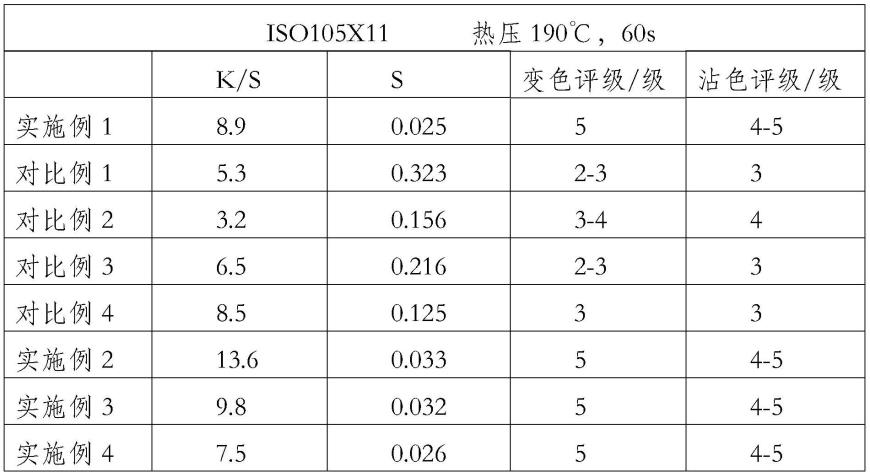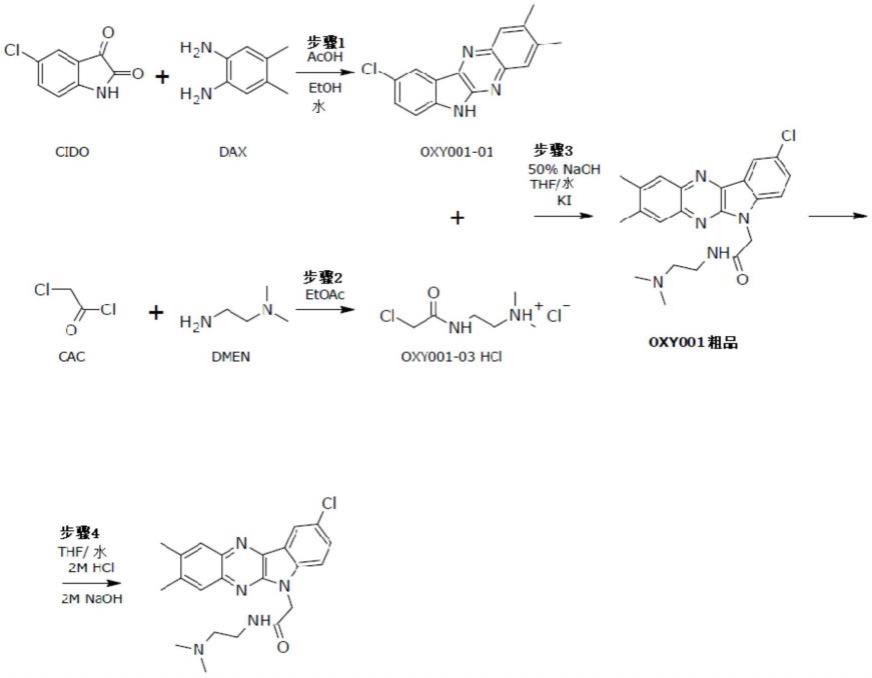
用于制备结晶形式的rabeximod的方法
技术领域
1.本发明涉及一种制备9-氯-2,3-二甲基-6-(n,n-二甲基氨基乙基氨基-2-氧代乙基)-6h-吲哚并-[2,3-b]喹喔啉(rabeximod)的方法,其中所述工艺适合用于大规模合成。该方法的参数稳定,并且该方法适合用于gmp。
背景技术:
[0002]
化合物rabeximod已被描述在欧洲专利申请公开ep1756111a1中,该申请后来以ep1756111b1被授权。rabeximod作为化合物e的制备作为小规模方法被具体描述在ep1756111a1中,但是就如何开发可用于gmp和放大的方法没有任何描述。在小规模实验室方法中以58%的产率制备了rabeximod,但没有公开用于放大的参数。
[0003]
本发明的目的是提供一种适合用于大规模合成、产率高、方法参数稳定且适合用于gmp生产的方法。
技术实现要素:
[0004]
本发明涉及一种制备9-氯-2,3-二甲基-6-(n,n-二甲基氨基乙基氨基-2-氧代乙基)-6h-吲哚并-[2,3-b]喹喔啉(也被称为rabeximod)的新方法,该方法可以放大到大规模和/或工业规模,例如10kg或更高。该方法也可用于较小规模,例如200g至10kg。
[0005]
本发明的更多目标和优点将从以下描述和权利要求中显现。
[0006]
发明描述
[0007]
在inn下被称为“rabeximod”的化合物具有的iupac名称为9-氯-2,3-二甲基-6-(n,n-二甲基氨基乙基氨基-2-氧代乙基)-6h-吲哚并-[2,3b]喹喔啉,并具有以下分子结构。
[0008][0009]
在本技术通篇中,术语“rabeximod”、“rabeximod”或“9-氯-2,3-二甲基-6-(n,n-二甲基氨基乙基氨基-2-氧代乙基)-6h-吲哚并-[2,3b]喹喔啉”可互换使用,并意指任何固体形式或液体形式的该化合物,除非在给定情况下另有说明或暗示。
[0010]
在第一方面,本发明涉及一种制备9-氯-2,3-二甲基-6-(n,n-二甲基氨基乙基氨基-2-氧代乙基)-6h-吲哚并-[2,3-b]喹喔啉(rabeximod)或其盐的方法,该方法适合用于大规模生产/合成,其中该方法包括以下步骤:
[0011]-在足以使吲哚n-h去质子化的碱水溶液和任选的催化剂的存在下,使9-氯-2,3-二甲基-6h-吲哚并[2,3-b]喹喔啉的溶液或悬浮液与2-氯-n-(2-二甲基氨基乙基)乙酰胺或其盐反应得到rabeximod或其盐。
[0012]
在一个实施方案中,存在催化剂。当存在时,催化剂通常是基于碱金属卤素的催化剂,例如ki(碘化钾)。
[0013]
在进一步的实施方案中,将9-氯-2,3-二甲基-6h-吲哚并[2,3-b]喹喔啉或其盐溶解在有机溶剂中。通常,此类溶剂是可水混溶性有机溶剂,例如极性水混溶性有机溶剂,例如极性非质子水混溶性有机溶剂,例如环醚,例如thf。典型的实施方案选自乙腈、乙酸异丙酯、乙酸乙酯、thf和甲苯中的一种或多种,任选地与水混合。
[0014]
在更进一步的实施方案中,将2-氯-n-(2-二甲氨基乙基)乙酰胺或其盐溶解在有机溶剂中,该有机溶剂例如水混溶性有机溶剂,例如极性水混溶性有机溶剂。通常,例如极性非质子水混溶性有机溶剂,例如环醚,例如thf。
[0015]
在该方法的过程中,2-氯-n-(2-二甲基氨基乙基)乙酰胺可以以游离碱或盐的形式使用。在进一步的实施方案中,2-氯-n-(2-二甲基-氨基乙基)乙酰胺是盐,优选盐酸盐。在另一个实施方案中,2-氯-n-(2-二甲基氨基乙基)乙酰胺以游离碱的形式使用。
[0016]
如上所述,该方法涉及使用碱水溶液,并且通常碱水溶液是基于碱金属的碱,例如koh或naoh水溶液。优选地,碱是naoh水溶液,例如50%的naoh水溶液。
[0017]
