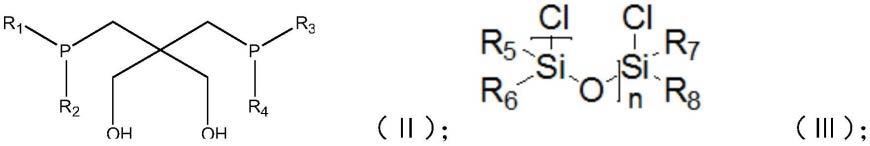
1.本发明涉及化工技术领域,尤其涉及一种新双齿膦配体、其制备方法及其应用。
背景技术:
2.聚酮(pok)是由一氧化碳、烯烃(乙烯、丙烯、苯乙烯)合成的新型绿色聚合物材料,聚酮具有光降解和生物降解特性,并且可进一步化学修饰,其优异和宽泛的性能表现,使它成为一款“天然”的热塑性工程塑料。聚酮主链上的酮基赋予了其优异的可光降解性能和可化学改性的性能,主要原料来源广泛,其中的co除可以从煤造气获得,还可以从含co工业废气中通过净化获得,这使聚酮的合成和应用名副其实地成为新世纪的绿色合成高分子材料。
3.目前,在本领域中,用于制备聚酮的催化剂是已知的。聚合催化剂通常由pd(ii)/双膦配体/酸的体系组成,所用的催化活性较高的双膦配体诸如1,3-双[双(2-甲氧基苯基)膦基]丙烷、((2,2-二甲基-1,3-二氧己环-5,5-二基)双(亚甲基))双(双(2-甲氧基苯基)膦、3,3-双-[双-(2-甲氧基苯基)膦甲基]-1,5-二氧杂-螺[5,5]十一烷等。这些催化剂在聚合过程中一般随反应的进行,催化活性越来越低,分子量分布较宽,对产品性能有一定的影响。
技术实现要素:
[0004]
本发明解决的技术问题在于提供一种新双齿膦配体,其与二价钯盐形成的络合物作为聚酮聚合的主催化剂,可高速聚合且反应过程不变,可有效解决结垢问题。
[0005]
有鉴于此,本技术提供了一种如式(ⅰ)所示的新双齿膦配体,
[0006][0007]
其中,r1~r4独立的选自c1~c10取代的或未取代的烃基;
[0008]
r5~r8独立的选自c1~c20取代的或未取代的烃基;
[0009]
n为1~4。
[0010]
优选的,所述r1~r4选自带供电子的单取代芳烃基或带供电子的多取代芳烃基。
[0011]
优选的,所述r1~r4独立的选自甲氧基苯基、乙氧基苯基或对甲氧基苯基。
[0012]
优选的,所述r5~r8独立的选自c1~c8的未取代的烷基。
[0013]
优选的,所述r5~r8独立的选自异丙基或叔丁基。
[0014]
本技术还提供了所述的新双齿膦配体的制备方法,包括以下步骤:
[0015]
a)将如式(ⅱ)所示的醇类化合物、缚酸剂和相转移催化剂在溶剂中混合,得到混
合液;
[0016]
b)在一定温度下,向混合液中加入如式(ⅲ)所示的二氯化物,反应后得到新双齿膦配体;
[0017][0018]
其中,r1~r4独立的选自c1~c10取代的或未取代的烃基;
[0019]
r5~r8独立的选自c1~c20取代的或未取代的烃基;
[0020]
n为1~4。
[0021]
优选的,所述缚酸剂选自三乙胺、吡啶、碳酸钾、碳酸铵和碳酸钠中的一种或多种,所述相转移催化剂选自苄基三乙基氯化铵、四丁基溴化铵、四丁基氯化铵、四丁基硫酸氢铵和三辛基甲基氯化铵中的一种或多种;所述醇类化合物和所述二氯化物的摩尔比为1:1。
[0022]
优选的,所述反应的温度为80~120℃,所述反应的时间为3~5h。
[0023]
本技术还提供了一种聚酮的制备方法,包括:
[0024]
将一氧化碳和烯烃在主催化剂、助催化剂的甲醇溶液中进行聚合;
[0025]
所述主催化剂为所述的配体或所述的制备方法所制备的配体和二价钯盐的络合物。
[0026]
优选的,所述二价钯盐选自四乙腈三氟甲磺酸钯、硝酸钯、硫酸钯、磺酸钯和醋酸钯中的一种或多种;所述助催化剂选自硝酸、对甲苯磺酸、氨基磺酸、三氟甲基磺酸、三氯乙酸和三氟乙酸中的一种或多种;所述主催化剂和所述助催化剂的摩尔比为(2~10):1。
[0027]
本技术提供了一种新双齿膦配体,其与二价钯盐形成的络合物作为主催化剂,其结构中的硅氧烷的界面张力小,具有表面活性,使其在甲醇界面进行聚合时,相当于对釜壁和搅拌桨表面起到保护作用,且整个聚合过程催化活性不变,因此该配体与二价钯盐形成的络合物作为主催化剂用于聚酮聚合,可实现高速聚合且反应过程不变,有效解决了结垢问题。
