶液的浓度为5%~80%,优选10%~60%。
[0020]
进一步地,不对称催化合成方法的反应温度为-100℃~180℃,优选为-80~ 80℃,更优选的反应温度为-80~30℃;反应时间为1~72小时,优选为5~60小时,进一步优选为5~36小时,更进一步优选为6~24小时,特别优选为10~24 小时。
[0021]
与现有技术相比,本发明具有如下的有益效果:
[0022]
1、本发明运用手性催化剂,在工业上较成熟的不对称技术,成功地高收率、高对映选择性地获得(s,s)-氨基磷酸酯;
[0023]
2、本发明采用设计合成的新型手性咪唑类化合物催化剂,合成路线简单,原料易得,降低了催化剂成本;
[0024]
3、本发明采用手性咪唑类化合物催化剂进行催化合成(s,s)-氨基磷酸酯,直接得到优势富集的手性(s,s)-氨基磷酸酯,使产物的收率,从现有的26%提高到63%; dr值从1:1提高到5.5:1;
[0025]
4、本发明合成方法原料易得,条件温和,操作简便,成本较低;产物易于分离,收率高,化学纯度和光学纯度高,易于工业化生产,具有好的工业应用前景。
具体实施方式
[0026]
下面结合具体实施例对本发明进行详细说明。以下实施例将有助于本领域的技术人员进一步理解本发明,但不以任何形式限制本发明。应当指出的是,对本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变化和改进。这些都属于本发明的保护范围。
equiv),分子筛(3.0g),手性咪唑类催化剂(c2:0.001mol,0.1equiv),体系通过真空线,用氮气置换3次,加入新蒸的脱气dcm(二氯甲烷,15ml),溶液在-78℃下,注入二氯化磷酸苯酯(0.011mol,1.1equiv),再滴加碱(三乙胺,0.011mol, 1.1equiv),搅拌反应3小时,吹拂氮气下,加入4-硝基苯酚(1.5g,0.011mol,1.1equiv),
ꢀ-
78℃搅拌反应3小时。tlc检测反应结束,滴水进行淬灭,过滤,萃取,浓缩,柱层析得到产物氨基磷酸酯。非对映体过量值(dr)为2.1/1。通过异丙醇重结晶,得到手性 (s,s)-氨基磷酸酯(ⅱ,1.9g,42%)1h nmr(400mhz,cdcl3):δ8.23(d,j=9.2hz,2h), 7.36(m,4h),7.24-7.22(m,3h),4.16-4.13(m,1h),4.10-4.08(m,2h),4.00-3.91(m,1h), 1.55-1.47(m,1h);1.45-1.40(m,3h),1.38-1.28(m,4h),0.87(t,j=7.6hz,6h);
31
p nmr (162mhz,cdcl3):δ-3.16。
[0037]
实施例3
[0038]
手性(s,s)-氨基磷酸酯的制备
[0039]
在一个50ml双口反应瓶中,加入(s)-氨基丙酸-(2)-乙基丁酯(2.1g,0.01mol,1 equiv),分子筛(3.0g),手性咪唑类催化剂(c3:0.001mol,0.1equiv),体系通过真空线,用氮气置换3次,加入新蒸的脱气dcm(二氯甲烷,15ml),溶液在
ꢀ-
78℃下,注入二氯化磷酸苯酯(0.011mol,1.1equiv),再滴加碱(三乙胺,0.011mol, 1.1equiv),搅拌反应3小时,吹拂氮气下,加入4-硝基苯酚(1.5g,0.011mol,1.1equiv),
ꢀ-
78℃搅拌反应3小时。tlc检测反应结束,滴水进行淬灭,萃取,浓缩,柱层析得到产物氨基磷酸酯。非对映体过量值(dr)为4.8/1。通过异丙醇重结晶,得到手性(s,s)
-ꢀ
氨基磷酸酯(ⅱ,2.6g,56%)1h nmr(400mhz,cdcl3):δ8.23(d,j=9.2hz,2h),7.36(m, 4h),7.24-7.22(m,3h),4.16-4.13(m,1h),4.10-4.08(m,2h),4.00-3.91(m,1h),1.55-1.47 (m,1h);1.45-1.40(m,3h),1.38-1.28(m,4h),0.87(t,j=7.6hz,6h);
31
p nmr(162mhz, cdcl3):δ-3.16。
[0040]
实施例4
[0041]
手性(s,s)-氨基磷酸酯的制备
[0042]
在一个50ml双口反应瓶中,加入(s)-氨基丙酸-(2)-乙基丁酯(2.1g,0.01mol,1 equiv),分子筛(3.0g),手性咪唑类催化剂(c4:0.001mol,0.1 equiv),体系通过真空线,用氮气置换3次,加入新蒸的脱气dcm(二氯甲烷,15ml),溶液在-78℃下,注入二氯化磷酸苯酯(0.011mol,1.1equiv),再滴加碱(三乙胺,0.011 mol,1.1equiv),搅拌反应3小时,吹拂氮气下,加入4-硝基苯酚(1.5g,0.011mol,1.1 equiv),-78℃搅拌反应3小时。tlc检测反应结束,滴水进行淬灭,萃取,浓缩,柱层析得到产物氨基磷酸酯。非对映体过量值(dr)为3.1/1。通过异丙醇重结晶,得到手性(s,s)-氨基磷酸酯(ⅱ,
equiv),分子筛(3.0g),手性咪唑类催化剂(c7:0.001mol,0.1 equiv),体系通过真空线,用氮气置换3次,加入新蒸的脱气dcm(二氯甲烷,15ml),溶液在-78℃下,注入二氯化磷酸苯酯(0.011mol,1.1equiv),再滴加碱(三乙胺,0.01 1mol,1.1equiv),搅拌反应3小时,吹拂氮气下,加入4-硝基苯酚(1.5g,0.011mol,1.1 equiv),-78℃搅拌反应3小时。tlc检测反应结束,滴水进行淬灭,萃取,浓缩,柱层析得到产物氨基磷酸酯。非对映体过量值(dr)为2.7/1。通过异丙醇重结晶,得到手性(s,s)-氨基磷酸酯(ⅱ,2.3g,51%)1h nmr(400mhz,cdcl3):δ8.23(d,j=9.2hz,2h), 7.36(m,4h),7.24-7.22(m,3h),4.16-4.13(m,1h),4.10-4.08(m,2h),4.00-3.91(m,1h), 1.55-1.47(m,1h);1.45-1.40(m,3h),1.38-1.28(m,4h),0.87(t,j=7.6hz,6h);
