1.本发明涉及化学技术领域,具体来说涉及含喹唑啉结构的糖苷类衍生物的制备方法及其组合物对猕猴桃溃疡病、黄瓜花叶病毒病、烟草花叶病毒病具有抑制作用的用途。
背景技术:
2.猕猴桃种植产业发展前景良好,就经济效益而言,是农民增收致富,带动农村产业发展的一个重要举措,但由于可引起猕猴桃溃疡病的丁香假单胞杆菌猕猴桃致病变种(pseudomonassyringaepv.actinidiae,psa)的存在,猕猴桃无法健康的生长,阻碍了我国乃至全世界的猕猴桃产业的发展。由于psa引起的猕猴桃溃疡病具有传播十分迅速、传播途径多、传播过程不易被发现的特点,在植株感病早期,植株一般不出现症状,等种植区出现溃疡病病症时,病情可能已经到难以控制的程度,随着保护地的继续发展和高附加值作物的连年栽培,呈现发展越来越难以控制的趋势,除此以外,由细菌和病毒引起的植物病害导致农作物的谷物产量显着下降,黄瓜花叶病毒、烟草花叶病毒等病毒或病菌引起的病害对农业造成了巨大的经济损失,目前,市面上常用的抗菌剂不仅有田间防效不佳,不利于环境的可持续发展的缺点,更使细菌的耐药性不断增强,因此,研发一种高效、低毒、对环境友好的抗病毒剂和抗菌剂已刻不容缓。
3.松脂酸铜是一种高效的光谱杀菌剂,在使用松脂酸铜药剂时,在喷洒至植物上后,会在植物表面形成药膜,对于落在药膜上的病原菌能有效杀灭,此外,松脂酸铜形成的药膜具有很像的粘着性、展布性和渗透性,药效持久,对植物上出现的细菌性病害和真菌性病害都有很好的防治效果。
4.在杂环化合物中,含氮杂环化合物占据着重要的位置,由于其存在着独特的生物活性,得到许多化学家的青睐,由于喹唑啉类杂环具有的良好生物活性和环境相容性,成为国内外研究的热点。近年来,发现含有喹唑啉杂环的化合物具有较好的抗植物细菌性活性。
5.2007年,徐广芳扥(徐广芳.新型喹唑啉类化合物的抑菌活性筛选及作用机理初步研究[d].贵州大学,2007.)采用室内生长速率法对合成的数十个喹唑啉类化合物进行了抑菌活性的初步筛选,通过筛选发现含喹唑啉母环结构的化合物具有较好的抗真菌活性,化合物6-氟-4-乙硫基喹唑啉抑菌效果最好,对小麦赤霉病原菌(g.zeae)、辣椒枯萎病原菌(f.oxysporum)、苹果腐烂病原菌(c.mandshurica)、半夏立枯病原菌(r.solani)、水稻纹枯病原菌(t.cucumeris)、马铃薯晚疫病原菌(p.infestans)、油菜菌核病原菌(s.sclerotiorum)、黄瓜灰霉病原菌(b.cinerea)、苹果炭疽病原菌(c.gloeosporioides)九种植物病原真菌都有较好的生物活性,生物活性甚至超过了商品药剂恶霉灵。
[0006]
2020年,张贵强等(张贵强,郭晴晴,易君明,等.新型含2,4-二氯苯基的1,2,4-三唑喹唑啉衍生物的合成及抑菌活性[j].合成化学,2020(6):491-499.)合成了含2,4-二氯苯基1,2,4-三唑1,3,4-噻二嗪喹唑啉类化合物,采用浊度法测试目标化合物对柑橘溃疡病菌(xac),烟草青枯病菌(rs),水稻百叶枯病菌(xoo)的抑制活性.结果表明,部分化合物具
有较好的抑菌活性,和对照药剂噻菌酮和叶枯唑的抑制率相当。
[0007]
综上所述,喹唑啉酮类衍生物表现出了一定的杀菌活性。为创制新型高效的抗病毒剂和杀菌剂,本发明在前期工作的基础上,设计合成了系列含喹唑啉结构的糖苷类衍生物化合物,期望筛选出高活性的抗病毒药物和抗菌药物。
技术实现要素:
[0008]
本发明目的在于提供一种具有杀菌活性和抗病毒活性的含喹唑啉结构的糖苷类衍生物及其组合物的制备方法。
[0009]
本发明的另一目的在于对猕猴桃溃疡病、黄瓜花叶病毒病、烟草花叶病毒病具有抑制作用的用途。
[0010]
本发明的技术方案:一种含喹唑啉结构的糖苷类衍生物,其通式为下式(i):
[0011][0012]
其中:r1为卤素、甲基、甲氧基或上述取代基团任意组合的二取代或三取代。
[0013]
优选的,r1为6-氯,3-氯,5-溴,5-甲基,5-氟,4-氯,3-甲氧基,3-甲基,3,4-二甲基,6-氟,3-氟,6-甲氧基,3-甲基-5-氯,5-碘,4-氟,4-甲基,5-甲氧基,4-溴或6-甲基。
[0014]
一种含喹唑啉结构的糖苷类衍生物的制备方法,包括以下步骤:
[0015]
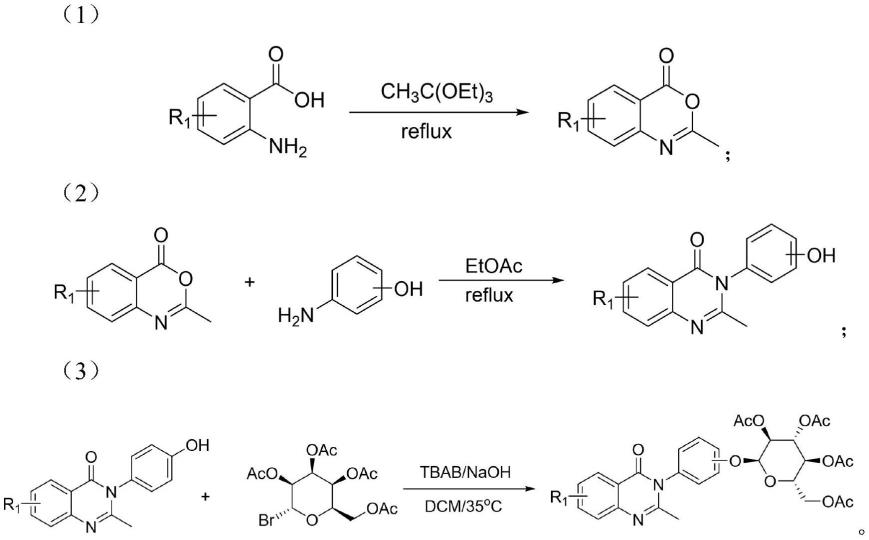
(1)按摩尔比取代苯甲酸:原乙酸三乙酯=1:20~50投料,加热回流4-6h,待反应结束,温度降到室温后,过滤收集固体,得到取代2-甲基-4h-苯并[d][1,3]噁嗪-4-酮。
[0016][0017]
(2)按摩尔比取代2-甲基-4h-苯并[d][1,3]噁嗪-4-酮:对氨基苯酚或3-氨基苯酚:冰乙酸=1:1.1:20~50投料,加热回流4-6h,待反应结束,将反应体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度降到室温后,过滤收集固体,得到取代2-甲基-3-苯基喹唑啉-4(3h)-酮。
[0018][0019]
(3)按摩尔比取代2-甲基-3-苯基喹唑啉-4(3h)-酮:糖苷:二氯甲烷=1:3:30投料,加热至35℃,待反应结束,将反应体系用氢氧化钠水溶液,饱和氯化钠溶液萃取,柱层析纯化,得到目标化合物含2-甲基-4-氧代喹唑啉结构的糖苷;
[0020][0021]
一种含喹唑啉结构的糖苷类衍生物在制备防治猕猴桃溃疡病、黄瓜花叶病毒病、烟草花叶病毒病的药物和药剂中的应用。
[0022]
一种包含所合成含喹唑啉结构的糖苷类衍生物、a23((2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(3-(7-氯-2-甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯)和松脂酸铜的复配组合物,其中,化合物a23具有如下式(2)的化合物:
[0023][0024]
所述的农药组合物,a23和松脂酸铜的质量比例为1:1;1:2;2:1。所述的组合物用于防治植物病害的用途,所述植物病害为植物细菌性病害。所述植物细菌性病害为猕猴桃溃疡病。
[0025]
本发明的有益效果:本发明合成了具有抗猕猴桃溃疡病菌活性的含喹唑啉结构的糖苷类衍生物。本发明的优点在于,原料易得,工艺简单,反应条件温和。并且本发明中的化合物a23具有较好的抗猕猴桃溃疡病菌活性,另外,本发明选定的组合物,化合物a23和松脂酸铜在防治猕猴桃溃疡病菌的过程中表现出协同增效的作用,能够进一步降低生产成本和使用成本,达到药减效增的目的。