在进一步的实施方案中,反应在常压下在惰性气体例如氮气或氩气下发生。优选地,惰性气体是氮气。
[0018]
在更进一步的实施方案中,将约1摩尔当量的9-氯-2,3-二甲基-6h-吲哚并[2,3-b]喹喔啉用至少约2体积的碱水溶液去质子化。通常,使用约8当量(相对于9-氯-2,3-二甲基-6h-吲哚并[2,3-b]喹喔啉)的碱水溶液。优选地,将9-氯-2,3-二甲基-6h-吲哚并[2,3-b]喹喔啉和碱水溶液在合适的温度例如约50-65℃下混合,直到形成澄清溶液,例如长达约1小时或更长时间。溶剂例如thf溶解所有的9-氯-2,3-二甲基-6h-吲哚并[2,3-b]喹喔啉是优选的,因为产率增加。
[0019]
优选地,存在催化剂。在一个实施方案中,在剧烈搅拌下添加合适量的催化剂并在合适的温度例如约50-65℃下混合约10至60分钟。合适的催化剂量应足以发挥催化作用,可以为约0.5至1.5摩尔当量。例如,ki通常以约0.7-0.9摩尔当量的量使用。
[0020]
在更进一步的实施方案中,将2-氯-n-(2-二甲基氨基乙基)乙酰胺或其盐添加到9-氯-2,3-二甲基-6fi-吲哚并[2,3-b]喹喔啉在碱水溶液中的溶液,例如thf溶液中,并在合适的温度例如约50-65℃下混合至少约1小时,例如至少约2小时。优选地,2-氯-n-(2-二甲基氨基乙基)乙酰胺的添加量为约1-3摩尔当量,例如约2摩尔当量。
[0021]
可根据已知技术或如本文后面和/或实验部分中所述纯化rabeximod化合物。在一个优选的实施方案中,将rabeximod纯化和分离为游离碱,例如结晶游离碱。
[0022]
在另一方面,本发明涉及rabeximod的结晶游离碱。优选地,rabeximod的结晶游离碱具有259-261℃的熔点。还通过图1和图2中显示的xrpd衍射图、dsc和x射线鉴定了rabeximod的结晶游离碱。分离得到高纯度的rabeximod的结晶游离碱,如通过hplc测量为高于98%,因此在进一步的实施方案中,rabeximod的结晶游离碱是纯度高于98%的分离的游离碱。
[0023]
结晶rabeximod游离碱化合物适合进一步加工成固体口服剂量组合物,用于治疗自身免疫性疾病,例如类风湿性关节炎和/或多发性硬化症。
[0024]
在进一步的实施方案中,本方法包括以下在先步骤:
[0025]-使4,5-二甲基-1,2-苯二胺的溶液或悬浮液与5-氯靛红在酸性条件下在升高的温度直至回流下反应,得到9-氯-2,3-二甲基-6h-吲哚并[2,3-b]喹喔啉或其盐。
[0026]
在更进一步的实施方案中,本方法包括以下在先步骤:
[0027]-使氯乙酰氯的溶液或悬浮液与n,n-二甲基乙二胺反应,得到2-氯-n-(2-二甲基氨基乙基)乙酰胺或其盐。
[0028]
优选地,本发明的用于制备rabeximod的方法包括以下两个在先步骤:
[0029]-使4,5-二甲基-1,2-苯二胺的溶液或悬浮液与5-氯靛红在酸性条件下在升高的温度直至回流下反应,得到9-氯-2,3-二甲基-6h-吲哚并[2,3-b]喹喔啉或其盐,和
[0030]-使氯乙酰氯的溶液或悬浮液与n,n-二甲基乙二胺反应,得到2-氯-n-(2-二甲基氨基乙基)乙酰胺或其盐。
[0031]
在又一方面,本发明涉及一种制备9-氯-2,3-二甲基-6h-吲哚并[2,3-b]喹喔啉的方法,该方法适合用于大规模生产/合成,其中该方法包括以下步骤:
[0032]-使4,5-二甲基-1,2-苯二胺的溶液或悬浮液与5-氯靛红在酸性条件下在升高的温度直至回流下反应,得到9-氯-2,3-二甲基-6h-吲哚并[2,3-b]喹喔啉。
[0033]
如下所述的实施方案独立地为制备rabeximod或其盐的方法和制备9-氯-2,3-二甲基-6h-吲哚并[2,3-b]喹喔啉或其盐的方法的实施方案。
[0034]
在进一步的实施方案中,酸性条件是有机酸,例如c2-c5羧酸,通常是乙酸。
[0035]
在更进一步的实施方案中,在反应前将4,5-二甲基-1,2-苯二胺和5-氯靛红都溶解在酸中。
[0036]