31
p nmr (162mhz,cdcl3):δ-3.16。
[0052]
实施例8
[0053]
手性(s,s)-氨基磷酸酯的制备
[0054]
在一个50ml双口反应瓶中,加入(s)-氨基丙酸-(2)-乙基丁酯(2.1g,0.01mol,1 equiv),分子筛(3.0g),手性咪唑类催化剂(c8:0.001mol,0.1 equiv),体系通过真空线,用氮气置换3次,加入新蒸的脱气dcm(二氯甲烷,15ml),溶液在-78℃下,注入二氯化磷酸苯酯(0.011mol,1.1equiv),再滴加碱(三乙胺,0.011 mol,1.1equiv),搅拌反应3小时,吹拂氮气下,加入4-硝基苯酚(1.5g,0.011mol,1.1 equiv),-78℃搅拌反应3小时。tlc检测反应结束,滴水进行淬灭,萃取,浓缩,柱层析得到产物氨基磷酸酯。非对映体过量值(dr)为2.4/1。通过异丙醇重结晶,得到手性(s,s)-氨基磷酸酯(ⅱ,2.2g,48%)1h nmr(400mhz,cdcl3):δ8.23(d,j=9.2hz,2h), 7.36(m,4h),7.24-7.22(m,3h),4.16-4.13(m,1h),4.10-4.08(m,2h),4.00-3.91(m,1h), 1.55-1.47(m,1h);1.45-1.40(m,3h),1.38-1.28(m,4h),0.87(t,j=7.6hz,6h);
31
p nmr (162mhz,cdcl3):δ-3.16。
[0055]
实施例9
[0056]
手性(s,s)-氨基磷酸酯的制备
[0057]
在一个50ml双口反应瓶中,加入(s)-氨基丙酸-(2)-乙基丁酯(2.1g,0.01mol,1 equiv),分子筛(3.0g),手性咪唑类催化剂(c9:0.001mol, 0.1equiv),体系通过真空线,用氮气置换3次,加入新蒸的脱气dcm(二氯甲烷,15ml),溶液在-78℃下,注入二氯化磷酸苯酯(0.011mol,1.1equiv),再滴加碱(三乙胺,0.01 1mol,1.1equiv),搅拌反应3小时,吹拂氮气下,加入4-硝基苯酚(1.5g,0.011mol,1.1 equiv),-78℃搅拌反应3小时。tlc检测反应结束,滴水进行淬灭,萃取,浓缩,柱层析得到产物氨基磷
equiv),分子筛(3.0g),手性咪唑类催化剂(c15:0.001mol, 0.1equiv),体系通过真空线,用氮气置换3次,加入新蒸的脱气dcm(二氯甲烷,15ml),溶液在-78℃下,注入二氯化磷酸苯酯(0.011mol,1.1equiv),再滴加碱(三乙胺,0.011 mol,1.1equiv),搅拌反应3小时,吹拂氮气下,加入4-硝基苯酚(1.5g,0.011mol,1.1 equiv),-78℃搅拌反应3小时。tlc检测反应结束,滴水进行淬灭,萃取,浓缩,柱层析得到产物氨基磷酸酯。非对映体过量值(dr)为5.5/1。通过异丙醇重结晶,得到手性(s,s)-氨基磷酸酯(ⅱ,2.7g,61%)1h nmr(400mhz,cdcl3):δ8.23(d,j=9.2hz,2h), 7.36(m,4h),7.24-7.22(m,3h),4.16-4.13(m,1h),4.10-4.08(m,2h),4.00-3.91(m,1h), 1.55-1.47(m,1h);1.45-1.40(m,3h),1.38-1.28(m,4h),0.87(t,j=7.6hz,6h);
31
p nmr (162mhz,cdcl3):δ-3.16。
[0076]
实施例16
[0077]
手性(s,s)-氨基磷酸酯的制备
[0078]
在一个50ml双口反应瓶中,加入(s)-氨基丙酸-(2)-乙基丁酯(2.1g,0.01mol,1 equiv),分子筛(3.0g),手性咪唑类催化剂(c16:0.001 mol,0.1equiv),体系通过真空线,用氮气置换3次,加入新蒸的脱气dcm(二氯甲烷, 15ml),溶液在-78℃下,注入二氯化磷酸苯酯(0.011mol,1.1equiv),再滴加碱(三乙胺,0.011mol,1.1equiv),搅拌反应3小时,吹拂氮气下,加入4-硝基苯酚(1.5g,0.011 mol,1.1equiv),-78℃搅拌反应3小时。tlc检测反应结束,滴水进行淬灭,萃取,浓缩,柱层析得到产物氨基磷酸酯。非对映体过量值(dr)为2.7/1。通过异丙醇重结晶,得到手性(s,s)-氨基磷酸酯(ⅱ,2.3g,52%)1h nmr(400mhz,cdcl3):δ8.23(d,j=9.2 hz,2h),7.36(m,4h),7.24-7.22(m,3h),4.16-4.13(m,1h),4.10-4.08(m,2h),4.00-3.91(m, 1h),1.55-1.47(m,1h);1.45-1.40(m,3h),1.38-1.28(m,4h),0.87(t,j=7.6hz,6h);
31
pnmr(162mhz,cdcl3):δ-3.16。
[0079]
实施例17
[0080]
手性(s,s)-氨基磷酸酯的制备
[0081]
在一个50ml双口反应瓶中,加入(s)-氨基丙酸-(2)-乙基丁酯(2.1g,0.01mol,1 equiv),分子筛(3.0g),手性咪唑类催化剂(c17:0.001mol,.1 equiv),体系通过真空线,用氮气置换3次,加入新蒸的脱气dcm(二氯甲烷,15ml),溶液在-78℃下,注入二氯化磷酸苯酯(0.011mol,1.1equiv),再滴加碱(三乙胺,0.011 mol,1.1equiv),搅拌反应3小时,吹拂氮气下,加入4-硝基苯酚(1.5g,0.011mol,1.1 equiv),-78℃搅拌反应3小时。tlc检测反应结束,滴水进行淬灭,萃取,浓缩,柱层析得到产物氨基磷酸酯。非对映体过量值(dr)为4.6/1。通过异丙醇重结晶,得到手性(s,s)-氨基磷酸酯(ⅱ,2.1g,46%)1h nmr(400mhz,cdcl3):δ8.23(d,j=9.2hz,2h), 7.36(m,4h),7.24-7.22(m,
