1.本发明属于医药技术领域,具体涉及一种抗肿瘤化合物的合成方法及其中间体。
背景技术:
2.肿瘤的生长和迁移除了与有丝分裂激酶(例如aurora激酶)的过表达相关之外,也依赖于大量新生血管的生成,其中vegf/vegfr(血管内皮生长因子/血管内皮生长因子受体)途径在肿瘤新生血管的生成中起关键作用。
3.专利wo2018108079a1公开了系列化合物,能够抑制、调节和/或调控一种或多种蛋白激酶例如aurora激酶和vegfr激酶的活性,通过抑制肿瘤的生长、迁移,逆转肿瘤微环境,发挥肿瘤免疫效应和抗肿瘤疗效。该专利中记载了化合物29的合成方法,通过进一步研究发现,其合成工艺仍需要优化,以得到工艺更简单,收率更高,纯度更高,成本更低,并适合工业化大生产的合成方法。
[0004][0005]
化合物29
技术实现要素:
[0006]
在第一方面中,本发明提供了下述技术方案1
‑
15:
[0007]
1.一种式7抗肿瘤化合物的合成方法,其特征在于,包括如下步骤:
[0008][0009]
(a)于有机溶剂中,将式1化合物与式2m化合物在碱的作用下发生芳香亲核取代反应得到式im化合物,式im化合物经分离或者不经分离,与式3化合物继续发生芳香亲核取代反应,例如与式3化合物在碱性条件下继续发生芳香亲核取代反应,得到式4m化合物;
[0010]
(b)于有机溶剂中,将所述式4m化合物在催化剂的作用下发生还原反应,得到式5m化合物;
[0011]
(c)于有机溶剂中,将所述式5m化合物在酸的作用下关环,得到式6m化合物;
[0012]
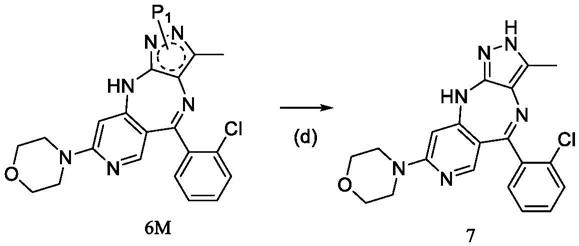
(d)于有机溶剂中,将所述式6m化合物脱去氨基保护基,得到式7抗肿瘤化合物;
[0013]
反应式如下:
[0014][0015]
其中,x1和x2分别选自卤素;p1为氨基保护基,优选地,p1选自boc、cbz、tos、fmoc、pmb、mom、eom、tbu、bn、ac、sem、trt或thp,例如boc、cbz、tos、fmoc、pmb、trt或thp;p1与所在环上的任一n原子相连,优选地p1与所在环上的甲基邻位的n原子相连。
[0016]
2.一种式7抗肿瘤化合物的合成方法,
[0017][0018]
其特征在于,包括如下步骤:
[0019]
(d)于有机溶剂中,将所述式6m化合物脱去氨基保护基,得到式7抗肿瘤化合物;
[0020]
反应式如下:
[0021][0022]
其中,p1为氨基保护基,优选地,p1选自boc、cbz、tos、fmoc、pmb、mom、eom、tbu、bn、ac、sem、trt或thp,例如boc、cbz、tos、fmoc、pmb、trt或thp;p1与所在环上的任一n原子相连,优选地p1与所在环上的甲基邻位的n原子相连。
[0023]
3.一种式6m中间体化合物的合成方法或者根据前述技术方案中任一项的方法,
[0024][0025]
其特征在于,包括如下步骤:
[0026]
(c)于有机溶剂中,将所述式5m化合物在酸的作用下关环,得到式6m化合物;
[0027]
反应式如下:
[0028][0029]
其中,p1为氨基保护基,优选地,p1选自boc、cbz、tos、fmoc、pmb、mom、eom、tbu、bn、ac、sem、trt或thp,例如boc、cbz、tos、fmoc、pmb、trt或thp;p1与所在环上的任一n原子相连,优选地p1与所在环上的甲基邻位的n原子相连。
[0030]
4.一种式5m中间体化合物的合成方法或者根据前述技术方案中任一项的方法,
[0031][0032]
其特征在于,包括如下步骤:
[0033]
(b)于有机溶剂中,将所述式4m化合物在催化剂的作用下发生还原反应,得到式5m化合物;
[0034]
反应式如下:
[0035][0036]
其中,p1为氨基保护基,优选地,p1选自boc、cbz、tos、fmoc、pmb、mom、eom、tbu、bn、ac、sem、trt或thp,例如boc、cbz、tos、fmoc、pmb、trt或thp;p1与所在环上的任一n原子相连,优选地p1与所在环上的甲基邻位的n原子相连。
[0037]
5.一种式4m中间体化合物的合成方法或者根据前述技术方案中任一项的方法,
[0038][0039]
其特征在于,包括如下步骤:
[0040]
(a)于有机溶剂中,将式1化合物与式2m化合物在碱的作用下发生芳香亲核取代反应得到式im化合物,式im化合物经分离或者不经分离,与式3化合物继续发生芳香亲核取代反应,例如与式3化合物在碱性条件下继续发生芳香亲核取代反应,得到式4m化合物;
[0041]
反应式如下:
[0042][0043]
其中,x1和x2分别选自卤素;p1为氨基保护基,优选地p1选自boc、cbz、tos、fmoc、pmb、mom、eom、tbu、bn、ac、sem、trt或thp,例如boc、cbz、tos、fmoc、pmb、trt或thp;p1与所在环上的任一n原子相连,优选地p1与所在环上的甲基邻位的n原子相连。
[0044]
6.根据前述技术方案中任一项的方法,其特征在于,步骤(a)中,所述有机溶剂选自2
‑
甲基四氢呋喃、乙腈、四氢呋喃和甲苯中的一种或几种;所述碱选自氢化钠、氢氧化钠、碳酸铯、三乙烯二胺、叔丁醇钠、叔丁醇钾、双三甲基硅基胺基锂和六甲基二硅基氮烷钠中的一种或几种;
[0045]
任选地步骤(a)中,
[0046]
反应压力为0至10mpa(表压),例如常压;
[0047]
反应时间为1
‑
96小时,例如17小时;反应温度可以为回流温度;
[0048]
特别地,在式1化合物与式2m化合物在碱的作用下发生芳香亲核取代反应中,反应
时间为3
‑
18小时,例如反应时间为3
‑
8小时;反应温度为40℃至70℃,例如反应温度为40℃至60℃;
[0049]
特别地,在与式3化合物继续发生芳香亲核取代反应中,反应时间为8小时至16小时,例如,13小时至16小时;反应温度可以为回流温度;
[0050]
比例为:
[0051]
溶剂∶式1化合物∶式2m化合物∶式3化合物∶碱=0.5
‑
60l∶0.1
‑
11摩尔∶1摩尔∶0.3
‑
30摩尔∶0.2
‑
25摩尔;例如,5.8l∶1.1摩尔∶1摩尔∶3摩尔∶2.5摩尔;
[0052]
特别地,在式1化合物与式2m化合物在碱的作用下发生芳香亲核取代反应中加入硫酸镁,在与式3化合物继续发生芳香亲核取代反应中加入醋酸,
[0053]
比例为:
[0054]
溶剂∶式1化合物∶式2m化合物∶式3化合物∶碱∶硫酸镁∶醋酸=0.5
‑
60l∶0.1
‑
11摩尔∶1摩尔∶0.3
‑
30摩尔∶0.2
‑
25摩尔∶0.2
‑
20摩尔∶0.1
‑
15摩尔,例如,5.8l∶1.1摩尔∶1摩尔∶3摩尔∶2.5摩尔∶1.6摩尔∶1.5摩尔。
[0055]
7.根据前述技术方案中任一项的方法,其特征在于,步骤(b)中,所述有机溶剂选自甲醇、乙醇、异丙醇、四氢呋喃、2
‑
甲基四氢呋喃、醋酸和乙腈中一种或几种;所述催化剂选自铁粉、氧化铂、pt/c、pd(oh)2/c、rh/c或pd/c;
[0056]
任选地步骤(b)中,当催化剂为铁粉时,反应时间为1
‑
96小时,例如15
‑
24小时;反应压力为0至10mpa(表压),例如常压;反应温度为回流温度;
[0057]
比例为:
[0058]
有机溶剂∶式4m化合物∶催化剂=1
‑
130l∶1摩尔∶1至150摩尔,例如13l∶1摩尔∶10
‑
18摩尔,例如13l∶1摩尔∶14
‑