在进一步的实施方案中,酸是过量的,例如至少2体积,例如至少4体积。通常,酸对于4,5-二甲基-1,2-苯二胺为至少4体积过量的,对于5-氯靛红为至少10体积过量的。
[0037]
在更进一步的实施方案中,升高的温度是回流温度。
[0038]
在进一步的实施方案中,4,5-二甲基-1,2-苯二胺或其盐在与5-氯靛红反应之前溶解在酸中,并且5-氯靛红或其盐在与4,5-二甲基-1,2-苯二胺反应之前溶解在酸中。
[0039]
在更进一步的实施方案中,4,5-二甲基-1,2-苯二胺的添加量为约1-3摩尔当量,例如约1-2摩尔当量,例如约1.1摩尔当量。
[0040]
在进一步的实施方案中,5-氯靛红的添加量为约1-3摩尔当量,例如约1-2摩尔当量,例如约1摩尔当量。
[0041]
在更进一步的实施方案中,在回流温度下将溶解的4,5-二甲基-1,2-苯二胺添加到溶解的5-氯靛红中。通常,在回流温度下历经至少约2小时例如2-4小时将4,5-二甲基-1,2-苯二胺添加到5-氯靛红中。
[0042]
在进一步的实施方案中,从反应混合物中蒸馏出酸,并在蒸馏过程中以相似的速率添加另外的酸。通常,反应混合物在蒸馏后在回流温度下搅拌至少约1小时,例如约2小时。
[0043]
在更进一步的实施方案中,9-氯-2,3-二甲基-6h-吲哚并[2,3-b]喹喔啉被纯化和分离为游离碱。
[0044]
另一方面,本发明涉及为固体形式的9-氯-2,3-二甲基-6h-吲哚并[2,3-b]喹喔啉,例如结晶游离碱。
[0045]
又一方面,本发明涉及一种制备2-氯-n-(2-二甲基氨基乙基)乙酰胺或其盐的方法,该方法适合用于大规模生产/合成,其中该方法包括以下步骤:
[0046]-使氯乙酰氯的溶液或悬浮液与n,n-二甲基乙二胺反应,得到2-氯-n-(2-二甲基氨基乙基)乙酰胺或其盐。
[0047]
如下所述的实施方案独立地为制备rabeximod或其盐的方法和制备2-氯-n-(2-二甲基氨基乙基)乙酰胺或其盐的方法的实施方案。
[0048]
在一个实施方案中,将氯乙酰氯溶解在有机溶剂中,该有机溶剂例如有机酯,例如c4至c6酯,例如乙酸乙酯。
[0049]
在进一步的实施方案中,将n,n-二甲基乙二胺溶解在有机溶剂中,该有机溶剂例如有机酯,例如c4至c6酯,例如乙酸乙酯。
[0050]
在更进一步的实施方案中,在反应前将氯乙酰氯和n,n-二甲基乙二胺都溶解。
[0051]
在进一步的实施方案中,将溶液形式的n,n-二甲基乙二胺以保持溶液中的温度低于约30℃的速率添加到溶液形式的氯乙酰氯中。
[0052]
在更进一步的实施方案中,n,n-二甲基乙二胺或其盐在与氯乙酰氯反应之前溶解在溶剂中,并且5-氯乙酰氯或其盐在与n,n-二甲基乙二胺反应之前溶解在溶剂中。
[0053]
在进一步的实施方案中,溶剂相对于氯乙酰氯是过量的,例如至少约2体积,例如至少约4体积。
[0054]
在更进一步的实施方案中,溶剂相对于n,n-二甲基乙二胺是当量或体积过量的,例如以当量比。
[0055]
在进一步的实施方案中,氯乙酰氯的添加量为约1-3摩尔当量,例如约1-2摩尔当量,例如约1摩尔当量。
[0056]
在更进一步的实施方案中,n,n-二甲基乙二胺的添加量为约1-3摩尔当量,例如约1-2摩尔当量,例如约1摩尔当量。
[0057]
在进一步的实施方案中,2-氯-n-(2-二甲基氨基乙基)乙酰胺或其盐被纯化和分离为其盐,例如hcl盐。
[0058]
在一个优选的实施方案中,使用本文所述的方法得到的rabeximod通过以下连续步骤进行纯化:将粗反应产物溶解在水混溶性有机溶剂(例如极性水混溶性有机溶剂,例如极性非质子水混溶性有机溶剂,例如环醚,通常是四氢呋喃)、水和酸(例如氢卤化物,通常是hcl)的混合物中;过滤并加热到35℃以上,优选到约50℃;通过添加碱水溶液(通常为naoh)调节ph到至少9,通常到10-12范围内的值;冷却到18至25℃之间的温度并用水稀释;搅拌至少10小时,通常至少12小时;通常在20至25℃下过滤;以及通常在过滤器上用水混溶性有机溶剂(例如极性水混溶性有机溶剂,例如极性非质子水混溶性有机溶剂,例如环醚,通常是四氢呋喃)和水的混合物洗涤,以提供纯化的rabeximod结晶游离碱。如果需要,药学上可接受的盐可由rabeximod的结晶游离碱的溶液制成。