具体实施方式
[0028]
为了进一步理解本发明,下面结合实施例对本发明优选实施方案进行描述,但是应当理解,这些描述只是为进一步说明本发明的特征和优点,而不是对本发明权利要求的限制。
[0029]
本发明提供了一种聚酮催化剂新配体结构,用于聚酮聚合反应,可使反应催化活性达到33kg/(g-pd
·
h)以上,反应过程活性不变,分子量分布窄,可解决结垢问题,满足工业化生产。具体的,本技术首先提供了如式(ⅰ)所示的新双齿膦配体,
[0030][0031]
其中,r1~r4独立的选自c1~c10取代的或未取代的烃基;
[0032]
r5~r8独立的选自c1~c20取代的或未取代的烃基;
[0033]
n为1~4。
[0034]
在本发明中,所述的r1、r2、r3、r4表示相同或不同的烃基,它们可以任选取代,这里主要为带取代基的芳烃基,取代基可以为甲氧基、乙氧基或对甲氧基,在本技术中,所述r1、r2、r3、r4优选为甲氧基苯基。
[0035]
所述的r5、r6、r7、r8表示相同或不同的烃基,优选表示相同的烃基,可以为异丙基或叔丁基。n=1~4,优选n=1,主要目的为赋予该配体与二价钯盐络合形成的催化剂具有表面活性,使单体在界面聚合。
[0036]
本技术提供的新双齿膦配体作为主催化剂,其结构的特殊性,硅氧烷的界面张力小,具有表面活性,使其在甲醇界面进行聚合,相当于对釜壁和搅拌桨表面起到保护作用,且整个聚合过程催化活性不变,因此该配体与二价钯盐配合形成催化剂用于聚酮聚合,可实现高速聚合且反应过程不变,有效解决了结垢问题。
[0037]
本技术还提供了所述新双齿膦配体的制备方法,包括以下步骤:
[0038]
a)将如式(ⅱ)所示的醇类化合物、缚酸剂和相转移催化剂在溶剂中混合,得到混合液;
[0039]
b)在一定温度下,向混合液中加入如式(ⅲ)所示的二氯化物,反应后得到新双齿膦配体;
[0040][0041]
其中,r1~r4独立的选自c1~c10取代的或未取代的烃基;
[0042]
r5~r8独立的选自c1~c20取代的或未取代的烃基;
[0043]
n为1~4。
[0044]
在所述新双齿膦配体的制备过程中,所述缚酸剂选自三乙胺、吡啶、碳酸钾、碳酸铵和碳酸钠中的一种或多种,所述相转移催化剂选自苄基三乙基氯化铵(teba)、四丁基溴化铵、四丁基氯化铵、四丁基硫酸氢铵(tbab)和三辛基甲基氯化铵中的一种或多种;所述醇类化合物具体可选自3,3-双-[双-(2-甲氧基苯基)膦甲基-1,5-丙二醇,所述3,3-双-[双-(2-甲氧基苯基)膦甲基-1,5-丙二醇和所述含硅氧的二氯化物的摩尔比为1:1。所述反应的温度为80~120℃,时间为3~5h。
[0045]
所述醇类化合物具体选自3,3-双-[双-(2-甲氧基苯基)膦甲基-1,5-丙二醇时,上
述制备过程中,具体反应过程如下式所示:
[0046][0047]
在本技术中,示例的,含硅氧的二氯化物以1,3-二氯-1,1,3,3-四异丙基-二硅氧烷为例,所述新配体可以用以下方法制备:
[0048]
a)以3,3-双-[双-(2-甲氧基苯基)膦甲基-1,5-丙二醇为原料,将原料、缚酸剂和相转移催化剂加入高沸点溶剂混合,得到二元醇的混合液;
[0049]
b)在一定温度下,向二元醇的混合液中滴加入1,3-二氯-1,1,3,3-四异丙基-二硅氧烷,于滴加温度下保温一段时间,然后进行高温反应,跟踪检测反应终点,经后处理后得到目标配体。
[0050]1h nmr(400mhz,chloroform-d):δ7.32-7.23(m,4h,ch),7.20(ddd,4h,ch),6.86(tt,4h,ch),6.80(ddd,4h,ch),3.73(s,12h,och3),3.71(s,4h,si-och2),1.8(m,2h,si-ch),1.31(s,4h,pch2),0.9(d,12h,ch(ch3)2)。