具体实施方式:
[0026]
实施例1:(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(5-氯-2-甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯的合成(化合物编号为a1),包括以下步骤:
[0027]
(1)将2-氨基-6-氯苯甲酸(10.0mmol),以原乙酸三乙酯50ml为溶剂,加入到100ml三口烧瓶中回流反应4-5h,待反应结束,冷却至室温后,过滤收集固体,得到中间体5-氯-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮。
[0028]
(2)将5-氯-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),对氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到5-氯-3-(4-羟基苯基)-2-甲基喹唑啉-4(3h)-酮。
[0029]
(3)将5-氯-3-(4-羟基苯基)-2-甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应结束。将反应体系用氢氧化钠水20ml
×
3溶液,饱和氯化钠溶液20ml
×
3萃取,柱层析纯化,得到固体(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(5-氯-2-甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯,收率为66.6%,熔点为219.2-220.1℃。
[0030]
实施例2:(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(8-氯-2-甲基-4-氧代喹
唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯的合成(化合物编号为a2),包括以下步骤:
[0031]
(1)将2-氨基-3-氯苯甲酸(10.0mmol),以原乙酸三乙酯50ml为溶剂,加入到100ml三口烧瓶中回流反应4-5h,待反应结束,冷却至室温后,过滤收集固体,得到中间体8-氯-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮。
[0032]
(2)将8-氯-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),对氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到8-氯-3-(4-羟基苯基)-2-甲基喹唑啉-4(3h)-酮。
[0033]
(3)将8-氯-3-(4-羟基苯基)-2-甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应结束。将反应体系用氢氧化钠水20ml
×
3溶液,饱和氯化钠溶液20ml
×
3萃取,柱层析纯化,得到固体(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(8-氯-2-甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯,收率为52.6%,熔点为206.3-207.6℃。
[0034]
实施例3:(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(6-溴-2-甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯的合成(化合物编号为a3),包括以下步骤:
[0035]
(1)将2-氨基-5-溴苯甲酸(10.0mmol),以原乙酸三乙酯50ml为溶剂,加入到100ml三口烧瓶中回流反应4-5h,待反应结束,冷却至室温后,过滤收集固体,得到中间体6-溴-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮。
[0036]
(2)将6-溴-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),对氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到6-溴-3-(4-羟基苯基)-2-甲基喹唑啉-4(3h)-酮。
[0037]
(3)将6-溴-3-(4-羟基苯基)-2-甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应结束。将反应体系用氢氧化钠水20ml
×
3溶液,饱和氯化钠溶液20ml
×
3萃取,柱层析纯化,得到固体(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(6-溴-2-甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯,收率为40.1%,熔点为198.1-199.3℃。
[0038]
实施例4:(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(2,6-二甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯的合成(化合物编号为a4),包括以下步骤:
[0039]
(1)将2-氨基-5-甲基苯甲酸(10.0mmol),以原乙酸三乙酯50ml为溶剂,加入到100ml三口烧瓶中回流反应4-5h,待反应结束,冷却至室温后,过滤收集固体,得到中间体2,6-二甲基-4h-苯并[d][1,3]噁嗪-4-酮。
[0040]
(2)将2,6-二甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),对氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到3-(4-羟基苯基)-2,6-二甲基喹唑啉-4(3h)-酮。
[0041]
(3)将3-(4-羟基苯基)-2,6-二甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应结束。将反应体系用氢氧化钠水20ml