18摩尔,例如13l∶1摩尔∶15摩尔;
[0059]
特别地,当催化剂为铁粉时:步骤(b)中还加入数量为0.05
‑
5摩尔,例如0.5摩尔的氯化铵;溶剂为乙醇与四氢呋喃的混合溶剂,例如,乙醇与四氢呋喃的体积比为(6至10)∶10;
[0060]
特别地,步骤(b)中,当催化剂为氧化铂、pt/c、pd(oh)2/c、rh/c或pd/c时,反应在氢气气氛下在30℃至80℃下进行20小时至80小时;例如,反应在氢气气氛下在60℃至80℃下进行20小时至72小时;反应压力为大于0至10mpa(表压),例如0.5
‑
2mpa;比例为溶剂∶式4m化合物∶催化剂=1
‑
50ml∶1克∶0.03至0.2克,例如22ml∶1克∶0.1克;
[0061]
任选地步骤(b)中,步骤(b)中还加入少量的水,例如,水与四氢呋喃的体积比为(0.2至2)∶10。
[0062]
8.根据前述技术方案中任一项的方法,其特征在于,步骤(c)中,所述酸选自三氟乙酸、甲磺酸、对甲苯磺酸、甲酸、乙酸和盐酸中的一种或几种;所述有机溶剂选自二氯甲烷、甲醇、乙醇和异丙醇中的至少一种;
[0063]
任选地步骤(c)中,反应时间为1
‑
96小时,例如1.5
‑
3小时;反应压力为0至10mpa(表压),例如常压;反应温度为回流温度;比例为溶剂∶式5m化合物∶酸=0.5
‑
70l∶1摩尔∶0.220摩尔,例如6.6l∶1摩尔∶2摩尔。
[0064]
9.根据前述技术方案中任一项的方法,其特征在于,步骤(c)具体为:式5m化合物于异丙醇中,在三氟乙酸的作用下回流反应1小时至3小时。
[0065]
10.根据前述技术方案中任一项的方法,其特征在于,步骤(d)中,所述有机溶剂选
自甲苯、二氯甲烷、乙腈、四氢呋喃、甲醇、乙醇和异丙醇中的一种或几种;
[0066]
步骤(d)中,还加入酸,所述酸选自甲酸、乙酸、盐酸、甲磺酸和三氟乙酸中的至少一种;
[0067]
步骤(d)中,还加入除溶剂以外的醇或酚;所述醇选自甲醇和/或乙醇,所述酚选自苯酚和/或对甲氧基苯酚;
[0068]
任选地步骤(d)中,反应时间为1
‑
96小时,例如17小时;反应压力为0至10mpa(表压),例如常压;反应温度为回流温度;比例为溶剂∶式6m化合物∶酸∶醇或酚=10
‑
800l∶1摩尔∶0.1至10摩尔∶0.2至20摩尔,例如84l∶1摩尔∶1摩尔∶2摩尔。
[0069]
11.根据前述技术方案中任一项的方法,其特征在于,还包括合成式2m化合物的步骤:
[0070]
(i)于溶剂中,如浓硫酸、醋酸酐、醋酸中,将式2
‑
1化合物(例如,1摩尔当量)与硝酸(例如,1
‑
2摩尔当量)发生硝化反应(反应时间:0.5
‑
2小时,反应温度;10
‑
20℃),得到式2
‑
2化合物;
[0071]
(ii)于有机溶剂如四氢呋喃、2
‑
甲基四氢呋喃、二氯甲烷和乙腈中的一种或几种中,将所述式2
‑
2化合物(例如,1摩尔当量)与氨基保护试剂(例如1
‑
1.5摩尔当量)发生氨基保护反应(反应时间1
‑
48小时,反应温度60
‑
100℃),得到式(2
‑
3)m化合物;
[0072]
(iii)于有机溶剂如乙醇、四氢呋喃和乙腈中的一种或几种中,将所述式(2
‑
3)m化合物(例如,1摩尔当量)脱去邻苯二甲酰基(例如,反应时间:1
‑
3小时,反应温度:回流温度),得到式2m化合物;
[0073]
反应式如下:
[0074][0075]
其中,所述氨基保护试剂为boc2o、cbzcl、toscl、fmoccl、pmbbr、momcl、eomcl、叔丁醇、异丁烯、bncl、乙酸酐、semcl、trtcl或dhp,例如boc2o、cbzcl、toscl、fmoccl、pmbbr、trtcl或dhp;p1选自boc、cbz、tos、fmoc、pmb、mom、eom、tbu、bn、ac、sem、trt或thp,例如boc、cbz、tos、fmoc、pmb、trt或thp;p1与所在环上的任一n原子相连,优选地p1与所在环上的甲基邻位的n原子相连。
[0076]
12.根据技术方案11的方法,其特征在于,
[0077]
当所述氨基保护试剂为trtcl,步骤(ii)的反应条件为:在碱的作用下,60℃至100℃下反应1小时至48小时;
[0078]
当所述氨基保护试剂为dhp,步骤(ii)的反应条件为:在对甲苯磺酸或吡啶对甲苯磺酸盐的作用下,60℃至100℃下回流反应3小时至48小时。
[0079]
13.根据前述技术方案中任一项的方法,其特征在于,还包括合成式1化合物的步骤:
[0080]
于有机溶剂如thf中,将式1
‑
1化合物(例如,1至2摩尔当量)与式1
‑
2化合物(例如,1至2摩尔当量)发生偶联反应(反应时间:2
‑
4小时,反应温度:于
‑
70至60℃),得到式1化合
物;
[0081]
反应式如下:
[0082][0083]
其中,x1、x2和x3分别选自卤素;优选地,x1、x2和x3分别选自氯。
[0084]
14.一种制备式7抗肿瘤化合物的中间体,其特征在于,具有以下结构式:
[0085][0086]
其中,x1和x2分别选自卤素;
[0087]
p1选自trt或thp;p1与所在环上的任一n原子相连,优选地p1与所在环上的甲基邻位的n原子相连。
[0088]
15.一种制备式7抗肿瘤化合物的中间体,其特征在于,具有以下结构式:
[0089][0090]
其中,p1选自trt或thp;p1与所在环上的任一n原子相连,优选地p1与所在环上的甲基邻位的n原子相连;
[0091]
该式6m为含有1分子三氟乙酸与1分子异丙醇的复合物。
[0092]
在第二方面中,本发明的目的是提供式7抗肿瘤化合物的合成方法,包括如下步骤:
[0093][0094]
(a)于有机溶剂中,将式1化合物与式2化合物在碱的作用下发生芳香亲核取代反应,然后与式3化合物继续发生芳香亲核取代反应,例如与式3化合物在碱性条件下继续发生芳香亲核取代反应,得到式4化合物;
[0095]
(b)于有机溶剂中,将所述式4化合物在催化剂的作用下发生还原反应,得到式5化合物;
[0096]
(c)于有机溶剂中,将所述式5化合物在酸的作用下关环,得到式6化合物;
[0097]
(d)于有机溶剂中,将所述式6化合物脱去氨基保护基,得到式7抗肿瘤化合物;
[0098]
反应式如下:
[0099][0100]
其中,x1和x2分别选自卤素;p1为氨基保护基,p1选自boc、cbz、tos、fmoc、pmb、mom、eom、tbu、bn、ac、sem、trt或thp,例如boc、cbz、tos、fmoc、pmb、trt或thp。
[0101]
在其中一个实施例中,步骤(a)中,所述有机溶剂选自2
‑
甲基四氢呋喃、乙腈、四氢呋喃、和甲苯中的一种或几种;所述碱选自氢化钠、氢氧化钠、碳酸铯、三乙烯二胺、叔丁醇钠、叔丁醇钾、双三甲基硅基胺基锂和六甲基二硅基氮烷钠中的一种或几种。
[0102]
在其中一个实施例中,步骤(b)中,所述有机溶剂选自甲醇、乙醇、异丙醇、四氢呋喃、2
‑
甲基四氢呋喃和乙腈中一种或几种;所述催化剂选自铁粉、氧化铂、pt/c或pd/c。
[0103]
在其中一个实施例中,步骤(c)中,所述酸选自三氟乙酸、甲磺酸、对甲苯磺酸、甲酸、乙酸和盐酸中的一种或几种;所述有机溶剂选自二氯甲烷、甲醇、乙醇和异丙醇中的至少一种。
[0104]
在其中一个实施例中,步骤(c)具体为:式5化合物于异丙醇中,在三氟乙酸的作用下回流反应1小时至3小时,例如1.5小时至3小时。
[0105]