[0051]
本技术还提供了一种聚酮的制备方法,包括:
[0052]
将一氧化碳和烯烃在主催化剂、助催化剂的甲醇溶液中进行聚合;
[0053]
所述主催化剂为上述方案所述的新双齿膦配体和二价钯盐的络合物。
[0054]
在本发明中,所述的二价钯盐与该新配体络合形成催化剂的方法可以为:二价钯盐与新配体以摩尔比1:1在二氯甲烷或丙酮中室温进行络合,
31
p谱跟踪反应终点,经后处理重结晶得目标催化剂。
[0055]
所述二价钯盐可以为四乙腈三氟甲磺酸钯、硝酸钯、硫酸钯、磺酸钯和醋酸钯,优选醋酸钯。
[0056]
在本发明中,以一氧化碳和一种或多种烯烃混合物为原料,在本发明中所定义的催化剂、助催化剂强酸和醌的甲醇溶液中进行聚酮聚合。
[0057]
所述烯烃化合物主要为乙烯,作为第三组分的烯烃可为丙烯、1-丁烯等a-烯烃,优选丙烯;第三组分主要调节聚酮的熔点,进而有利于后加工,但第三组分的加入会降低催化活性,故考虑生产效率,优选丙烯;丙烯在反应釜中的气体占比优选为20%~60%,具体可为20%、25%、30%、35%、40%、45%、50%、55%、60%。
[0058]
所述的一氧化碳和乙烯的摩尔比为1:(1~1.1),可以为1:1、1:1.05、1:1.1,优选1:1.05。由于一氧化碳与烯烃为严格交替的共聚反应,但是一氧化碳于甲醇中几乎无溶解度,所以一氧化碳选择摩尔量大于乙烯摩尔量,新配体与二价钯盐络合形成的催化剂具有表面活性,可界面进行聚合,降低一氧化碳溶解性差的问题,更优选1:1.05。
[0059]
所述助催化剂为硝酸、对甲苯磺酸、氨基磺酸、三氟甲基磺酸、三氯乙酸和三氟乙
酸中的一种或多种,优选三氟甲磺酸。所述助催化剂与主催化剂的摩尔比优选为(10~2):1,具体可为10:1、9:1、8:1、7:1、6:1、5:1、4:1、3:1、2:1。
[0060]
在本发明提供的制备方法中,所述助催化剂中还含有醌和/或pka<6的酸的阴离子。其中,所述醌优选包括苯醌和/或萘醌,所述苯醌包括但不限于1,2-苯醌(邻苯醌)、1,4-苯醌(对苯醌),所述萘醌包括但不限于1,2-萘醌和/或1,4-萘醌;所述醌与所述催化剂的摩尔比优选为(5~20):1,具体可为5:1、6:1、7:1、8:1、9:1、10:1、11:1、12:1、13:1、14:1、15:1、16:1、17:1、18:1、19:1或20:1,更优选10:1。
[0061]
在本发明中,所述pka<6的酸的阴离子包括但不限于硝酸根、对甲苯磺酸根、氨基磺酸根、三氟甲基磺酸根、三氯乙酸根和三氟乙酸根中的一种或多种;所述阴离子对应的金属盐包括但不限于钠盐、钾盐、镁盐、锌盐和铁盐中的一种或多种,具体可为三氟甲磺酸镁、三氟甲磺酸锌和/或三氟甲磺酸铁。在本发明中,所述金属盐与所述催化剂的摩尔比优选为(2~10):1,具体可为2:1、3:1、4:1、5:1、6:1、7:1、8:1、9:1或10:1,更优选4:1。
[0062]
在本发明中,所述聚合的温度为87℃~93℃,压力为4.0mpa~5.5mpa。
[0063]
本发明涉及到一种聚酮催化剂新配体结构,用于聚酮聚合反应,反应催化活性达到33kg/(g-pd
·
h)以上,反应过程活性不变,分子量分布窄,可解决结垢问题,满足工业化生产需求。
[0064]
为了进一步理解本发明,下面结合实施例对本发明提供的新双齿膦配体、其制备方法及其应用进行详细说明,本发明的保护范围不受以下实施例的限制。
[0065]
以下实施例中的新双齿膦配体的结构如下所示:
[0066][0067]
实施例1
[0068]