[0059]
在一个特定的实施方案中,提供了制备如本文所定义的9-氯-2,3-二甲基-6-(n,n-二甲基氨基乙基氨基-2-氧代乙基)-6h-吲哚并-[2,3-b]喹喔啉(rabeximod)或其盐的方法,所述方法包括以下步骤:
[0060]
a)使4,5-二甲基-1,2-苯二胺的溶液或悬浮液与5-氯靛红在酸性条件下在升高的温度直至回流下反应,得到9-氯-2,3-二甲基-6h-吲哚并[2,3-b]喹喔啉或其盐;
[0061]
b)使氯乙酰氯的溶液或悬浮液与n,n-二甲基乙二胺反应,得到2-氯-n-(2-二甲基氨基乙基)乙酰胺或其盐;
[0062]
c)使步骤a)中得到的9-氯-2,3-二甲基-6h-吲哚并[2,3-b]喹喔啉的溶液或悬浮液在足够强以使吲哚n-h去质子化的碱水溶液和任选的催化剂的存在下与步骤b)中得到的2-氯-n-(2-二甲基氨基乙基)乙酰胺或其盐反应,得到rabeximod或其盐;
[0063]
d)使步骤c)中得到的rabeximod或其盐经历一个或多个纯化步骤,优选经历如上文所定义的纯化程序。
[0064]
基于本公开,对于本领域技术人员而言显而易见的是,上述方法中的步骤a)和b)可以(并且通常是)独立地并且以任何顺序或次序进行,包括同时进行。
[0065]
本发明的另一方面涉及可通过本文定义的任何一种方法得到的rabeximod化合物。
[0066]
在本文的实验部分中描述了该方法的其他实施方案,并且每个单独的方法以及每种起始材料构成可以形成实施方案的一部分的实施方案。
[0067]
本文所用的术语“和/或”旨在表示两个备选方案以及单独的备选方案中的每一个。例如,表达式“xxx和/或yyy”意指“xxx和yyy”;“xxx”;或“yyy”,所有三个备选方案都受制于个别实施方案。
[0068]
上述实施方案应被视为是指本文描述的任何一个方面(例如“rabeximod的结晶游离碱”和/或“适合用于大规模合成以制备rabeximod的方法”)以及本文描述的任何一个实施方案,除非指明实施方案涉及本发明的某个方面或某些方面。
[0069]
本文引用的所有参考文献,包括出版物、专利申请和专利,均以引用的方式并入本文,其程度与每篇参考文献均单独且具体指明以引用方式并入并在本文中完整阐述的程度相同。
[0070]
本文使用的所有标题和副标题只是为了方便,而不应被解释为以任何方式限制本发明。
[0071]
除非本文另有说明或与上下文明显矛盾,否则本发明涵盖上述要素的所有可能变体的任何组合。
[0072]
在描述本发明的上下文中使用的术语“一(a和an)”和“该(the)”以及类似的指示物应被解释为涵盖单数和复数,除非本文另有说明或与上下文明显矛盾。
[0073]
除非本文另有说明,否则本文数值范围的列举仅旨在用作单独指代落入该范围内的每个单独值的速记方法,并且每个单独的值都被并入说明书中,如同其在本文中被单独列举一样。
[0074]
除非另有说明,否则本文提供的所有精确值均表示相应的近似值(例如,针对特定因素或测量值提供的所有精确示例性值可以被视为也提供相应的近似值,在适当的地方由“约”修饰)。
[0075]
除非本文另有说明或与上下文明显矛盾,否则本文描述的所有方法可以任何合适的顺序进行。
[0076]
除非另有说明,否则本文提供的任何和所有实例或示例性语言(例如,“例如”)的
使用仅旨在更好地阐明本发明并且不对本发明的范围构成限制。说明书中的任何语言都不应被解释为表明任何元素对于本发明的实践是必要的,除非有明确说明。
[0077]
此处引用和并入专利文件仅为方便起见,并不反映此类专利文件的有效性、可专利性和/或可执行性的任何观点。
[0078]