在其中一个实施例中,步骤(d)中,所述有机溶剂选自甲苯、二氯甲烷、乙腈、四氢呋喃、甲醇、乙醇和异丙醇中的一种或几种;
[0106]
步骤(d)中,还加入催化剂,所述催化剂选自甲酸、乙酸、盐酸、甲磺酸和三氟乙酸中的至少一种;
[0107]
步骤(d)中,还加入醇或酚;所述醇选自甲醇和/或乙醇,所述酚选自苯酚和/或对甲氧基苯酚。
[0108]
在其中一个实施例中,还包括合成式2化合物的步骤:
[0109]
(i)于溶剂中,将式2
‑
1化合物与硝酸发生硝化反应,得到式2
‑
2化合物;
[0110]
(ii)于有机溶剂中,将所述式2
‑
2化合物与氨基保护试剂发生氨基保护反应,得到式2
‑
3化合物;
[0111]
(iii)于有机溶剂中,将所述式2
‑
3化合物脱去邻苯二甲酰基,得到式2化合物;
[0112]
反应式如下:
[0113][0114]
其中,所述氨基保护试剂为boc2o、cbzcl、toscl、fmoccl、pmbbr、momcl、eomcl、叔丁醇、异丁烯、bncl、乙酸酐、semcl、trtcl或dhp,例如boc2o、cbzcl、toscl、fmoccl、pmbbr、trtcl或dhp;p1选自boc、cbz、tos、fmoc、pmb、mom、eom、tbu、bn、ac、sem、trt或thp,例如boc、cbz、tos、fmoc、pmb、trt或thp。
[0115]
在其中一个实施例中,当所述氨基保护试剂为trtcl,步骤(ii)的反应条件为:在碱的作用下,60℃至100℃下反应1小时至48小时;
[0116]
当所述氨基保护试剂为dhp,步骤(ii)的反应条件为:在对甲苯磺酸或吡啶对甲苯磺酸盐的作用下,60℃至100℃下回流反应3小时至48小时。
[0117]
在其中一个实施例中,还包括合成式1化合物的步骤:
[0118]
于有机溶剂中,将式1
‑
1化合物与式1
‑
2化合物发生偶联反应,得到式1化合物;
[0119]
反应式如下:
[0120][0121]
其中,x1、x2和x3分别选自卤素;优选地,x1、x2和x3分别选自氯。
[0122]
在第三方面中,本发明还提供一种制备式7抗肿瘤化合物的中间体,具有以下结构式:
[0123][0124]
其中,x1和x2分别选自卤素;
[0125]
p1选自trt或thp;
[0126]
p2选自trt或thp,并且p3不存在;或者p3选自trt或thp,并且p2不存在;
[0127]
pg1为乙酰基或者pg1‑
nh
‑
为
[0128]
表示可能存在的单键或者双键。
[0129]
在第四方面中,本发明提供一种制备式7抗肿瘤化合物的中间体,具有以下结构式:
[0130][0131]
其中,p1选自trt或thp;
[0132]
该式6为含有1分子三氟乙酸与1分子异丙醇的复合物。
[0133]
与现有技术相比,本发明具有以下有益效果:
[0134]
(1)本发明不同于现有合成工艺先脱保护后成三并环的工艺,采用了先合成三并环后脱保护的方式,以中间体(式2化合物)为原料,经过芳香亲核取代反应、还原反应后得到式5化合物,由此式5化合物可实现先成三并环再脱保护的工艺,故而避免现有工艺中的不足,
①
在先脱保护时需采用大量的三氟乙酸而产生废水的问题,
②
在后成三并环采用氯化亚锡还原成环,可操作性及后处理复杂的问题,
③
整体工艺复杂需要靠过柱子实现。
[0135]
(2)本发明的合成方法可操作性增强,简化了工艺,合成的式7化合物无需再进行复杂的重结晶工艺,即可获得更高的纯度,减少了三废的产生,更适合工业化大生产。
[0136]
(3)本发明提供的中间体用于制备抗肿瘤化合物,有效的减少反应过程中的副产物,提高了反应的总收率,较前述专利wo2018108079a1中化合物29的合成方法提高了至少一倍。
具体实施方式
[0137]
以下通过具体实施方式对本发明的上述内容作进一步的详细说明,但不应该理解为本发明上述主题的范围仅限于以下实施例。凡是基于本发明上述内容所实现的技术均属于本发明的范围。
[0138]
本文中使用的缩写具有本领域中通常理解的含义,如下:
[0139]“卤素”是指氟、氯、溴、碘等;
[0140]“boc2o”是指:二碳酸二叔丁酯;
[0141]“boc”是指:叔丁氧羰基;
[0142]“cbzcl”是指:氯甲酸苄酯;
[0143]“cbz”是指:苄氧羰基;
[0144]“toscl”是指:对甲苯磺酰氯;
[0145]“tos”是指:对甲苯磺酰基;
[0146]“fmoccl”是指:氯甲酸
‑9‑
芴甲酯;
[0147]“fmoc”是指:9
‑
芴基甲氧基羰基;
[0148]“pmbbr”是指:对甲氧基苄基溴;
[0149]“pmb”是指:对甲氧基苄基;
[0150]“momcl”是指:氯甲基甲基醚;
[0151]“mom”是指:甲氧基甲基;
[0152]“eomcl”是指:氯甲基乙基醚;
[0153]“eom”是指:乙氧基甲基;
[0154]“tbu”是指:叔丁基;
[0155]“bncl”是指:苄基氯;
[0156]“bn”是指:苄基;
[0157]“ac”是指:乙酰基;
[0158]“semcl”是指:2
‑
(三甲基硅烷基)乙氧甲基氯;
[0159]“sem”是指:2
‑
(三甲基硅烷基)乙氧甲基;
[0160]“trtcl”是指:三苯基氯甲烷;
[0161]“trt”是指:三苯基甲基;
[0162]“dhp”是指:3,4
‑
二氢
‑
2h
‑
吡喃;
[0163]“thp”是指:2
‑
四氢吡喃基;
[0164]“thf”是指:四氢呋喃;
[0165]“2
‑
methf”是指:二甲基四氢呋喃;
[0166]“mtbe”是指:甲基叔丁基醚;
[0167]“acn”是指:乙腈;
[0168]“tea”是指:三乙胺;
[0169]“tfa”是指:三氟乙酸;
[0170]“ipa”是指:异丙醇;
[0171]“tempo”是指:2,2,6,6
‑
四甲基哌啶
‑1‑
氧化物;
[0172]“dcm”是指:二氯甲烷;
[0173]“tlc”是指:薄层色谱法;
[0174]“hnmr”是指:氢核磁共振波谱法;
[0175]“dmso”是指:二甲基亚砜;
[0176]“hplc”是指:高效液相色谱法;
[0177]“pe”是指:石油醚;
[0178]“ea”是指:乙酸乙酯;
[0179]“lc
‑
ms”是指:液相色谱
‑
质谱联用。
[0180]
在本发明中,催化剂包括常规意义的能够改变反应速率的物质,还包括在反应中起氧化还原作用或者起酸碱作用的物质。
[0181]
在本发明中,在没有指明的情况下,反应是在常压下进行的。
[0182]
在本发明中,反应温度是指反应过程中所达到的最高温度。反应的全部过程或反应的部分过程在该最高温度下进行。
[0183]
在本发明中,在提及“比例为”,例如溶剂∶式1化合物∶式2m化合物∶式3化合物∶碱=5.8l∶1.1摩尔∶1摩尔∶3摩尔∶2.5摩尔时,这种表述是指反应过程中各个物料的进料量的比例关系,即进料1摩尔的式2m化合物,相应地,溶剂、式1化合物、式3化合物和碱的进料量分别是5.8l、1.1摩尔、3摩尔和2.5摩尔,如果式2m化合物的进料量增加或减少的话,其他物料的进料量成比例地增加或减少。每一种物料的进料量是反应过程中该物料的进料量的总和,例如如果某种物料一次性进料,进料量就是该一次性进料的量,如果某种物料分多次进料,进料量就是该若干次的进料量的总和。比例所提及的各个物料可以同时或分开进料。
[0184]
在本发明中,所述的化学结构式代表了该式所表示的化合物的游离碱或游离酸形式,以及水合物、溶剂化物、酸式盐、碱式盐、以及其组合的形式,例如下式a可以包括式a的游离碱形式,以及式a的各种复合物,例如含有1分子三氟乙酸与1分子异丙醇的复合物(如下式b所示):