×
3溶液,饱和氯化钠溶液20ml
×
3萃取,柱层析纯化,得到固体(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(2,6-二甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯,收率为20.8%,熔点为213.2-214.8℃。
[0042]
实施例5:(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(6-氟-2-甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯的合成(化合物编号为a5),包括以下步骤:
[0043]
(1)将2-氨基-5-氟-苯甲酸(10.0mmol),以原乙酸三乙酯50ml为溶剂,加入到100ml三口烧瓶中回流反应4-5h,待反应结束,冷却至室温后,过滤收集固体,得到中间体6-氟-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮。
[0044]
(2)将6-氟-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),对氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到6-氟-3-(4-羟基苯基)-2-甲基喹唑啉-4(3h)-酮。
[0045]
(3)将6-氟-3-(4-羟基苯基)-2-甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应结束。将反应体系用氢氧化钠水20ml
×
3溶液,饱和氯化钠溶液20ml
×
3萃取,柱层析纯化,得到固体(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(6-氟-2-甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯,收率为56.3%,熔点为204.5-204.9℃。
[0046]
实施例6:(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(7-氯-2-甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯的合成(化合物编号为a6),包括以下步骤:
[0047]
(1)将2-氨基-4-氯-苯甲酸(10.0mmol),以原乙酸三乙酯50ml为溶剂,加入到100ml三口烧瓶中回流反应4-5h,待反应结束,冷却至室温后,过滤收集固体,得到中间体7-氯-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮。
[0048]
(2)将7-氯-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),对氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到7-氯-3-(4-羟基苯基)-2-甲基喹唑啉-4(3h)-酮。
[0049]
(3)将7-氯-3-(4-羟基苯基)-2-甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应结束。将反应体系用氢氧化钠水20ml
×
3溶液,饱和氯化钠溶液20ml
×
3萃取,柱层析纯化,得到固体(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(7-氯-2-甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯,收率为54.2%,熔点为223.1-224.7℃。
[0050]
实施例7:(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(8-甲氧基-2-甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯的合成(化合物编号为a7),包括以下步骤:
[0051]
(1)将2-氨基-3-甲氧基-苯甲酸(10.0mmol),以原乙酸三乙酯50ml为溶剂,加入到
100ml三口烧瓶中回流反应4-5h,待反应结束,冷却至室温后,过滤收集固体,得到中间体8-甲氧基-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮。
[0052]
(2)将8-甲氧基-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),对氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到3-(4-羟基苯基)-8-甲氧基-2-甲基喹唑啉-4(3h)-酮。
[0053]
(3)将3-(4-羟基苯基)-8-甲氧基-2-甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应结束。将反应体系用氢氧化钠水20ml
×
3溶液,饱和氯化钠溶液20ml
×
3萃取,柱层析纯化,得到固体(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(8-甲氧基-2-甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯,收率为28.8%,熔点为205.1-206.4℃。
[0054]
实施例8:(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(2,8-二甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯的合成(化合物编号为a8),包括以下步骤:
[0055]
(1)将2-氨基-3-甲基-苯甲酸(10.0mmol),以原乙酸三乙酯50ml为溶剂,加入到100ml三口烧瓶中回流反应4-5h,待反应结束,冷却至室温后,过滤收集固体,得到中间体2,8-二甲基-4h-苯并[d][1,3]噁嗪-4-酮。
[0056]
(2)将2,8-二甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),对氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到3-(4-羟基苯基)-2,8-二甲基喹唑啉-4(3h)-酮。