本文在提及一个或多个要素时使用诸如“包含”、“具有”、“包括”或“含有”等术语对本发明的任何方面或实施方案的描述旨在为本发明的“由该特定一个或多个要素组成”、“基本上由其组成”或“基本上包含其”的类似方面或实施方案提供支持,除非另有说明或与上下文明显矛盾(例如,本文描述为包含特定元素的组合物应被理解为还描述由该元素组成的组合物,除非另有说明或与上下文明显矛盾)。
[0079]
本发明包括在可适用的法律允许的最大范围内在本文中提出的方面或权利要求中列举的主题的所有修改和等同物。
[0080]
本发明通过以下实施例进一步说明,然而,这些实施例不应被解释为限制保护范围。在前面的描述和下面的实施例中公开的特征,无论是单独的还是以它们的任何组合,对于以不同形式实现本发明都可能是关键的。
[0081]
编号的本发明实施方案
[0082]
1.一种制备9-氯-2,3-二甲基-6-(n,n-二甲基氨基乙基-氨基-2-氧代乙基)-6h-吲哚并-[2,3-b]喹喔啉(rabeximod)或其盐的方法,其中该方法包括以下步骤:
[0083]-在足够强以使吲哚n-h去质子化的碱水溶液和任选的催化剂的存在下,使9-氯-2,3-二甲基-6h-吲哚并[2,3-b]喹喔啉的溶液或悬浮液与2-氯-n-(2-二甲基氨基乙基)乙酰胺或其盐反应,得到rabeximod或其盐。
[0084]
2.实施方案1所述的方法,其中存在催化剂。
[0085]
3.实施方案1-2中任一项所述的方法,其中将9-氯-2,3-二甲基-6h-吲哚并[2,3-b]喹喔啉或其盐溶解在有机溶剂中。
[0086]
4.实施方案1-3中任一项所述的方法,其中将2-氯-n-(2-二甲基氨基乙基)乙酰胺或其盐溶解在有机溶剂中。
[0087]
5.实施方案1-4中任一项所述的方法,其中2-氯-n-(2-二甲基氨基乙基)乙酰胺是盐。
[0088]
6.实施方案1-5中任一项所述的方法,其中碱水溶液是naoh。
[0089]
7.实施方案1-6中任一项所述的方法,其中反应在惰性气体下发生。
[0090]
8.实施方案1-7中任一项所述的方法,其中1摩尔当量的9-氯-2,3-二甲基-6h-吲哚并[2,3-b]喹喔啉用至少2体积的碱水溶液去质子化。
[0091]
9.实施方案8所述的方法,其中在合适的温度下将9-氯-2,3-二甲基-6h-吲哚并[2,3-b]喹喔啉和碱水溶液混合直至形成澄清溶液。
[0092]
10.实施方案2-9中任一项所述的方法,其中在剧烈搅拌下添加合适量的催化剂并在合适的温度下混合10至60分钟。
[0093]
11.实施方案1-10中任一项所述的方法,其中将2-氯-n-(2-二甲基氨基乙基)乙酰胺或其盐添加到9-氯-2,3-二甲基-6h-吲哚并[2,3-b]喹喔啉在碱水溶液中的溶液,并在合适的温度下混合至少1小时。
[0094]
12.实施方案11所述的方法,其中以1-3倍的量添加2-氯-n-(2-二甲基氨基乙基)
乙酰胺。
[0095]
13.实施方案1-12中任一项所述的方法,其中rabeximod被纯化和分离为游离碱。
[0096]
14.实施方案13所述的方法,其中rabeximod被纯化和分离为结晶游离碱。
[0097]
15.实施方案1-14中任一项所述的方法,其中该方法包括以下在先步骤:
[0098]-使4,5-二甲基-1,2-苯二胺的溶液或悬浮液与5-氯靛红在酸性条件下在升高的温度直至回流下反应,得到9-氯-2,3-二甲基-6h-吲哚并[2,3-b]喹喔啉或其盐。
[0099]
16.实施方案1-14中任一项所述的方法,其中该方法包括以下在先步骤:
[0100]-使氯乙酰氯的溶液或悬浮液与n,n-二甲基乙二胺反应,得到2-氯-n-(2-二甲基氨基乙基)乙酰胺或其盐。
[0101]
17.实施方案1-14中任一项所述的方法,其中该方法包括实施方案15的在先步骤和实施方案16的在先步骤。
[0102]
18.一种制备9-氯-2,3-二甲基-6h-吲哚并[2,3-b]喹喔啉的方法,其中该方法包括以下步骤:
[0103]-使4,5-二甲基-1,2-苯二胺的溶液或悬浮液与5-氯靛红在酸性条件下在升高的温度直至回流下反应,得到9-氯-2,3-二甲基-6h-吲哚并[2,3-b]喹喔啉。