[0185][0186]
本发明提供一种式7抗肿瘤化合物的合成方法,包括如下步骤:
[0187][0188]
(a)于有机溶剂中,将式1化合物与式2m化合物在碱的作用下发生芳香亲核取代反应得到式im化合物,式im化合物经分离或者不经分离,与式3化合物继续发生芳香亲核取代反应,例如与式3化合物在碱性条件下继续发生芳香亲核取代反应,得到式4m化合物;
[0189]
(b)于有机溶剂中,将式4m化合物在催化剂的作用下发生还原反应,得到式5m化合物;
[0190]
(c)于有机溶剂中,将式5m化合物在酸的作用下关环,得到式6m化合物;
[0191]
(d)于有机溶剂中,将式6m化合物脱去氨基保护基,得到式7抗肿瘤化合物;
[0192]
反应式如下:
[0193][0194]
其中,x1和x2分别选自卤素;p1为氨基保护基,优选地p1选自boc、cbz、tos、fmoc、pmb、mom、eom、tbu、bn、ac、sem、trt或thp,例如boc、cbz、tos、fmoc、pmb、trt或thp;p1与所在环上的任一n原子相连,优选地p1与所在环上的甲基邻位的n原子相连。
[0195]
在一些实施例中,式2m化合物包括如下式2化合物和式2m化合物:
[0196]
优选式2m化合为式2化合物。
[0197]
式im化合物包括如下式i化合物和式im化合物:
[0198]
优选式im化合物为式i化合物。
[0199]
式4m化合物包括如下式4化合物和式4m化合物:
[0200]
优选式4m化合物为式4化合物。
[0201]
式5m化合物包括如下式5化合物和式5m化合物:
[0202]
优选式5m化合物为式5化合物。
[0203]
式6m化合物包括如下式6化合物和式6m化合物:
[0204]
优选式6m化合物为式6化合物。
[0205]
优选地,p1选自trt或thp。
[0206]
在一些实施例中,式4化合物的合成方法为步骤(a):于有机溶剂中,将式1化合物与式2化合物在碱的作用下发生芳香亲核取代反应得到式i化合物,式im化合物不经分离,与式3化合物继续发生芳香亲核取代反应,例如与式3化合物在碱性条件下继续发生芳香亲核取代反应,得到式4化合物。
[0207]
本发明合成式4化合物时,无需将式1化合物与式2化合物反应后的产物i进行分
离,而直接与式3化合物进行反应,简化了工艺,且提高了收率。
[0208]
在一些实施例中,步骤(a)中,有机溶剂选自2
‑
甲基四氢呋喃、乙腈、四氢呋喃和甲苯中的一种或几种。
[0209]
优选地,有机溶剂选自2
‑
甲基四氢呋喃或乙腈。
[0210]
在一些实施例中,步骤(a)中,碱选自氢化钠、氢氧化钠、碳酸铯、三乙烯二胺、叔丁醇钠、叔丁醇钾、双三甲基硅基胺基锂和六甲基二硅基氮烷钠中的一种或几种。
[0211]
在一些实施例中,步骤(a)中所述地碱性条件,可以不额外加入碱,以吗啉作碱,也可以额外加入碱性试剂,如氢化钠、氢氧化钠、碳酸铯、三乙烯二胺、叔丁醇钠、叔丁醇钾、双三甲基硅基胺基锂和六甲基二硅基氮烷钠中的一种或几种。在一些实施例中,步骤(a)中,碱为氢化钠,溶剂为2
‑
甲基四氢呋喃。
[0212]
在一些实施例中,步骤(a)中,式1化合物与式2化合物发生芳香亲核取代反应的条件为:在40℃至70℃下反应3小时至18小时;例如,在40℃至60℃下反应3小时至8小时。
[0213]
在一些实施例中,步骤(a)中,与式3化合物继续发生芳香亲核取代反应的条件为:回流反应8小时至16小时;例如,回流反应8小时至12小时;或者例如,回流反应13小时至16小时。
[0214]
在一些实施例中,步骤(a)中,式1化合物、式2化合物与式3化合物的摩尔比为(1至1.2)∶1∶3。
[0215]
在一些实施例中,步骤(a)中,还可以加入硫酸镁、分子筛或活性碳作催化剂。
[0216]
特别地,加入硫酸镁作催化剂,硫酸镁的加入量为0.2至3摩尔当量的式2化合物;例如,硫酸镁的加入量为2至3摩尔当量的式2化合物;或者例如,硫酸镁的加入量为1至3摩尔当量的式2化合物。
[0217]
进一步地,步骤(a)反应结束后,对式4化合物进行提纯,提纯操作可采用本领域常规的方法进行即可。
[0218]
本发明还提供另一种合成式4化合物的方法,包括如下步骤:
[0219]
于有机溶剂中,将式1化合物与式2化合物在碱的作用下发生芳香亲核取代反应,得到式i化合物;
[0220]
于有机溶剂中,将式i化合物与上述式3化合物在碱的作用下发生芳香亲核取代反应,得到式4化合物;
[0221]
反应式如下:
[0222][0223]
其中,x1和x2分别选自卤素;p1为氨基保护基,优选地p1选自boc、cbz、tos、fmoc、pmb、mom、eom、tbu、bn、ac、sem、trt或thp,例如boc、cbz、tos、fmoc、pmb、trt或thp。
[0224]
在该方法中,有机溶剂选自2
‑
甲基四氢呋喃、乙腈、四氢呋喃和甲苯中的一种或几种。碱选自氢化钠、氢氧化钠、碳酸铯、三乙烯二胺、叔丁醇钠、叔丁醇钾、双三甲基硅基胺基
锂和六甲基二硅基氮烷钠中的一种或几种。
[0225]
在一些实施例中,式4m化合物的合成步骤和反应条件如上述式4化合物的合成方法所述,区别在于将式2化合物替换为式2m化合物,将式i化合物替换为式im化合物。
[0226]
在一些实施例中,式5化合物的合成方法为步骤(b):于有机溶剂中,将式4化合物在催化剂的作用下发生还原反应,得到式5化合物。
[0227]
在一些实施例中,步骤(b)中,有机溶剂选自甲醇、乙醇、异丙醇、四氢呋喃、2
‑
甲基四氢呋喃、醋酸和乙腈中一种或几种。
[0228]
在一些实施例中,步骤(b)中,催化剂选自铁粉、氧化铂、pt/c、pd(oh)2/c、rh/c或pd/c。
[0229]
在一些实施例中,pt/c和rh/c的质量分数为5%;pd/c的质量分数为5%、10%;pd(oh)2/c的质量分数为10%、20%。
[0230]
当催化剂为铁粉时,铁粉的加入量为10至18摩尔当量的式4化合物;例如,铁粉的加入量为14至18摩尔当量的式4化合物。
[0231]
特别地,步骤(b)中还加入氯化铵,氯化铵的加入量为0.4至0.6摩尔当量的式4化合物;溶剂为乙醇与四氢呋喃的混合溶剂;例如,乙醇与四氢呋喃的体积比为(6至10)∶10。特别地,步骤(b)中还加入少量的水,水与四氢呋喃的体积比为(0.2至0.5)∶10。进一步地,发生还原反应的条件为:回流反应15小时至24小时。
[0232]
当催化剂为氧化铂时,发生还原反应的条件为:于氢气氛围(例如在0.5
‑
0.8mpa下)中,在35℃至80℃下反应20小时至70小时。例如,发生还原反应的条件为:于氢气氛围中,在35℃至50℃下反应40小时至50小时。
[0233]
进一步地,步骤(b)反应结束后对式5化合物进行提纯,提纯操作可采用本领域常规的方法进行即可。
[0234]
在一些实施例中,式5m化合物的合成步骤和反应条件如上述式5化合物的合成方法所述,区别在于将式4化合物替换为式4m化合物。
[0235]
在一些实施例中,式6化合物的合成方法为步骤(c):于有机溶剂中,将式5化合物在酸的作用下关环,得到式6化合物
[0236]
在一些实施例中,步骤(c)中,酸选自三氟乙酸、甲磺酸、对甲苯磺酸、甲酸、乙酸和盐酸中的一种或几种。有机溶剂选自二氯甲烷、甲醇、乙醇和异丙醇中的至少一种。
[0237]
特别地,酸选自三氟乙酸,有机溶剂为异丙醇。