0.1equiv),体系通过真空线,用氮气置换3次,加入新蒸的脱气dcm(二氯甲烷,15ml),溶液在-78℃下,注入二氯化磷酸苯酯(0.011mol,1.1equiv),搅拌反应3小时,吹拂氮气下,加入4-硝基苯酚(1.5g,0.011mol,1.1equiv),-78℃搅拌反应3小时。tlc 检测反应结束,滴水进行淬灭,萃取,浓缩,柱层析得到产物氨基磷酸酯。非对映体过量值(dr)为2.8/1。通过异丙醇重结晶,得到手性(s,s)-氨基磷酸酯(ⅱ,1.6g,36%) 1
h nmr(400mhz,cdcl3):δ8.23(d,j=9.2hz,2h),7.36(m,4h),7.24-7.22(m,3h), 4.16-4.13(m,1h),4.10-4.08(m,2h),4.00-3.91(m,1h),1.55-1.47(m,1h);1.45-1.40(m, 3h),1.38-1.28(m,4h),0.87(t,j=7.6hz,6h);
31
p nmr(162mhz,cdcl3):δ-3.16。
[0100]
实施例24
[0101]
手性(s,s)-氨基磷酸酯的制备
[0102]
在一个50ml双口反应瓶中,加入(s)-氨基丙酸-(2)-乙基丁酯(2.1g,0.01mol,1 equiv),分子筛(3.0g),手性咪唑类催化剂(c15:0.001mol, 0.1equiv),体系通过真空线,用氮气置换3次,加入新蒸的脱气dcm(二氯甲烷,15ml),溶液在-78℃下,注入二氯化磷酸苯酯(0.011mol,1.1equiv),再滴加碱(n,n-二甲基乙二胺,0.0055mol,0.55equiv),搅拌反应3小时,吹拂氮气下,加入4-硝基苯酚(1.5 g,0.011mol,1.1equiv),-78℃搅拌反应3小时。tlc检测反应结束,滴水进行淬灭,萃取,浓缩,柱层析得到产物氨基磷酸酯。非对映体过量值(dr)为5.5/1。通过异丙醇重结晶,得到手性(s,s)-氨基磷酸酯(ⅱ,2.8g,63%)1h nmr(400mhz,cdcl3):δ8.23 (d,j=9.2hz,2h),7.36(m,4h),7.24-7.22(m,3h),4.16-4.13(m,1h),4.10-4.08(m,2h), 4.00-3.91(m,1h),1.55-1.47(m,1h);1.45-1.40(m,3h),1.38-1.28(m,4h),0.87(t,j=7.6 hz,6h);
31
p nmr(162mhz,cdcl3):δ-3.16。
[0103]
实施例25
[0104]
手性(s,s)-氨基磷酸酯的制备
[0105]
在一个50ml双口反应瓶中,加入(s)-氨基丙酸-(2)-乙基丁酯(2.1g,0.01mol,1 equiv),分子筛(3.0g),手性咪唑类催化剂(c15:0.001mol, 0.1equiv),体系通过真空线,用氮气置换3次,加入新蒸的脱气dcm(二氯甲烷,15ml),溶液在-78℃下,注入二氯化磷酸苯酯(0.011mol,1.1equiv),再滴加碱(乙二胺,0.011 mol,1.1equiv),搅拌反应5小时,吹拂氮气下,加入4-硝基苯酚(1.5g,0.011mol,1.1 equiv),-78℃搅拌反应2小时。tlc检测反应结束,滴水进行淬灭,萃取,浓缩,柱层析得到产物氨基磷酸酯。非对映体过量值(dr)为3.0/1。通过异丙醇重结晶,得到手性(s,s)-氨基磷酸酯(ⅱ,2.4g,53%)1h nmr(400mhz,cdcl3):δ8.23(d,j=9.2hz,2h), 7.36(m,4h),7.24-7.22(m,3h),4.16-4.13(m,1h),4.10-4.08(m,2h),4.00-3.91(m,1h), 1.55-1.47(m,1h);1.45-1.40(m,3h),1.38-1.28(m,4h),0.87(t,j=7.6hz,6h);
31
p nmr (162mhz,cdcl3):δ-3.16。
[0106]
实施例26
[0107]
手性(s,s)-氨基磷酸酯的制备
[0108]
在一个50ml双口反应瓶中,加入(s)-氨基丙酸-(2)-乙基丁酯(2.1g,0.01mol,1 equiv),分子筛(3.0g),手性咪唑类催化剂(c15:0.001mol, 0.1equiv),体系通过真空线,用氮气置换3次,加入新蒸的脱气dcm(二氯甲烷,15ml),溶液在-78℃下,注入二氯化磷酸苯酯(0.011mol,1.1equiv),再滴加碱(乙二胺,0.0055 mol,0.55equiv),搅拌反应5小时,吹拂氮气下,加入4-硝基苯酚(1.5g,0.011mol,1.1 equiv),-78℃搅拌反应2小时。tlc检测反应结束,滴水进行淬灭,萃取,浓缩,柱层析得到产物氨基磷酸酯。非对映体过量值(dr)为3.2/1。通过异丙醇重结晶,得到手性(s,s)-氨基磷酸酯(ⅱ,2.5g,56%)1h nmr(400mhz,cdcl3):δ8.23(d,j=9.2hz,2h), 7.36(m,4h),7.24-7.22(m,3h),4.16-4.13(m,1h),4.10-4.08(m,2h),4.00-3.91(m,1h), 1.55-1.47(m,1h);1.45-1.40(m,3h),1.38-1.28(m,4h),0.87(t,j=7.6hz,6h);
31
p nmr (162mhz,cdcl3):δ-3.16。
[0109]
实施例27
[0110]
手性(s,s)-氨基磷酸酯的制备
[0111]