[0104]
19.实施方案15或18所述的方法,其中酸性条件是有机酸。
[0105]
20.实施方案15、18和19中任一项所述的方法,其中4,5-二甲基-1,2-苯二胺和5-氯靛红在反应前都溶解在酸中。
[0106]
21.实施方案15和18-20中任一项所述的方法,其中酸是过量的。
[0107]
22.实施方案15或21所述的方法,其中所述酸对于4,5-二甲基-1,2-苯二胺是至少4体积过量的并且对于5-氯靛红是至少10体积过量的。
[0108]
23.实施方案15和18-22中任一项所述的方法,其中升高的温度是回流温度。
[0109]
24.实施方案15和18-23中任一项所述的方法,其中4,5-二甲基-1,2-苯二胺或其盐在与5-氯靛红反应之前溶解在酸中,并且5-氯靛红或其盐在与4,5-二甲基-1,2-苯二胺反应之前溶解在酸中。
[0110]
25.实施方案15和18-24中任一项所述的方法,其中4,5-二甲基-1,2-苯二胺的添加量为1-3摩尔当量。
[0111]
26.实施方案15和18-25中任一项所述的方法,其中5-氯靛红的添加量为1-3摩尔当量。
[0112]
27.实施方案15和23-26中任一项所述的方法,其中将溶解的4,5-二甲基-1,2-苯二胺在回流温度下添加到溶解的5-氯靛红中。
[0113]
28.实施方案15或27所述的方法,其中在回流温度下历经至少2小时将4,5-二甲基-1,2-苯二胺添加到5-氯靛红中。
[0114]
29.实施方案15和27-28中任一项所述的方法,其中从反应混合物中蒸馏出酸,并且在蒸馏过程中以相似的速率添加另外的酸。
[0115]
30.实施方案15或29所述的方法,其中反应混合物在蒸馏后在回流温度下搅拌至少1小时。
[0116]
31.实施方案15和18-30中任一项所述的方法,其中9-氯-2,3-二甲基-6h-吲哚并[2,3-b]喹喔啉被纯化和分离为游离碱。
[0117]
32.一种制备2-氯-n-(2-二甲基氨基乙基)乙酰胺或其盐的方法,其中该方法包括以下步骤:
[0118]-使氯乙酰氯的溶液或悬浮液与n,n-二甲基乙二胺反应,得到2-氯-n-(2-二甲基氨基乙基)乙酰胺或其盐。
[0119]
33.实施方案16或32所述的方法,其中将氯乙酰氯溶解在有机溶剂中。
[0120]
34.实施方案16和32-33中任一项所述的方法,其中将n,n-二甲基乙二胺溶解在有机溶剂中。
[0121]
35.实施方案16和32-34中任一项所述的方法,其中氯乙酰氯和n,n-二甲基乙二胺在反应前均被溶解。
[0122]
36.实施方案16和32-34中任一项所述的方法,其中将溶液形式的n,n-二甲基乙二胺以保持溶液中的温度低于30℃的速率添加到溶液形式的氯乙酰氯中。
[0123]
37.实施方案16和32-36中任一项所述的方法,其中n,n-二甲基乙二胺或其盐在与氯乙酰氯反应之前溶解在溶剂中,并且5氯乙酰氯或其盐在与n,n-二甲基乙二胺反应之前溶解在溶剂中。
[0124]
38.实施方案16和32-37中任一项所述的方法,其中溶剂相对于氯乙酰氯是过量的,例如至少约2体积。
[0125]
39.实施方案16和32-38中任一项所述的方法,其中溶剂相对于n,n-二甲基乙二胺是当量或摩尔过量的,例如以当量比。
[0126]
40.实施方案16和32-39中任一项所述的方法,其中以1-3摩尔当量的量添加氯乙酰氯。
[0127]
41.实施方案16和32-40中任一项所述的方法,其中以1-3摩尔当量的量添加n,n-二甲基乙二胺。
[0128]
42.实施方案16和32-41中任一项所述的方法,其中2-氯-n-(2-二甲基氨基乙基)乙酰胺或其盐被纯化和分离为其盐。
[0129]
43.rabeximod的结晶游离碱,其熔点为259-261℃。
[0130]
44.可通过如实施方案1-17、19-31和33-42中任一项所定义的方法得到的rabeximod的结晶游离碱。
[0131]
附图简要说明