[0238]
在一些实施例中,步骤(c)中,式5化合物与酸的物质的量比为1∶(1.5至2)。
[0239]
在一些实施例中,步骤(c)具体为:式5化合物于异丙醇中,在三氟乙酸的作用下回流反应1小时至3小时;例如,反应时间为1.5小时至3小时。
[0240]
进一步地,步骤(c)反应结束后对式6化合物进行提纯,即将反应液抽滤,滤饼用异丙醇淋洗,干燥,得到提纯后的式6化合物。
[0241]
在一些实施例中,式6m化合物的合成步骤和反应条件如上述式6化合物的合成方法所述,区别在于将式5化合物替换为式5m化合物。
[0242]
在一些实施例中,式7化合物的合成方法为步骤(d):于有机溶剂中,将式6化合物脱去氨基保护基,得到式7抗肿瘤化合物。
[0243]
在一些实施例中,步骤(d)中,有机溶剂选自甲苯、二氯甲烷、乙腈、四氢呋喃、甲
醇、乙醇和异丙醇中的一种或几种。特别地,有机溶剂为甲苯。
[0244]
在一些实施例中,步骤(d)中,还可以加入催化剂,催化剂选自甲酸、乙酸、盐酸、甲磺酸和三氟乙酸中的至少一种。特别地,催化剂选自三氟乙酸。
[0245]
进一步地,步骤(d)中还加入醇或酚,醇选自甲醇和/或乙醇;酚选自苯酚和/或对甲氧基苯酚。进一步地,例如,醇或酚的加入量为1至2摩尔当量的式6化合物。
[0246]
进一步地,步骤(d)反应结束后对式7化合物进行提纯,提纯操作可采用本领域常规的方法进行即可。
[0247]
在一些实施例中,将式6化合物替换为式6m化合物合成式7化合物的合成步骤和反应条件如上述式7化合物的合成方法所述。
[0248]
在一些实施例中,式7化合物还可以通过式5m化合物直接一步合成得到。反应条件为:于有机溶剂中,将式5m化合物在酸的作用下发生反应,得到式7化合物。例如,在三氟乙酸的作用下,回流反应20
‑
40小时。有机溶剂选自甲苯、二氯甲烷、乙腈、2
‑
甲基四氢呋喃、四氢呋喃、甲醇、乙醇和异丙醇中的一种或几种。
[0249]
在一些实施例中,式7抗肿瘤化合物的合成方法还包括合成式2m化合物的步骤:
[0250]
(i)于溶剂中,将式2
‑
1化合物与硝酸发生硝化反应,得到式2
‑
2化合物;
[0251]
(ii)于有机溶剂中,将式2
‑
2化合物与氨基保护试剂发生氨基保护反应,得到式(2
‑
3)m化合物;
[0252]
(iii)于有机溶剂中,将式(2
‑
3)m化合物脱去邻苯二甲酰基,得到式2m化合物;
[0253]
反应式如下:
[0254][0255]
其中,氨基保护试剂为boc2o、cbzcl、toscl、fmoccl、pmbbr、momcl、eomcl、叔丁醇、异丁烯、bncl、乙酸酐、semcl、trtcl或dhp,例如boc2o、cbzcl、toscl、fmoccl、pmbbr、trtcl或dhp;p1选自boc、cbz、tos、fmoc、pmb、mom、eom、tbu、bn、ac、sem、trt或thp,例如boc、cbz、tos、fmoc、pmb、trt或thp;p1与所在环上的任一n原子相连,优选地p1与所在环上的甲基邻位的n原子相连。优选地,氨基保护试剂为trtcl或dhp;p1选自trt或thp。
[0256]
在一些实施例中,式(2
‑
3)m化合物包括如下式2
‑
3化合物和式(2
‑
3)m化合物:
[0257][0258]
在一些实施例中,步骤(i)中的溶剂如浓硫酸、醋酸酐、醋酸;
[0259]
在一些实施例中,步骤(i)发生硝化反应的条件为:于浓硫酸中,10℃至20℃下反应0.5小时至2小时。。
[0260]
进一步地,式2
‑
1化合物与浓硫酸的质量体积比为100g:(150至250)ml。
[0261]
进一步地,硝化反应结束后,得到的式2
‑
2化合物容易过滤,且在四氢呋喃,二氯甲烷,乙腈等溶剂中溶解性较好,使得反应溶剂的选取更加多样化。
[0262]
在一些实施例中,步骤(ii)根据氨基保护试剂的性质设置不同的反应条件。特别地,当氨基保护试剂为trtcl,步骤(ii)的反应条件为:在碱的作用下,60℃至100℃下反应1小时至48小时;当氨基保护试剂为dhp,步骤(ii)的反应条件为:在对甲苯磺酸或吡啶对甲苯磺酸盐的作用下,60℃至100℃下回流反应3小时至48小时。
[0263]
在一些实施例中,步骤(ii)反应的有机溶剂选自四氢呋喃、2
‑
甲基四氢呋喃、二氯甲烷和乙腈中一种或几种。
[0264]
特别地,当氨基保护试剂为trtcl时,步骤(ii)的反应条件为:在三乙胺的催化下,于乙腈中,回流反应1小时至3小时。
[0265]
在一些实施例中,步骤(iii)脱去邻苯二甲酰基的反应条件为:在水合肼的作用下,于有机溶剂中回流反应1小时至3小时,有机溶剂选自乙醇、四氢呋喃和乙腈中的一种或几种。
[0266]
在一些实施例中,式7抗肿瘤化合物的合成方法还包括式1化合物的合成步骤:
[0267]
于有机溶剂中,将式1
‑
1化合物与式1
‑
2化合物发生偶联反应,得到式1化合物;
[0268]
反应式如下:
[0269][0270]
其中,x1、x2和x3分别选自卤素。优选地,x1、x2和x3分别选自氯。
[0271]
进一步地,式1
‑
1化合物与式1
‑
2化合物发生偶联反应的条件为:在乙酰丙酮铁的作用下,于
‑
70至
‑
60℃下进行反应。
[0272]
本发明还提供制备式7抗肿瘤化合物的中间体,具有以下结构式:
[0273]
[0274]
优选地,所述中间体为以下结构:
[0275][0276]
其中,x1和x2分别选自卤素;
[0277]
p1选自trt或thpp1与所在环上的任一n原子相连,优选地p1与所在环上的甲基邻位的n原子相连。
[0278]
本发明还提供制备式7抗肿瘤化合物的中间体,其特征在于,具有以下结构式:
[0279][0280]
优选为以下结构:
[0281][0282]
其中,p1选自trt或thp;
[0283]
该式6m为含有1分子三氟乙酸与1分子异丙醇的复合物。
[0284]
以下通过实施例对本发明做进一步阐述,然而本发明的保护范围不受这些实施例的限制。
[0285]
本发明的实施例中所使用的原料均为市销产品,纯度为化学纯。
[0286]
实施例1
[0287]
式1
′
化合物的合成
[0288][0289]
配制(2
‑
氯苯基)氯化镁的thf溶液:将(2
‑
氯苯基)氯化镁(27.34mol)配成thf溶液(50.44l,0.542m),在
‑
5至5℃隔氧保温备用。
[0290]
配制4,6
‑
二氯烟酰氯的thf溶液:将4,6
‑
二氯烟酰氯(3.85kg,18.32mol)加入thf(25.0l)和乙酰丙酮铁(193.16g,0.55mol,0.03eq)配成溶液(28.0l,0.651m),备用。
[0291]
配制稀盐酸:在100l反应釜中加入浓盐酸2.8l和水5.6l,控制温度为5至15℃,制备好淬灭用。
[0292]
设置微反应器的反应温度为
‑
70至
‑
60℃,将(2
‑
氯苯基)氯化镁的thf溶液和4,6
‑
二氯烟酰氯的thf溶液,经微反应器反应得到(2
‑
氯苯基)(4,6
‑
二氯吡啶
‑3‑
基)甲酮的反应液,将反应液导入配制好的稀盐酸中淬灭。分液,得到黄色清澈有机相,浓缩。向浓缩残留物中加入mtbe(17.5l),分液;将得到的有机相浓缩除去溶剂后加入2l正庚烷,得到棕黄色粗品,粗品加入3.3l乙醇和9.9l正庚烷,加热至60摄氏度溶清后降温至5至10℃,过滤,滤饼干燥得到2.78kg,淡黄色粉末,收率:53.3%。
[0293]1hnmr(400mhz,dmso