在一个50ml双口反应瓶中,加入(s)-氨基丙酸-(2)-乙基丁酯(2.1g,0.01mol,1 equiv),分子筛(3.0g),手性咪唑类催化剂(c15:0.0005mol, 0.05equiv),体系通过真空线,用氮气置换3次,加入新蒸的脱气dcm(二氯甲烷,15 ml),溶液在-78℃下,注入二氯化磷酸苯酯(0.011mol,1.1equiv),再滴加碱(乙二胺, 0.0055mol,0.55equiv),搅拌反应2小时,吹拂氮气下,加入4-硝基苯酚(1.5g,0.011 mol,1.1equiv),-78℃搅拌反应2小时。tlc检测反应结束,滴水进行淬灭,萃取,浓缩,柱层析得到产物氨基磷酸酯。非对映体过量值(dr)为3.4/1。通过异丙醇重结晶,得到手性(s,s)-氨基磷酸酯(ⅱ,2.6g,58%)1h nmr(400mhz,cdcl3):δ8.23(d,j=9.2 hz,2h),7.36(m,4h),7.24-7.22(m,3h),4.16-4.13(m,1h),4.10-4.08(m,2h),4.00-3.91(m, 1h),1.55-1.47(m,1h);1.45-1.40(m,3h),1.38-1.28(m,4h),0.87(t,j=7.6hz,6h);
31
pnmr(162mhz,cdcl3):δ-3.16。
[0112]
实施例28
[0113]
手性(s,s)-氨基磷酸酯的制备
[0114]
在一个50ml双口反应瓶中,加入(s)-氨基丙酸-(2)-乙基丁酯(2.1g,0.01mol,1 equiv),分子筛(3.0g),手性咪唑类催化剂(c15:0.0005mol, 0.05equiv),体系通过真空线,用氮气置换3次,加入15ml新蒸的脱气甲苯,溶液在
ꢀ-
90℃下,注入二氯化磷酸苯酯(0.011mol,1.1equiv),再滴加碱(四甲基乙二胺,0.005 5mol,0.55equiv),搅拌反应2小时,吹拂氮气下,加入4-硝基苯酚(1.5g,0.011mol,1.1 equiv),-90℃搅拌反应2小时。tlc检测反应结束,滴水进行淬灭,萃取,浓缩,柱层析得到产物氨基磷
1.47(m,1h);1.45-1.40(m,3h),1.38-1.28(m, 4h),0.87(t,j=7.6hz,6h);
31
p nmr(162mhz,cdcl3):δ-3.16。
[0130]
实施例34
[0131]
手性(s,s)-氨基磷酸酯的制备
[0132]
在一个50ml双口反应瓶中,加入(s)-氨基丙酸-(2)-乙基丁酯(2.1g,0.01mol,1 equiv),分子筛(3.0g),手性咪唑类催化剂(c15:0.0005mol, 0.05equiv),体系通过真空线,用氮气置换3次,加入15ml丁基三甲基铵双(三氟甲磺酰)亚胺离子液体,溶液在60℃下,注入二氯化磷酸苯酯(0.015mol,1.5equiv),再滴加碱(碳酸钾,0.0055mol,0.55equiv),搅拌反应18小时,吹拂氮气下,加入4-硝基苯酚(1.5g,0.011mol,1.1equiv),60℃搅拌反应18小时。tlc检测反应结束,滴水进行淬灭,萃取,浓缩,柱层析得到产物氨基磷酸酯。非对映体过量值(dr)为2.8/1。通过异丙醇重结晶,得到手性(s,s)-氨基磷酸酯(ⅱ,1.9g,42%)1h nmr(400mhz, cdcl3):δ8.23(d,j=9.2hz,2h),7.36(m,4h),7.24-7.22(m,3h),4.16-4.13(m,1h), 4.10-4.08(m,2h),4.00-3.91(m,1h),1.55-1.47(m,1h);1.45-1.40(m,3h),1.38-1.28(m, 4h),0.87(t,j=7.6hz,6h);
31
p nmr(162mhz,cdcl3):δ-3.16。
[0133]
上述实施例中采用的手性咪唑类催化剂的制备方法如下:
[0134]
实施例35:手性咪唑类催化剂c1的制备
[0135]
在一个干燥的反应瓶中加入(s)-6,7-二氢-5h-吡咯并[1,2-a]咪唑-7-醇(300mg,2.4 mmol,1.0eq)、20ml二氯甲烷和三乙胺(1.08ml,7.2mmol,3.0eq)并搅拌5分钟。之后加入乙酸酐(0.34ml,3.6mmol,1.5eq),于20℃反应12小时。然后用30ml水淬灭反应,用30ml二氯甲烷萃取,合并有机相用无水硫酸镁干燥。之后旋干,乙酸乙酯/甲醇体积比10/1为流动相进行硅胶柱层析(颗粒度100~200目,比表面积300~400 m2/g)分离得产物c1(305mg,产率76%)。1h nmr(400mhz,cdcl3)δ7.19(s,1h),6.97 (s,1h),5.99(dd,j=7.2hz,2.4hz,1h),4.22-4.11(m,1h),4.05-3.95(m,1h),3.14-3.01(m, 1h),2.61-2.49(m,1h),2.11(s,3h).
13
c nmr(100mhz,cdcl3)δ169.8,150.5,134.0, 115.2,66.6,42.4,34.3,20.5。
[0136]
实施例36:手性咪唑类催化剂c2的制备
[0137]
在一个干燥的反应瓶中加入(s)-6,7-二氢-5h-吡咯并[1,2-a]咪唑-7-醇(300mg,2.4 mmol,1.0eq)和30ml四氢呋喃,于0℃下分批加入质量分数60%的氢化钠(116mg,2.9 mmol,1.2eq)并搅拌30分钟。之后加入溴化苄(0.43ml,3.6mmol,1.5eq),于20℃反应12小时。然后用30ml水淬灭反应,用30ml二氯甲烷萃取,合并有机相用无水硫酸镁干燥。之后旋干,纯乙酸乙酯为流动相进行硅胶柱层析(颗粒度100~200目,比表面积300~400m2/g)分离得产物c2(378mg,产率73%)。1h nmr(400mhz,cdcl3) δ7.41-7.27(m,5h),7.16(d,j=1.2hz,1h),6.93(d,j=1.2hz,1h),4.90(d,j=11.6hz, 1h),4.83(dd,j=7.2hz,2.0hz,1h),4.73(d,j=11.6hz,1h),4.21-4.13(m,1h), 3.96-3.89(m,1h),2.92-2.82(m,1h),2.67-2.59(m,1h).