[0057]
(3)将3-(4-羟基苯基)-2,8-二甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应结束。将反应体系用氢氧化钠水20ml
×
3溶液,饱和氯化钠溶液20ml
×
3萃取,柱层析纯化,得到固体(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(2,8-二甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯,收率为55.1%,熔点为193.3-194.8℃。
[0058]
实施例9:(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(2,7,8-三甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯的合成(化合物编号为a9),包括以下步骤:
[0059]
(1)将2-氨基-3,4-二甲基-苯甲酸(10.0mmol),以原乙酸三乙酯50ml为溶剂,加入到100ml三口烧瓶中回流反应4-5h,待反应结束,冷却至室温后,过滤收集固体,得到中间体2,7,8-三甲基-4h-苯并[d][1,3]噁嗪-4-酮。
[0060]
(2)将2,7,8-三甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),对氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到3-(4-羟基苯基)-2,7,8-三甲基喹唑啉-4(3h)-酮。
[0061]
(3)将3-(4-羟基苯基)-2,7,8-三甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应
甲氧基-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮。
[0072]
(2)将5-甲氧基-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),对氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到3-(4-羟基苯基)-5-甲氧基-2-甲基喹唑啉-4(3h)-酮。
[0073]
(3)将3-(4-羟基苯基)-5-甲氧基-2-甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应结束。将反应体系用氢氧化钠水20ml
×
3溶液,饱和氯化钠溶液20ml
×
3萃取,柱层析纯化,得到固体(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(5-甲氧基-2-甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯,收率为39.9%,熔点为241.2-242.8℃。
[0074]
实施例13:(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(6-氯-2,8-二甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯的合成(化合物编号为a13),包括以下步骤:
[0075]
(1)将2-氨基-3-甲基-5-氯-苯甲酸(10.0mmol),以原乙酸三乙酯50ml为溶剂,加入到100ml三口烧瓶中回流反应4-5h,待反应结束,冷却至室温后,过滤收集固体,得到中间体6-氯-2,8-二甲基-4h-苯并[d][1,3]噁嗪-4-酮。
[0076]
(2)将6-氯-2,8-二甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),对氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到6-氯-3-(4-羟基苯基)-2,8-二甲基喹唑啉-4(3h)-酮。
[0077]
(3)将6-氯-3-(4-羟基苯基)-2,8-二甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应结束。将反应体系用氢氧化钠水20ml
×
3溶液,饱和氯化钠溶液20ml
×
3萃取,柱层析纯化,得到固体(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(6-氯-2,8-二甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯,收率为21.2%,熔点为195.8-196.3℃。
[0078]
实施例14:(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(6-碘-2-甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯的合成(化合物编号为a14),包括以下步骤:
[0079]
(1)将2-氨基-5-碘-苯甲酸(10.0mmol),以原乙酸三乙酯50ml为溶剂,加入到100ml三口烧瓶中回流反应4-5h,待反应结束,冷却至室温后,过滤收集固体,得到中间体6-碘-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮。
[0080]
(2)将6-碘-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),对氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到3-(4-羟基苯基)-6-碘-2-甲基喹唑啉-4(3h)-酮。
[0081]
(3)将3-(4-羟基苯基)-6-碘-2-甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应结
束。将反应体系用氢氧化钠水20ml
×
3溶液,饱和氯化钠溶液20ml
×
3萃取,柱层析纯化,得到固体(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(6-碘-2-甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯,收率为35.6%,熔点为123.4-124.2℃。