‑
d6)δ(ppm):8.61
‑
8.57(m,1h),8.02(s,1h),7.70
‑
7.62(m,3h),7.55
‑
7.51(m,1h)。
[0294]
式1
″
化合物的合成
[0295]
步骤1:(6
‑
溴吡啶
‑3‑
基)(2
‑
氯苯基)甲醇的合成
[0296][0297]
向2l四口烧瓶中加入无水四氢呋喃(500ml)和2,5
‑
二溴吡啶(100.0g,0.42mol,1.0eq),搅拌下冰水浴降温至2℃,滴加异丙基氯化镁(210.5ml,2.0m,0.42mol,1.0eq),控制温度不超过10℃,约0.5h滴完。室温(20℃)搅拌1h,再用冰水浴降温至10℃,滴加2
‑
氯苯甲醛(62.3g,0.443mol,1.05eq)的四氢呋喃(200ml)溶液,约0.5h滴完。10℃搅拌2h,tlc显示反应结束。向反应体系中加入饱和氯化铵水溶液(300ml),搅拌10分钟后,分液,有机相浓缩得黄色油状物。水相用乙酸乙酯萃取(1.0l
×
2),与前面所得的黄色油状物合并,水洗(500ml),饱和食盐水洗涤(500ml),无水na2so4干燥,浓缩得到棕色油状物(140g粗品)。
[0298]
步骤2:(6
‑
溴吡啶
‑3‑
基)(2
‑
氯苯基)甲酮的合成
[0299][0300]
将(6
‑
溴吡啶
‑3‑
基)(2
‑
氯苯基)甲醇(140g粗品)于dcm(1.3l)中,加入tempo
(1.51g,9.4mmol)和nabr(1.92g,18.8mmol)。冰水浴降温至3℃,滴加用nahco3(45.0g)中和的naclo水溶液(1.34mol/l,600l,0.71mol),滴加过程中温度不超过20℃。滴完后搅拌10分钟,tlc显示反应结束。分液,水相用dcm萃取(1.0l),合并有机相,水洗(1.0l),饱和食盐水洗涤(1.0l),无水na2so4干燥,浓缩后得到黄色油状物,粗品用(150ml甲基叔丁基醚/500ml石油醚)打浆后得到黄色固体(50.3g,产率:两步39.7%)。
[0301]
步骤3:(6
‑
溴
‑4‑
碘
‑
吡啶
‑3‑
基)(2
‑
氯苯基)甲酮的合成
[0302][0303]
氮气保护下,向2l四口烧瓶中加入四甲基哌啶锂/氯化镁溶液(281ml,1.5mol/l,0.43mol,2.5eq),干冰/乙醇浴降温至
‑
65℃,滴加(6
‑
溴吡啶
‑3‑
基)(2
‑
氯苯基)甲酮(50.0g,0.17mol,1.0eq)的四氢呋喃(50ml)溶液,约0.5h加毕。然后升温至
‑
45℃搅拌1h,再降温至
‑
65℃,滴加i2(129.3g,0.51mol,3.0eq)的四氢呋喃(400ml)溶液,约1h加毕。搅拌20分钟后,tlc显示反应结束。向反应体系中加入饱和氯化铵水溶液(500ml)和饱和nahso3水溶液(500ml),搅拌15分钟,过滤,不溶物用乙酸乙酯洗涤(500ml
×
2),合并滤液,分液,水相用乙酸乙酯萃取(1.0l
×
2),合并所有有机相,水洗(800ml),饱和食盐水洗涤(800ml),无水na2so4干燥,浓缩得到黄色固体,用甲基叔丁基醚(500ml)/石油醚(500ml)打浆,干燥后得到黄色固体(30g,产率:41.8%)。
[0304]
实施例2
[0305]
化合物2
′
的合成
[0306]
步骤1:式2
‑
2化合物的合成
[0307][0308]
于四口反应瓶中加入浓硫酸(7.5l)搅拌均匀,冰水浴中体系温度降至20℃以下,分批次加入式2
‑
1化合物(3590g,15.80mol,1.0eq),体系温度控制10至20℃,向反应体系中缓慢滴加质量分数为63%的浓硝酸(1896.4g,18.96mol,1.2eq)。滴加完毕,撤去冰水浴,10至20℃反应1小时。tlc(dcm∶meoh=20∶1)检测原料转化完。将反应液缓慢倒入36kg冰水中,抽滤,抽干后再将滤饼加入50l广口桶中,加入36l水,搅拌半小时,抽滤,滤饼用水(6l)漂洗。滤饼干燥至恒重(约72小时)得式2
‑
2化合物,类白色固体(4160g,收率:96.7%,hplc纯度95.5%)。
[0309]1hnmr(400mhz,dmso
‑
d6)δ(ppm):14.23(br,1h),8.08
‑
7.97(m,4h),2.64(s,3h).
[0310]
步骤2:式2
‑3′
化合物的合成
[0311][0312]
于反应釜中加入acn(38l),搅拌下加入式2
‑
2化合物(4140g,15.21mol,1.0eq),tea(1847g,18.25mol,1.2eq),搅拌均匀,加入三苯基氯甲烷(4664g,16.73mol,1.1eq)。体系升温至回流(82℃)2小时,反应完全。降至25℃,抽滤,滤饼用acn淋洗2次(4l*2次)。滤干后滤饼用30l水打浆2小时,抽滤,滤饼用水漂洗2次(4l*2次),滤干后滤饼于50℃鼓风干燥烘箱中干燥24小时,得式2
‑3′
化合物,白色固体(6920g,hplc:99.7%,收率:88.4%)。
[0313]1hnmr(400mhz,cdcl3)δ(ppm):7.97
‑
7.92(m,2h),7.82
‑
7.78(m,2h),7.41
‑
7.33(m,9h),7.22
‑
7.19(m,6h),2.08(s,3h).
[0314]
步骤3:式2
′
化合物的合成
[0315][0316]
将thf(55l)平均分在4个20l四口瓶中,搅拌下均分别加入式2
‑3′
化合物(6900g,13.41mol,1.0eq),质量分数为85%的n2h4·
h2o(1580g,26.82mol,2.0eq),升温至回流,3小时反应完全。自然降温,抽滤,滤饼用thf漂洗两次(2l*2次)。滤液于50℃水浴减压浓缩除去24l thf,剩余滤液转移至50l反应釜中,升温至回流,缓慢加入95%乙醇(36l),黄绿色固体缓慢析出,搅拌2小时。降至室温析晶搅拌过夜。抽滤,滤饼用2l 95%乙醇淋洗一次,滤饼干燥得式2
′
化合物,黄绿色粉末状固体(4480g,hplc:100.0%,收率:86.9%)。
[0317]1hnmr(400mhz,cdcl3)δ(ppm):7.38
‑
7.36(m,9h),7.25
‑
7.22(m,6h),5.43(s,2h),2.46(s,3h).
[0318]
化合物2
″
的合成
[0319]
步骤1:2
‑
(5
‑
甲基
‑4‑
硝基
‑1‑
(四氢
‑
2h
‑
吡喃
‑2‑
基)
‑
1h
‑
吡唑
‑3‑
基)异吲哚啉
‑
1,3
‑
二酮的合成
[0320][0321]
向反应瓶中加入无水乙腈(270mol),对甲苯磺酸(1.72g,0.01mol,0.1eq),吡啶
(0.79g,0.01mol,0.1eq),升温至50℃搅拌2小时。加入式2
‑
2化合物(27.2g,0.1mol,1.0eq),升温至回流,1小时后滴加dhp(16.8g,0.2mol,2.0eq)。回流约20小时后,将反应液浓缩,残留物加入乙酸乙酯(90ml)打浆1小时,过滤,滤饼烘干得式2
‑
3〞化合物28.6g,收率=80.3%。
[0322]1hnmr(400mhz,dmso
‑
d6)δ(ppm):8.08
‑
7.98(m,4h),5.76
‑
5.71(m,1h),4.01
‑
3.75(m,2h),2.76(s,3h),2.30
‑
2.16(m,1h),1.98
‑
1.94(m,2h),1.72
‑
1.67(m,1h),1.57
‑
1.56(m,2h).