13
c nmr(100mhz,cdcl3)δ 153.5,137.9,133.8,128.4,128.1,127.7,115.0,71.1,70.8,43.1,35.3。
[0138]
实施例37:手性咪唑类催化剂c3的制备
[0139]
在一个干燥的反应瓶中加入(s)-6,7-二氢-5h-吡咯并[1,2-a]咪唑-7-醇(300mg,2.4 mmol,1.0eq)和30ml四氢呋喃,于0℃下分批加入质量分数60%的氢化钠(116mg,2.9 mmol,1.2eq)并搅拌30分钟。之后加入二碳酸二叔丁酯(0.83ml,3.6mmol,1.5eq),于20℃反应12小时。然后用30ml水淬灭反应,用30ml二氯甲烷萃取,合并有机相用无水硫酸镁干燥。之后旋干,乙酸乙酯/甲醇体积比10/1为流动相进行硅胶柱层析 (颗粒度100~200目,比表面积300~400m2/g)分离得产物c3(385.0mg,产率71%)。1h nmr(500mhz,cdcl3)δ7.18(d,j=1.2hz,1h),6.93(d,j=1.3hz,1h),5.89(dd,j= 6.8,1.9hz,1h),4.22
–
4.14(m,1h),4.03
–
3.95(m,1h),3.08
–
2.99(m,1h),2.69
–
2.61 (m,1h),1.50(s,9h).
13
c nmr(126mhz,cdcl3)δ152.7,150.8,134.9,115.4,82.9,69.3, 42.9,34.9,27.8。
[0140]
实施例38:手性咪唑类催化剂c4的制备
[0141]
在一个干燥的反应瓶中加入(s)-6,7-二氢-5h-吡咯并[1,2-a]咪唑-7-醇(300mg,2.4 mmol,1.0eq)、20ml二氯甲烷和三乙胺(1.68ml,12.1mmol,5.0eq)并搅拌5分钟。之后加入金刚烷酰氯(497mg,2.5mmol,1.05eq),于20℃反应12小时。然后用30ml 水淬灭反应,用30ml二氯甲烷萃取,合并有机相用无水硫酸镁干燥。之后旋干,乙酸乙酯/甲醇体积比10/1为流动相进行硅胶柱层析(颗粒度100~200目,比表面积300~400 m2/g)分离得产物c4(392mg,产率57%)。1h nmr(500mhz,cdcl3)δ7.20(s,1h),6.96 (s,1h),5.96(dd,j=7.4,2.9hz,1h),4.20
–
4.10(m,1h),4.03
–
3.94(m,1h),3.14
–
3.03 (m,1h),2.50
–
2.40(m,1h),2.03
–
1.97(m,3h),1.92
–
1.86(m,6h),1.74
–
1.64(m,6h). 13
c nmr(126mhz,cdcl3)δ177.3,151.4,134.8,115.5,66.9,43.0,40.9,39.0,38.8,36.7 36.5,35.3,28.2,28.0。
[0142]
实施例39:手性咪唑类催化剂c5的制备
[0143]
在一个干燥的反应瓶中加入(s)-6,7-二氢-5h-吡咯并[1,2-a]咪唑-7-醇(200mg,1.6 mmol,1.0eq)和20ml四氢呋喃,于0℃下分批加入质量分数60%的氢化钠(77mg,1.9 mmol,1.2eq)并搅拌30分钟。之后加入2,6-二异丙基苯异氰酸酯(0.52ml,2.4mmol,1.5 eq),于20℃反应12小时。然后用30ml水淬灭反应,用30ml二氯甲烷萃取,合并有机相用无水硫酸镁干燥。之后旋干,乙酸乙酯/甲醇体积比10/1为流动相进行硅胶柱层析(颗粒度100~200目,比表面积300~400m2/g)分离得产物c5(402mg,产率76%)。1h nmr(500mhz,cdcl3)δ7.31
–
7.27(m,1h),7.21(s,1h),7.17(s,1h),7.16(s,1h), 6.98(s,1h),6.46(s,1h),5.94(dd,j=7.1,2.8hz,1h),4.24
–
4.16(m,1h),4.04
–
3.96(m, 1h),3.22
–
3.09(m,3h),2.69
–
2.60(m,1h),1.23(d,j=6.9hz,6h),1.19(d,j=6.9hz, 6h).
13
c nmr(126mhz,cdcl3)δ154.7,151.2,146.8,134.8,130.5,128.4,123.5,115.6, 68.4,43.0,35.7,28.6,23.8,23.5。
[0144]
实施例40:手性咪唑类催化剂c6的制备
[0145]
在一个干燥的反应瓶中加入(s)-6,7-二氢-5h-吡咯并[1,2-a]咪唑-7-醇(200mg,1.6 mmol,1.0eq)和20ml四氢呋喃,于0℃下分批加入质量分数60%的氢化钠(77mg,1.9 mmol,1.2eq)并搅拌30分钟。之后加入异氰酸-2,4,6-三氯苯酯(538mg,2.4mmol,1.5eq),于20℃反应12小时。然后用30ml水淬灭反应,用30ml二氯甲烷萃取,合并有机相用无水硫酸镁干燥。之后旋干,乙酸乙酯/甲醇体积比10/1为流动相进行硅胶柱层析 (颗粒度100~200目,比表面积300~400m2/g)分离得产物c6(229mg,产率41%)。1h nmr(500mhz,cdcl3)δ8.18
(s,1h),7.38(s,2h),7.16(s,1h),6.94(s,1h),5.98(dd, j=7.3,2.7hz,1h),4.22
–
4.14(m,1h),4.03
–
3.93(m,1h),3.20
–
3.07(m,1h),2.74
–ꢀ
2.64(m,1h).