[0132]
图1显示了rabeximod的差示扫描量热法(dsc)热分析图和热重分析(tga)曲线。
[0133]
图2显示了rabeximod的x射线粉末图。
[0134]
实验
[0135]
如下面的反应流程所示,并如下文所详细描述的,制造rabeximod的当前方法包括几个方法步骤。
[0136][0137]
oxy001-01中间体的制造方法
[0138][0139]
起始原料:5-氯靛红(cido)和4,5-二甲基-1,2-苯二胺(dax)
[0140]
表1:步骤1所需的原材料和量的概述
[0141]
项目描述mwmol所需的量eq.5-氯靛红(cido)181.5866.112.0kg1.04,5-二甲基-1,2-苯二胺(dax)136.1972.79.9kg1.1a乙酸,acoh
‑‑
282l23.5c乙醇,etoh
‑‑
150l12.5c饮用水
‑‑
504.2c[0142]
a)mol//mol的cido;b)kg/kg的cido;c)l/kg的cido
[0143]
表2:步骤1的原材料的规范
[0144][0145]
得到的产物(中间体):oxy001-01
[0146]
批量大小:13.03kg的oxy001-01
[0147]
方法描述:将4,5-二甲基-1,2-苯二胺(1.1当量)添加到反应器(反应器在氮气和大气压力下运行)中的乙酸(4.7体积)中并在 20至 25℃下以中等速率搅拌至多3小时,直至形成澄清的深棕色溶液。将4,5-二甲基-1,2-苯二胺在乙酸溶液中的溶液转移到中间进料容器中。将5-氯靛红(1.0当量)添加到反应器中的乙酸(14.3体积)中并搅拌,同时将反应器的夹套温度调节到大约 150℃以达到溶剂活跃回流的回流温度。当达到回流温度时,历经2-3小时缓慢添加4,5-二甲基-1,2-苯二胺的乙酸溶液,同时从反应混合物中蒸馏出乙酸(4.7体积)。以与发生蒸馏(4.7体积)大致相同的速率将新鲜的乙酸部分(4.7体积)添加到反应器中。蒸馏后,将反应混合物在回流温度下再搅拌至少2小时。反应器中内容物的预期外观为深黄色至橙色浆液。将反应混合物冷却到 65至 70℃并使用nutsche过滤器使用聚酯滤布(27μm)或类似物作为过滤介质进行过滤。将滤饼用新鲜乙醇洗涤3次(3
×
4.2体积)并用水洗涤1次(1
×
4.2体积)。洗涤后,将滤饼在 40至 45℃下干燥12小时,并另外在 40℃下在真空盘式干燥器中干燥12小时,得到黄色至橙色/棕色固体。取过程中的控制样品并分析干燥失重(lod)。lod应为《2%(w/w)。如果lod为》2%,则重复真空托盘干燥器步骤。
[0148]
理论产量:18.62kg
[0149]
产率:70
±
5%(13.03
±
0.96kg)
[0150]
最大体积:216l oxy001-03 hcl中间体的制造方法
[0151][0152]
起始原料:氯乙酰氯(cac)和n,n-二甲基乙二胺(dmen)
[0153]
表3:步骤2所需的原材料和量的概述
[0154]
项目描述mwmol所需的量eq.n,n-二甲基乙二胺(dmen)88.15124.811.0kg1.0氯乙酰氯(cac)112.94128.514.5kg1.03a乙酸乙酯,etoac
‑‑
341l31c[0155]
a)mol//mol的dmen;b)kg/kg的dmen;c)l/kg的dmen
[0156]
表4:步骤2的原材料的规范
[0157][0158][0159]
得到的产物(中间体):oxy001-03 hcl
[0160]
批量大小:22.6kg的oxy001-03 hcl
[0161]
方法描述:在 20℃下,将氯乙酰氯(1.03当量)溶解在反应器(反应器在氮气和大气压力下运行)中的乙酸乙酯(15体积)中。将溶液搅拌并冷却到 10℃。当温度达到 10至 25℃的范围时,历经1-2小时以使内部温度不超过 25℃的速率,将n,n-二甲基乙二胺(1.00当量)在乙酸乙酯(1.0体积)中的溶液缓慢添加到反应器中。在 20至 25℃下搅拌浆料5至30分钟并使用nutch过滤器使用聚酰胺滤布(25μm)或类似物作为过滤介质进行过滤。将产物在过滤器上用乙酸乙酯洗涤3次(3