[0323]
步骤2:5
‑
甲基
‑4‑
硝基
‑1‑
(四氢
‑
2h
‑
吡喃
‑2‑
基)
‑
1h
‑
吡唑
‑3‑
胺的合成
[0324][0325]
向反应瓶中加入式2
‑3″
化合物(569.4g,1.6mol,1.0eq),thf(5.7l),升温至回流,滴加水合肼(85%)(141.1g,2.4mol,1.5eq),约半小时滴完。继续回流反应约8小时,tlc(pe∶ea=2∶1)反应完全,降至20
‑
25℃,抽滤,滤液浓缩得303g粗品。粗品加入95%乙醇3l,升温至回流,降至室温,过滤,得式2〞化合物,黄绿色固体。收率=62.5%。
[0326]1hnmr(400mhz,dmso
‑
d6)δ(ppm):6.15(s,2h),5.42
‑
5.38(m,1h),3.91
‑
3.87(m,1h),3.69
‑
3.65(m,1h),2.58(s,3h),2.17
‑
1.98(m,1h),1.98
‑
1.93(m,1h),1.81
‑
1.76(m,1h),1.66
‑
1.52(m,3h).
[0327]
实施例3
[0328]
式4
′
化合物[(2
‑
氯苯基)(4
‑
((5
‑
甲基
‑4‑
硝基
‑1‑
三苯甲基
‑
1h
‑
吡唑
‑3‑
基)氨基)
‑6‑
吗啉代吡啶
‑3‑
基)甲酮]的合成
[0329]
步骤1:(6
‑
氯
‑4‑
((5
‑
甲基
‑4‑
硝基
‑1‑
三苯甲基
‑
1h
‑
吡唑
‑3‑
基)氨基)吡啶
‑3‑
基)(2
‑
氯苯基)甲酮的合成
[0330][0331]
将nah(10.0g)加入至2
‑
methf(600ml)中,温度升至50℃后加入2
′
化合物(38.4g,0.1mol),搅拌1小时后加入式1
′
化合物(28.5g,0.1mol),反应6
‑
8小时反应完全,降温至20
‑
25℃后,滴加醋酸(9g),加水200ml搅拌分层,有机相浓缩至约100ml后,加入乙醇(300ml),回流4
‑
6小时后,关闭加热,冷至20
‑
25℃后过滤,烘干得式i
′
化合物,黄色固体41.4g,收率=65.4%,直接用于下一步。
[0332]
步骤2:(2
‑
氯苯基)(4
‑
((5
‑
甲基
‑4‑
硝基
‑1‑
三苯甲基
‑
1h
‑
吡唑
‑3‑
基)氨基)
‑6‑
吗啉代吡啶
‑3‑
基)甲酮的合成
[0333][0334]
将式i
′
化合物(41.4g),吗啉(17.1g),2
‑
methf(400ml)加入至反应瓶中,加热至80℃反应12
‑
16小时,反应液浓缩至约200ml后,回流下加入乙醇(300ml),回流1小时后冷却至20
‑
25℃,过滤,烘干得式4
′
化合物,黄色固体41g,收率=91.7%。
[0335]
实施例4
[0336]
步骤1:式4
″
化合物(2
‑
氯苯基)(4
‑
((5
‑
甲基
‑4‑
硝基
‑1‑
(四氢
‑
2h
‑
吡喃
‑2‑
基)
‑
1h
‑
吡唑
‑3‑
基)氨基)
‑6‑
吗啉代吡啶
‑3‑
基甲酮的合成
[0337][0338]
将质量分数为60%的钠氢(87.36g,2.18mol)置于10l三口瓶,加入2
‑
methf(4.3l)体系乳白色浑浊,45
‑
50℃体系搅拌1小时,分批加入式2〞化合物(197.4g,0.87mol),体系由黄色变为棕色,搅拌1小时后,加入式1
′
化合物(231.5g,0.81mol),45
‑
50℃下反应2.5小时,hplc中控,反应结束,温度降至12℃,分批加入醋酸(78.61g,1.31mol),内温升至18℃,再加入吗啉(228.3g,2.62mol),90℃反应15小时,hplc中控反应结束,降温至20
‑
25℃,加入2l水,分液,有机相浓缩,加入1l乙酸乙酯打浆2
‑
4小时,过滤,滤饼鼓风干燥,得式4〞化合物,黄色固体218g,收率51.2%。
[0339]
步骤2:式7化合物4
‑
(5
‑
(2
‑
氯苯基)
‑3‑
甲基
‑
2,10
‑
二氢吡唑[4,3
‑
b]吡啶并[4,3
‑
e][1,4]二氮杂卓
‑8‑
基)吗啉的合成
[0340][0341]
将4”(1g,1.90mmol)溶于醋酸60ml,加入水6ml,300mgpd/c,氢气下反应24小时,过滤,母液浓缩,加入水50ml,大量黄色固体析出,过滤烘干得720mg的式6〞,收率80%。
[0342]
将6”(1g,2.09mmol)溶于甲醇,加入对甲苯磺酸(0.43g,2.50mmol),室温搅拌16小时,升温至55℃,补加对甲苯磺酸(0.28g,1.6mmol),反应3小时,浓缩,柱层析得产物7黄色固体。
[0343]
分子式:c
20
h
19
cln6o分子量:394.86lc
‑
ms(pos,m/z)=395.22[m h]
.
[0344]
实施例5
[0345]
式7化合物的合成
[0346]
步骤1:式4
′
化合物(2
‑
氯苯基)(4
‑
((5
‑
甲基
‑4‑
硝基
‑1‑
三苯甲基
‑
1h
‑
吡唑
‑3‑
基)氨基)
‑6‑
吗啉代吡啶
‑3‑
基)甲酮的合成
[0347][0348]
向反应瓶加入2
‑
methf(15l),搅拌下加入硫酸镁(500g,4.15mol),质量分数为60%的钠氢(260g,6.50mol),50℃搅拌2小时,分批加入式2
′
化合物(1000g,2.60mol),体系变为棕黄色,搅拌1小时后,体系变为棕色,加入式1
′
化合物(820g,2.86mol)。46
‑
60℃反应3.5小时,hplc中控式1
′
化合物反应完毕。分批加入醋酸(234g,3.90mol),体系变棕色悬浊,加入吗啉(680g,7.80mol),回流反应15小时,hplc中控反应结束,体系冷至15
‑
25℃。加入7.5l水,分液,有机相用无水硫酸镁干燥,过滤,滤液浓缩至剩余约1
‑
2l溶剂,加入5l甲苯,15l乙醇,78℃打浆2小时,降至室温,过滤,干燥得1250g黄色固体,收率70.3%。
[0349]1hnmr(400mhz,dmso
‑
d6)δ(ppm):12.55(s,1h),7.89(s,1h),7.60
‑
7.52(m,2h),7.50
‑
7.49(m,2h),7.42
‑
7.33(m,9h),7.29
‑
7.27(m,6h),7.21(s,1h),3.60
‑
3.50(m,4h),3.70(s,4h),2.07(s,3h).
[0350]
步骤2:式5
′
化合物(4
‑
((4
‑
氨基
‑5‑
甲基
‑1‑
三苯甲基
‑
1h
‑
吡唑
‑3‑
基)氨基)
‑6‑
吗啉代吡啶
‑3‑
基)(2
‑
氯苯基)甲酮的合成
[0351][0352]
(示例1)向20l反应瓶加入水(300ml),乙醇(95%,7.5l),氯化铵(30.30g,0.57mol),铁粉(950g,17mol),55
‑
60℃搅拌1.5小时,加入thf(7.5l),式4
′
化合物(775g,1.13mol),回流反应5小时,补加铁粉(320g,5.66mol),回流反应15小时,hplc中控反应结束。反应液通过硅藻土(1.5kg)热过滤,滤饼用thf(3l)淋洗,滤液浓缩至剩余约5l溶剂,加入5l乙醇,65℃打浆3小时,冷却至室温,过滤,滤饼用4l乙醇淋洗,干燥得590g黄色固体,收率79.6%。
[0353]1hnmr(300mhz,cdcl3)δ(ppm):11.42(s,1h),8.04(s,1h),7.47
‑
7.23(m,19h),7.11
(s,1h),3.67
‑
3.63(m,4h),3.27
‑
3.24(m,4h),1.59(s,3h).