13
c nmr(126mhz,cdcl3)δ153.3,150.9,134.9,134.7,133.4,131.2,128.6, 115.6,100.1,68.9,43.1,35.5。
[0146]
实施例41:手性咪唑类催化剂c7的制备
[0147]
在一个干燥的反应瓶中加入(s)-6,7-二氢-5h-吡咯并[1,2-a]咪唑-7-醇(200mg,1.6 mmol,1.0eq)和20ml四氢呋喃,于0℃下分批加入质量分数60%的氢化钠(77mg,1.9 mmol,1.2eq)并搅拌30分钟。之后加入异氰酸2-联苯酯(472mg,2.4mmol,1.5eq),于20℃反应12小时。然后用30ml水淬灭反应,用30ml二氯甲烷萃取,合并有机相用无水硫酸镁干燥。之后旋干,乙酸乙酯/甲醇体积比10/1为流动相进行硅胶柱层析 (颗粒度100~200目,比表面积300~400m2/g)分离得产物c7(423mg,产率82%)。1h nmr(500mhz,cdcl3)δ8.19(d,j=8.3hz,1h),7.48
–
7.41(m,2h),7.41
–
7.30(m, 4h),7.22
–
7.10(m,3h),6.94(s,1h),6.75(s,1h),5.98(dd,j=7.3,2.6hz,1h),4.17
–ꢀ
4.09(m,1h),4.02
–
3.93(m,1h),3.17
–
3.04(m,1h),2.68
–
2.57(m,1h).
13
c nmr(126 mhz,cdcl3)152.8,151.0,138.0,135.0,134.7,130.4,129.4,129.3,128.6,128.1,123.6, 115.7,68.3,43.1,35.3。
[0148]
实施例42:手性咪唑类催化剂c8的制备
[0149]
在一个干燥的反应瓶中加入双环咪唑c7(319mg,1.0mmol,1.0eq)和10ml四氢呋喃,于0℃下分批加入质量分数60%的氢化钠(48mg,1.2mmol,1.2eq)并搅拌30 分钟。之后加入碘甲烷(62.3μl,1.0mmol,1.0eq),于20℃反应12小时。然后用20ml 水淬灭反应,用20ml二氯甲烷萃取,合并有机相用无水硫酸镁干燥。之后旋干,乙酸乙酯/甲醇体积比10/1为流动相进行硅胶柱层析(颗粒度100~200目,比表面积300~400 m2/g)分离得产物c8(185mg,产率55%)。1h nmr(500mhz,cdcl3)δ7.47
–
7.19(m, 20h),7.17
–
7.12(m,1h),6.93
–
6.88(m,1h),5.93
–
5.80(m,2h),4.19
–
3.80(m,4h), 3.08(s,3h),2.98
–
2.80(m,5h),2.46
–
2.36(m,1h),2.00
–
1.91(m,1h).
13
c nmr(126 mhz,cdcl3)δ174.0,154.6,151.1,150.9,140.8,139.9,139.8,139.6,138.9,134.1,133.8, 133.6,130.9,130.6,128.5,128.4,128.3,128.3,128.2,127.8,127.6,127.3,115.6,115.4, 115.3,68.2,68.1,43.3,43.0,38.3,37.7,35.6,34.6,21.4。
[0150]
实施例43:手性咪唑类催化剂c9的制备
[0151]
在一个干燥的反应瓶中加入(s)-6,7-二氢-5h-吡咯并[1,2-a]咪唑-7-醇(300mg,2.4 mmol,1.0eq)和30ml四氢呋喃,于0℃下分批加入质量分数60%的氢化钠(116mg,2.9 mmol,1.2eq)并搅拌30分钟。之后加入3-异丙基-二甲基苄基异氰酸酯(0.72ml,3.6 mmol,1.5eq),于20℃反应12小时。然后用30ml水淬灭反应,用30ml二氯甲烷萃取,合并有机相用无水硫酸镁干燥。之后旋干,乙酸乙酯/甲醇体积比10/1为流动相进行硅胶柱层析(颗粒度100~200目,比表面积300~400m2/g)分离得产物c9(415mg,产率53%)。1h nmr(400mhz,cdcl3)δ7.48(s,1h),7.36
–
7.27(m,3h),7.18(s,1h),6.94 (s,1h),5.82(dd,j=7.4,2.8hz,1h),5.38
–
5.25(m,2h),5.11
–
5.05(m,1h),4.17
–
4.07 (m,1h),3.99
–
3.89(m,1h),3.08
–
2.95(m,1h),2.62
–
2.49(m,1h),2.15(s,3h),1.70(s, 3h),1.66(s,3h).
13
c nmr(101mhz,cdcl3)δ153.7,151.4,146.7,143.5,141.3,134.7, 128.3,124.1,124.0,122.0,115.4,112.6,67.4,55.4,42.9,35.4,29.5,28.9,22.0。
[0152]
实施例44:手性咪唑类催化剂c10的制备
[0153]
在一个干燥的反应瓶中加入(s)-6,7-二氢-5h-吡咯并[1,2-a]咪唑-7-醇(300mg,2.4 mmol,1.0eq)、20ml二氯甲烷和三乙胺(1.68ml,12.1mmol,5.0eq)并搅拌5分钟。之后加入二异丙基甲氨酰氯(415mg,2.5mmol,1.05eq),于20℃反应12小时。然后用30ml水淬灭反应,用30ml二氯甲烷萃取,合并有机相用无水硫酸镁干燥。之后旋干,乙酸乙酯/甲醇体积比10/1为流动相进行硅胶柱层析(颗粒度100~200目,比表面积300~400m2/g)分离得产物c9(161mg,产率27%)。1h nmr(500mhz,cdcl3)δ7.19 (d,j=1.2hz,1h),6.95(d,j=1.3hz,1h),5.96(dd,j=7.1,2.8hz,1h),4.18
–
4.11(m, 1h),4.09
–
3.95(m,2h),3.76(s,1h),3.12
–
3.03(m,1h),2.64
–
2.56(m,1h),1.19(s, 12h).