×
5体积)并在过滤器上干燥至少16小时,并另外在 40℃下在真空盘式干燥器中干燥12小时,得到灰白色至米色固体。
[0162]
理论产量:25.09kg
[0163]
产率:90
±
5%(22.6
±
1.25kg)
[0164]
最大体积:202l oxy001粗品的制造方法
[0165][0166]
起始原料:oxy001-01和oxy001-03 hcl
[0167]
表5:步骤3所需的原材料和量的概述
[0168]
项目描述mwmol所需的量eq.oxy001-01281.7446.313.0kg1.0oxy001-03hcl201.0992.518.6kg2.0a50%naoh水溶液40.00370.129.6kg8.0a碘化钾,ki166.0037.56.2kg0.81a四氢呋喃,thf
‑‑
705l54.2c饮用水
‑‑
395l30.4c[0169]
a)mol//mol的oxy001-01;b)kg/kg的oxy001-01;c)l/kg的oxy001-01
[0170]
表6:步骤3的原材料/中间材料的规范
[0171][0172]
得到的产物:oxy001粗品(粗rabeximod)
[0173]
批量大小:11.38kg的oxy001粗品
[0174]
方法描述:将oxy001-01(1.0当量)溶解在反应器(反应器在氮气和大气压下运行)中的四氢呋喃(15.4体积)和50% naoh水溶液(相对于oxy001-01为8.0当量)中,并在 55至 60℃下混合直至大约1小时,直至形成透明的深红色溶液。在剧烈搅拌下添加碘化钾(0.81当量)并在 55至 60℃下混合10至30分钟。将oxy001-03 hcl(2.0当量)添加到溶液中并在 55至 60℃下混合至少2小时。反应完成后,将混合物用水(15.4体积)淬灭并通过减压蒸发除去四氢呋喃(15.4体积)。将浆液冷却到 20至 25℃并搅拌1小时,并使用nutch过滤器使用聚酰胺滤布(25μm)或类似物作为过滤介质进行过滤。将所得滤饼用水洗涤3次(3
×
5体积),直到滤液的ph值在8-7之间,然后在 40至 45℃下通过空气抽吸在过滤器上干燥至少12小时,并另外在 40℃下在真空盘式干燥机中干燥12小时。然后将所得材料在 45至 50℃下悬浮在四氢呋喃(25体积)中至少1小时。通过使用nutch过滤器使用聚酰胺滤布(25μm)或类似物作为过滤介质进行过滤来分离oxy001粗品,并将其在过滤器上用四氢呋喃洗涤2次(2
×
7体积)。将所得滤饼在 40至 45℃下在过滤器上干燥至少12小时,并另外在 40℃下在真空盘式干燥器中干燥12小时。
[0175]
理论产量:18.96kg
[0176]
产率:60
±
5%(11.38
±
0.95kg)
[0177]
最大体积:500l
[0178]
rabeximod粗品的纯化:
[0179]
将oxy001粗品(1.0当量)溶解在四氢呋喃(10体积)、水(3体积)和2m hcl(1.4体积)的混合物中。将溶液澄清过滤并加热到 50℃。通过添加2m naoh(1.3体积)将混合物的ph调节到10-12。将形成的浆液冷却到 20至 25℃,并用水(12体积)稀释。
[0180]
搅拌至少12小时后,将浆液在 20至 25℃下过滤,并在过滤器上用四氢呋喃:水(5:2)的混合物洗涤(2
×
3体积)。rabeximod的分子量为409.92g/mol,被分离为结晶游离碱,其熔点为259-261℃。
[0181]
用于2期和1期临床研究的批次放行结果提供在表7中。
[0182]
通过hplc测量,纯度等于或高于98%。
[0183]
表7:1期和2期临床研究中使用的rabeximod原料药的批次放行结果
[0184]
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。