在500ml高压反应釜中加入250ml甲醇,主催化剂(新双齿膦配体(r5=r6=r7=r8=-ipr,n=1)与乙酸钯的有机金属络合物)6.23mg,三氟甲磺酸的甲醇溶液2ml(7.5mmol/l),8.1mg对苯醌,9.67mg三氟甲磺酸镁;加入上述物质后,高压釜内充入氮气保压、置换,然后充入丙烯20g,充入co和c2h4质量比为1:1.05的混合气30g,开始升温,设定温度93℃,搅拌速度400r/min,当温度升至93℃时,持续通入co和c2h4质量比为1:1的混合气,维持反应压力为5.5mpa,反应时间4h;
[0069]
反应结束后产品与甲醇分层,在搅拌桨以及釜壁无结垢,从高压釜中直接放出,将所得聚酮产品过滤,用甲醇洗涤,然后真空干燥箱80℃干燥4h,得到的产品量为120g,催化活性为37.73kg/(g-pd
·
h),分子量分布为1.5。
[0070]
实施例2
[0071]
在500ml高压反应釜中加入250ml甲醇,新双齿膦配体(r5=r6=r7=r8=-ipr,n=1)与乙酸钯的有机金属络合物)6.23mg,三氟甲磺酸的甲醇溶液4ml(7.5mmol/l),9.72mg对苯醌,19.34mg三氟甲磺酸镁;
[0072]
加入上述物质后,高压釜内充入氮气保压、置换,然后充入丙烯30g,充入co和c2h4质量比为1:1.05的混合气30g,开始升温,设定温度90℃,搅拌速度400r/min,当温度升至90℃时,持续通入co和c2h4质量比为1:1的混合气,维持反应压力为5.0mpa,反应时间4h;
[0073]
反应结束后产品与甲醇分层,从高压釜中直接放出,将所得聚酮产品过滤,用甲醇洗涤,然后真空干燥箱80℃干燥4h,得到的产品量为112g,催化活性为35.22kg/(g-pd
·
h),分子量分布为1.6。
[0074]
实施例3
[0075]
在500ml高压反应釜中加入250ml甲醇,新双齿膦配体(r5=r6=r7=r8=-tbu,n=1)与乙酸钯的有机金属络合物)8.86mg,三氟甲磺酸的甲醇溶液13.33ml(7.5mmol/l),9.72mg对苯醌,25.79mg三氟甲磺酸镁;
[0076]
加入上述物质后,高压釜内充入氮气保压、置换,然后充入丙烯25g,充入co和c2h4质量比为1:1.05的混合气37.5g,开始升温,设定温度93℃,搅拌速度400r/min,当温度升至93℃时,持续通入co和c2h4质量比为1:1的混合气,维持反应压力为5.5mpa,反应时间4h;
[0077]
反应结束后产品与甲醇分层,从高压釜中直接放出,将所得聚酮产品过滤,用甲醇洗涤,然后真空干燥箱80℃干燥4h,得到的产品量为150g,催化活性为35.73kg/(g-pd
·
h),分子量分布为1.5。
[0078]
实施例4
[0079]
在500ml高压反应釜中加入250ml甲醇,新双齿膦配体(r5=r6=r7=r8=-tbu,n=1)与乙酸钯的有机金属络合物)4.43mg,三氟甲磺酸的甲醇溶液1.33ml(7.5mmol/l),8.1mg对苯醌,16.1mg三氟甲磺酸镁;
[0080]
加入上述物质后,高压釜内充入氮气保压、置换,然后充入丙烯15g,充入co和c2h4质量比为1:1.05的混合气45g,开始升温,设定温度87℃,搅拌速度400r/min,当温度升至87℃时,持续通入co和c2h4质量比为1:1的混合气,维持反应压力为4.5mpa,反应时间6h;
[0081]
反应结束后产品与甲醇分层,从高压釜中直接放出,将所得聚酮产品过滤,用甲醇洗涤,然后真空干燥箱80℃干燥4h,得到的产品量为105g,催化活性为33.02kg/(g-pd
·
h),分子量分布为1.45。
[0082]
以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
[0083]
对所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。