13
c nmr(126mhz,cdcl3)δ154.8,151.9,134.7,115.3,67.6,46.6,45.7,43.0,35.7, 21.5,20.6。
[0154]
实施例45:手性咪唑类催化剂c11的制备
[0155]
在一个干燥的反应瓶中加入(s)-6,7-二氢-5h-吡咯并[1,2-a]咪唑-7-醇(300mg,2.4 mmol,1.0eq)、20ml二氯甲烷和三乙胺(1.68ml,12.1mmol,5.0eq)并搅拌5分钟。之后加入1-哌啶酰氯(0.32ml,2.5mmol,1.05eq),于20℃反应12小时。然后用30ml 水淬灭反应,用30ml二氯甲烷萃取,合并有机相用无水硫酸镁干燥。之后旋干,乙酸乙酯/甲醇体积比10/1为流动相进行硅胶柱层析(颗粒度100~200目,比表面积300~400 m2/g)分离得产物c11(271mg,产率48%)。1h nmr(500mhz,cdcl3)δ7.19(d,j=1.2 hz,1h),6.95(d,j=1.2hz,1h),5.91(dd,j=7.3,2.9hz,1h),4.20
–
4.11(m,1h),4.01
–ꢀ
3.93(m,1h),3.47
–
3.32(m,4h),3.14
–
3.03(m,1h),2.65
–
2.55(m,1h),1.61
–
1.45(m, 6h).
13
c nmr(126mhz,cdcl3)δ154.7,151.7,134.6,115.3,68.1,44.9,42.9,35.4,25.6, 24.3。
[0156]
实施例46:手性咪唑类催化剂c12的制备
[0157]
在一个干燥的反应瓶中加入(s)-6,7-二氢-5h-吡咯并[1,2-a]咪唑-7-醇(300mg,2.4 mmol,1.0eq)和30ml四氢呋喃,于0℃下分批加入质量分数60%的氢化钠(116mg,2.9 mmol,1.2eq)并搅拌30分钟。之后加入叔丁基异氰酸酯(0.41ml,3.6mmol,1.5eq),于20℃反应12小时。然后用30ml水淬灭反应,用30ml二氯甲烷萃取,合并有机相用无水硫酸镁干燥。之后旋干,乙酸乙酯/甲醇体积比10/1为流动相进行硅胶柱层析 (颗粒度100~200目,比表面积300~400m2/g)分离得产物c12(460mg,产率85%)。1h nmr(500mhz,cdcl3)δ7.18(s,1h),6.94(s,1h),5.88(dd,j=7.4,2.7hz,1h),4.78 (s,1h),4.19
–
4.09(m,1h),4.05
–
3.93(m,1h),3.14
–
3.00(m,1h),2.68
–
2.54(m,1h), 1.32(s,9h).
13
c nmr(126mhz,cdcl3)δ155.0,151.7,134.9,115.5,67.2,50.7,43.1,35.5, 29.0。
[0158]
实施例47:手性咪唑类催化剂c13的制备
[0159]
在一个干燥的反应瓶中加入双环咪唑c12(223mg,1.0mmol,1.0eq)和10ml四氢呋喃,于0℃下分批加入质量分数60%的氢化钠(48mg,1.2mmol,1.2eq)并搅拌30 分钟。之后加入碘甲烷(62.3μl,1.0mmol,1.0eq),于20℃反应12小时。然后用20ml 水淬灭反应,用20ml二氯甲烷萃取,合并有机相用无水硫酸镁干燥。之后旋干,乙酸乙酯/甲醇体积比10/1为流动相进行硅胶柱层析(颗粒度100~200目,比表面积300~400 m2/g)分离得产物c13(193mg,产率81%)。1h nmr(400mhz,cdcl3)δ7.19(d,j=1.2 hz,1h),6.95(d,j=1.2hz,1h),5.91(dd,j=7.2,2.8hz,1h),4.19
–
4.10(m,1h),4.02
–ꢀ
3.93(m,1h),3.13
–
3.02(m,1h),2.88(s,3h),2.63
–
2.54(m,1h),1.38(s,9h).
13
c nmr (101mhz,cdcl3)δ155.6,151.9,134.6,115.4,67.7,55.9,43.0,35.6,31.5,28.7。
[0160]
实施例48:手性咪唑类催化剂c14的制备
[0161]
在干燥的反应瓶a中加入三光气(474.8mg,1.6mmol,1.0eq)与10ml二氯甲烷。在滴液漏斗中加入叔辛胺(0.48ml,1.6mmol,1.0eq)与3ml二氯甲烷。在0℃下,将叔辛胺的二氯甲烷溶液缓慢滴加到三光气的二氯甲烷溶液中。10分钟后,在滴液漏斗中加入三乙胺(0.89ml,6.4mmol,4.0eq)和3ml二氯甲烷,并将其滴入上述反应液中。之后于20℃反应2小时。在另一个干燥的反应瓶b中加入(s)-6,7-二氢-5h-吡咯并[1,2-a]咪唑-7-醇(200mg,1.6mmol,1.0eq)和20ml四氢呋喃,于0℃下分批加入质量分数60%的氢化钠(193mg,4.8mmol,3.0eq)并搅拌30分钟。之后将反应瓶a中的溶液转移至反应瓶b中,于20℃反应12小时。然后用30ml水淬灭反应,用30ml 二氯甲烷萃取,合并有机相用无水硫酸镁干燥。之后旋干,乙酸乙酯/甲醇体积比10/1 为流动相进行硅胶柱层析(颗粒度100~200目,比表面积300~400m2/g)分离得产物 c14(164.8mg,产率43%)。1h nmr(400mhz,cdcl3)δ7.19(d,j=1.3hz,1h),6.95(d, j=1.2hz,1h),5.86(dd,j=7.4,2.7hz,1h),4.75(s,1h),4.19
–
4.08(m,1h),4.03
–
3.92 (m,1h),3.14
–
3.01(m,1h),2.64
–
2.52(m,1h),1.79(d,j=14.9hz,1h),1.59(d,j=14.9 hz,1h),1.38(s,3h),1.34(s,3h),0.99(s,9h).