[0082]
实施例15:(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(7-氟-2-甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯的合成(化合物编号为a15),包括以下步骤:
[0083]
(1)将2-氨基-4-氟-苯甲酸(10.0mmol),以原乙酸三乙酯50ml为溶剂,加入到100ml三口烧瓶中回流反应4-5h,待反应结束,冷却至室温后,过滤收集固体,得到中间体7-氟-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮。
[0084]
(2)将7-氟-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),对氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到7-氟-3-(4-羟基苯基)-2-甲基喹唑啉-4(3h)-酮。
[0085]
(3)将7-氟-3-(4-羟基苯基)-2-甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应结束。将反应体系用氢氧化钠水20ml
×
3溶液,饱和氯化钠溶液20ml
×
3萃取,柱层析纯化,得到固体(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(7-氟-2-甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯,收率为33.0%,熔点为209.1-210.3℃。
[0086]
实施例16:(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(2,7-二甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯的合成(化合物编号为a16),包括以下步骤:
[0087]
(1)将2-氨基-4-甲基-苯甲酸(10.0mmol),以原乙酸三乙酯50ml为溶剂,加入到100ml三口烧瓶中回流反应4-5h,待反应结束,冷却至室温后,过滤收集固体,得到中间体2,7-二甲基-4h-苯并[d][1,3]噁嗪-4-酮。
[0088]
(2)将2,7-二甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),对氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到3-(4-羟基苯基)-2,7-二甲基喹唑啉-4(3h)-酮。
[0089]
(3)将3-(4-羟基苯基)-2,7-二甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应结束。将反应体系用氢氧化钠水20ml
×
3溶液,饱和氯化钠溶液20ml
×
3萃取,柱层析纯化,得到固体(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(2,7-二甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯,收率为26.7%,熔点为211.5-212.2℃。
[0090]
实施例17:(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(6-甲氧基-2-甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯的合成(化合物编号为a17),包括以下步骤:
[0091]
(1)将2-氨基-5-甲氧基-苯甲酸(10.0mmol),以原乙酸三乙酯50ml为溶剂,加入到100ml三口烧瓶中回流反应4-5h,待反应结束,冷却至室温后,过滤收集固体,得到中间体6-甲氧基-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮。
[0092]
(2)将6-甲氧基-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),对氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到3-(3-羟基苯基)-6-甲氧基-2-甲基喹唑啉-4(3h)-酮。
[0093]
(3)将3-(4-羟基苯基)-6-甲氧基-2-甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应结束。将反应体系用氢氧化钠水20ml
×
3溶液,饱和氯化钠溶液20ml
×
3萃取,柱层析纯化,得到固体(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(6-甲氧基-2-甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯,收率为34.2%,熔点为116.0-117.6℃。
[0094]
实施例18:(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(6-甲氧基-2-甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯的合成(化合物编号为a18),包括以下步骤:
[0095]
(1)将2-氨基-5-甲氧基-苯甲酸(10.0mmol),以原乙酸三乙酯50ml为溶剂,加入到100ml三口烧瓶中回流反应4-5h,待反应结束,冷却至室温后,过滤收集固体,得到中间体6-甲氧基-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮。
[0096]
(2)将6-甲氧基-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),对氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到3-(4-羟基苯基)-6-甲氧基-2-甲基喹唑啉-4(3h)-酮。
[0097]