[0354]
(示例2)将式4
′
化合物(6.8g,10.0mmol),thf(150ml)加入高压釜中,加入氧化铂(700mg),通入0.5
‑
0.8mpa氢气,40℃反应46小时,hplc检测反应完全。反应液过滤,滤液浓缩至剩余约50ml,回流下滴加100ml正庚烷,冷至室温,过滤,烘干得黄色固体5.5g,收率84.6%。
[0355]
步骤3:式6
′
化合物4
‑
(5
‑
(2
‑
氯苯基)
‑3‑
甲基
‑2‑
三苯甲基
‑
2,10
‑
二氢吡唑[4,3
‑
b]吡啶并[4,3
‑
e][1,4]二氮杂卓
‑8‑
基)吗啉异丙醇合物三氟乙酸盐的合成
[0356][0357]
将20l异丙醇加入50l反应釜中,加入式5
′
化合物(1980g,3.03mol),tfa(690g,6.05mol),升温至回流,反应2小时后,hplc中控反应结束,降温至20℃,抽滤,滤饼用异丙醇淋洗至滤液澄清,干燥得黄色固体2300g,收率93.8%。
[0358]1hnmr(400mhz,dmso
‑
d6)δ(ppm):9.24(s,1h),7.51
‑
7.48(m,1h),7.43
‑
7.35(m,9h),7.32
‑
7.29(m,3h),7.17
‑
7.15(m,6h),6.79(s,1h),6.17(s,1h),3.81
‑
3.74(m,1h),3.66
‑
3.64(m,4h),3.36
‑
.3.33(m,4h),1.36(s,3h),1.48
‑
1.32(d,6h,j=6.12hz).
[0359]
步骤4:式7化合物4
‑
(5
‑
(2
‑
氯苯基)
‑3‑
甲基
‑
2,10
‑
二氢吡唑[4,3
‑
b]吡啶并[4,3
‑
e][1,4]二氮杂卓
‑8‑
基)吗啉的合成
[0360][0361]
将式6
′
化合物(58.0g,71.5mmol),甲醇(4.6g,143.7mmol)加入至甲苯(600ml)中,加入三氟乙酸(8.2g,71.9mmol),加热至回流反应17小时,降温至50℃,抽滤,滤饼用甲苯(400ml)打浆(50℃)1小时后,抽滤,100℃鼓风干燥得32g黄色固体,所得固体溶于甲醇(300ml)和水(100ml),氨水(稀释一倍)调ph=7至8,大量黄色固体析出,50℃搅拌1小时,抽滤,滤饼干燥得黄色固体24g,纯度:99.7%,收率:85.4%。
[0362]1h
‑
nmr(dmso
‑
d6,400mhz)11.57(s,1h),8.29(s,1h),7.32
‑
7.47(m,4h),6.89(s,1h),5.95(s,1h),3.61(m,4h),3.31(m,4h),1.97(s,3h).
[0363]
实施例6
[0364]
步骤1:式7化合物4
‑
(5
‑
(2
‑
氯苯基)
‑3‑
甲基
‑
2,10
‑
二氢吡唑[4,3
‑
b]吡啶并[4,3
‑
e][1,4]二氮杂卓
‑8‑
基)吗啉的合成
[0365][0366]
将式5
′
化合物(1g,1.53mmol)溶于dcm(10ml),加入tfa(350mg,3.06mmol)。搅拌2.5小时体系析出黄色固体,加热至回流,体系有黄色浑浊逐渐变为橘黄色澄清。搅拌过夜,共反应24小时,体系棕黑色澄清,加入ea(100ml),饱和碳酸氢钠水溶液,分液,有机相浓缩,20ml ea打浆,过滤得到式7化合物。
[0367]
实施例7
[0368]
本实施例的步骤类似于实施例5的步骤1,将2
‑
methf替换为甲苯,制备得到式4
′
化合物。
[0369]
实施例8
[0370]
本实施例的步骤类似于实施例5的步骤1,将2
‑
methf替换为乙腈,制备得到式4
′
化合物。
[0371]
实施例9
[0372]
本实施例的步骤类似于实施例5的步骤1,将2
‑
methf替换为四氢呋喃,制备得到式4
′
化合物。
[0373]
实施例10
[0374][0375]
将式i
′
化合物(1.3g,2mmol),吗啉(5ml,57mmol),碳酸钾(2.0g,14.5mmol),加入至dcm(10ml)中,加热至40℃,反应2h后,hplc检测反应完毕,关闭加热,过滤,滤液加入etoh(10ml),加热回流2小时后,关闭加热,自然冷至20
‑
25℃,过滤,滤饼烘干得1.2g。
[0376]
实施例11
[0377][0378]
向反应瓶加入2
‑
methf(10l),搅拌下加入60%的钠氢(300g,7.50mol),升温至50℃,分批加入式2
′
化合物(1153.2g,3mol),搅拌1小时后,加入式1
′
化合物(940.3g,3.3mol)的2
‑
methf(2l)溶液。50℃反应6小时,tlc点板式1
′
化合物反应完毕。降温至20℃,缓慢滴加
醋酸(270g,4.5mol),搅拌10分钟后,加入吗啉(784.8g,9mol),回流反应15小时,hplc中控反应结束,关闭加热,体系冷至15
‑
25℃。水洗(10l
×
2),有机相无水na2so4干燥,抽滤,滤饼用2
‑
methf(2l)淋洗。滤液加入200g活性炭,回流1小时,趁热过滤,滤饼用2
‑
methf(1l)淋洗,滤液常压蒸出6l溶剂,再加入etoh(8l),回流1小时,关闭加热,自然降至20
‑
25℃,过滤,滤饼用etoh(1l)淋洗,所得湿品转移至dcm(15l)中,升温至回流,加入活性碳(300g),搅拌0.5小时,趁热过滤,滤饼用dcm(1l)淋洗,滤液常压蒸出2l溶剂,加入etoh(12l),回流搅拌4小时,关闭加热,自然降至20
‑
25℃,抽滤,烘干得黄色固体1234.5g,收率60.1%。
[0379]
实施例12
[0380]
本实施例的步骤类似于实施例5的步骤2(示例2),将氧化铂替换为pt/c,进行反应后制备得到式5
′
化合物,收率达到80%以上。
[0381]
实施例13
[0382]
本实施例的步骤类似于实施例5的步骤2(示例2),将氧化铂替换为pd/c,进行反应后制备得到式5
′
化合物。
[0383]
实施例14
[0384]
本实施例的步骤类似于实施例5的步骤2(示例2),将氧化铂替换为pd(oh)2/c,进行反应后制备得到式5
′
化合物。
[0385]
实施例15
[0386]
本实施例的步骤类似于实施例5的步骤2(示例2),将氧化铂替换为rh/c,进行反应后制备得到式5
′
化合物。
[0387]
实施例16
[0388]
本实施例的步骤类似于实施例5的步骤2,将thf(示例2)替换为thf和异丙醇的混合物,进行反应后制备得到式5
′
化合物。
[0389]
实施例17
[0390]
本实施例的步骤类似于实施例5的步骤3,将三氟乙酸(tfa)替换为对甲苯磺酸,进行反应后制备得到式6
′
化合物。
[0391]
实施例18
[0392]
本实施例的步骤类似于实施例5的步骤3,将三氟乙酸(tfa)替换为甲磺酸,进行反应后制备得到式6
′
化合物。
[0393]
实施例19
[0394]
本实施例的步骤类似于实施例5的步骤4,将甲醇替换为乙醇,进行反应后制备得到式7化合物,收率达到80%以上。
[0395]
实施例20
[0396]
将式6
′
化合物(385g,0.47mol),tfa(138g,1.21mol),etoh(55.66g,1.21mol),加入至dcm(4l)中,加热至回流反应,1.5小时后,关闭加热冷至20
‑
25℃,加入naoh(66g)的水(2l)溶液,有固体析出,搅拌1小时后抽滤。滤饼溶于12l etoh 2l水中,加入活性碳15g,搅拌0.5小时后,抽滤,滤液浓缩至1.5l后,继续回流1小时,冷却至20
‑
25℃后,抽滤,烘干得到114g黄色固体,收率=61.0%。
[0397]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本明保护的范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。