13
c nmr(101mhz,cdcl3)δ153.7,151.7, 134.7,115.5,67.3,54.4,51.7,43.1,35.7,31.7,31.6,29.5,29.5。
[0162]
实施例49:手性咪唑类催化剂c15的制备
[0163]
在一个干燥的反应瓶中加入(s)-6,7-二氢-5h-吡咯并[1,2-a]咪唑-7-醇(200mg,1.6 mmol,1.0eq)和20ml四氢呋喃,于0℃下分批加入质量分数60%的氢化钠(77mg,1.9 mmol,1.2eq)并搅拌30分钟。之后加入异氰酸1-金刚烷酯(428mg,2.4mmol,1.5eq),于20℃反应12小时。然后用30ml水淬灭反应,用30ml二氯甲烷萃取,合并有机相用无水硫酸镁干燥。之后旋干,石油醚/乙酸乙酯体积比1/10为流动相进行硅胶柱层析(颗粒度100~200目,比表面积300~400m2/g)分离得产物c15(285mg,产率59%)。1h nmr(500mhz,cdcl3)δ7.18(d,j=1.3hz,1h),6.94(d,j=1.2hz,1h),5.86(dd,j= 7.2,2.7hz,1h),4.72(s,1h),4.19
–
4.06(m,1h),4.03
–
3.91(m,1h),3.11
–
2.98(m,1h), 2.66
–
2.53(m,1h),2.11
–
2.05(m,3h),1.95
–
1.90(m,6h),1.68
–
1.64(m,6h).
13
c nmr (126mhz,cdcl3)δ153.5,151.7,134.8,115.5,67.1,51.0,43.1,42.6,41.8,36.6,36.3,35.4, 29.7,29.5。
[0164]
实施例50:手性咪唑类催化剂c16的制备
[0165]
在一个干燥的反应瓶中加入(s)-6,7-二氢-5h-吡咯并[1,2-a]咪唑-7-醇(200mg,1.6 mmol,1.0eq)和20ml四氢呋喃,于0℃下分批加入质量分数60%的氢化钠(77mg,1.9 mmol,1.2eq)并搅拌30分钟。之后加入2-(乙氧基羰基)苯基异氰酸酯(462mg,2.4mmol, 1.5eq),于20℃反应12小时。然后用30ml水淬灭反应,用30ml二氯甲烷萃取,合并有机相用无水硫酸镁干燥。之后旋干,乙酸乙酯/甲醇体积比10/1为流动相进行硅胶柱层析(颗粒度100~200目,比表面积300~400m2/g)分离得产物c16(298mg,产率 59%)。1h nmr(500mhz,cdcl3)δ10.60(s,1h),8.47(d,j=8.5hz,1h),8.01(dd,j=8.1, 1.6hz,1h),7.59
–
7.50(m,1h),7.21(s,1h),7.04(t,j=7.6hz,1h),6.97(s,1h),6.07(d, j=6.3hz,1h),4.39
–
4.29(m,2h),4.25
–
4.17(m,1h),4.06
–
3.98(m,1h),3.13
–
3.03 (m,1h),2.74
–
2.66(m,1h),1.38(t,j=7.1hz,3h).
13
c nmr(126mhz,cdcl3)δ168.0, 152.7,141.4,134.5,130.8,121.7,118.9,115.5,115.0,67.8,61.3,43.0,35.1,14.2。
[0166]
实施例51:手性咪唑类催化剂c17的制备
[0167]
在一个干燥的反应瓶中加入(s)-6,7-二氢-5h-吡咯并[1,2-a]咪唑-7-醇(200mg,1.6 mmol,1.0eq)和20ml四氢呋喃,于0℃下分批加入质量分数60%的氢化钠(77mg,1.9 mmol,1.2eq)并搅拌30分钟。之后加入异氰酸异丙酯(0.18ml,2.4mmol,1.5eq),于 20℃反应12小时。然后用30ml水淬灭反应,用30ml二氯甲烷萃取,合并有机相用无水硫酸镁干燥。之后旋干,乙酸乙酯/甲醇体积比10/1为流动相进行硅胶柱层析(颗粒度100~200目,比表面积300~400m2/g)分离得产物c17(158mg,产率47%)。1h nmr (500mhz,cdcl3)δ7.19(s,1h),6.95(s,1h),5.91(dd,j=7.2,2.7hz,1h),4.63(d,j=7.3 hz,1h),4.18
–
4.10(m,1h),4.01
–
3.94(m,1h),3.89
–
3.78(m,1h),3.12
–
3.02(m,1h), 2.66
–
2.56(m,1h),1.15(d,j=6.4hz,6h).
13
c nmr(126mhz,cdcl3)δ154.7,151.5, 134.8,115.4,67.5,43.2,42.9,35.3,23.0。
[0168]
实施例52:手性咪唑类催化剂c18的制备
[0169]
在一个干燥的反应瓶中加入(s)-6,7-二氢-5h-吡咯并[1,2-a]咪唑-7-醇(200mg,1.6 mmol,1.0eq)、10ml二氯甲烷和2,6-二甲基吡啶(0.56ml,4.8mmol,3.0eq)并搅拌5 分钟。之后在0℃下加入叔丁基二甲硅基三氟甲磺酸酯(0.74ml,3.2mmol,2.0eq),并于20℃反应24小时。然后直接旋干,乙酸乙酯/甲醇体积比10/1为流动相进行硅胶柱层析(颗粒度100~200目,比表面积300~400m2/g)分离得产物c18(287mg,产率75%)。1h nmr(500mhz,cdcl3)δ7.13(d,j=1.3hz,1h),6.85(d,j=1.2hz,1h),5.10(dd,j= 6.8,2.9hz,1h),4.18
–
4.11(m,1h),3.91
–
3.83(m,1h),2.90
–
2.79(m,1h),2.50
–
2.42 (m,1h),0.92(s,9h),0.21(s,3h),0.13(s,3h).
13
c nmr(126mhz,cdcl3)δ155.0,134.0, 114.6,66.3,42.8,38.3,25.9,18.4,-4.6,-4.8。
[0170]
以上对本发明的具体实施例进行了描述。需要理解的是,本发明并不局限于上述特定实施方式,本领域技术人员可以在权利要求的范围内做出各种变化或修改,这并不影响本发明的实质内容。在不冲突的情况下,本技术的实施例和实施例中的特征可以任意相互组合。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。