(3)将3-(4-羟基苯基)-6-甲氧基-2-甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应结束。将反应体系用氢氧化钠水20ml
×
3溶液,饱和氯化钠溶液20ml
×
3萃取,柱层析纯化,得到固体(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(6-甲氧基-2-甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯,收率为56.8%,熔点为196.6-197.3℃。
[0098]
实施例19:(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(7-溴-2-甲基-4-氧代喹唑啉)-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯的合成(化合物编号为a19),包括以下步骤:
[0099]
(1)将2-氨基-4-溴-苯甲酸(10.0mmol),以原乙酸三乙酯50ml为溶剂,加入到100ml三口烧瓶中回流反应4-5h,待反应结束,冷却至室温后,过滤收集固体,得到中间体7-溴-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮。
[0100]
(2)将7-溴-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),对氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到7-溴-3-(4-羟基苯基)-2-甲基喹唑啉-4(3h)-酮。
[0101]
(3)将7-溴-3-(4-羟基苯基)-2-甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应结束。将反应体系用氢氧化钠水20ml
×
3溶液,饱和氯化钠溶液20ml
×
3萃取,柱层析纯化,得
到固体(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(4-(7-溴-2-甲基-4-氧代喹唑啉)-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯,收率为45.1%,熔点为221.4-222.6℃。
[0102]
实施例20:(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(3-(2,7,8-三甲基-4-氧代喹唑啉)-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯的合成(化合物编号为a20),包括以下步骤:
[0103]
(1)同实施例9的第(1)步
[0104]
(2)将2,7,8-三甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),3-氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到3-(3-羟基苯基)-2,7,8-三甲基喹唑啉-4(3h)-酮。
[0105]
(3)将3-(3-羟基苯基)-2,7,8-三甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应结束。将反应体系用氢氧化钠水20ml
×
3溶液,饱和氯化钠溶液20ml
×
3萃取,柱层析纯化,得到固体(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(3-(2,7,8-三甲基-4-氧代喹唑啉)-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯,收率为73.5%,熔点为238.2-239.5℃。
[0106]
实施例21:(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(3-(6-氟-2-甲基-4-氧代喹唑啉)-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯的合成(化合物编号为a21),包括以下步骤:
[0107]
(1)同实施例5的第(1)步
[0108]
(2)将6-氟-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),3-氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到6-氟-3-(3-羟基苯基)-2-甲基喹唑啉-4(3h)-酮。
[0109]
(3)将6-氟-3-(3-羟基苯基)-2-甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应结束。将反应体系用氢氧化钠水20ml
×
3溶液,饱和氯化钠溶液20ml
×
3萃取,柱层析纯化,得到固体(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(3-(6-氟-2-甲基-4-氧代喹唑啉)-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯,收率为69.8%,熔点为117.5-118.8℃。
[0110]
实施例22:(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(3-(6-碘-2-甲基-4-氧代喹唑啉)-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯的合成(化合物编号为a22),包括以下步骤:
[0111]
(1)同实施例14的第(1)步
[0112]
(2)将6-碘-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),3-氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到3-(3-羟基苯基)-6-碘-2-甲基喹唑啉-4(3h)-酮,收率为42.4%。
[0113]
(3)将3-(3-羟基苯基)-6-碘-2-甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应结束。将反应体系用氢氧化钠水20ml
×
3溶液,饱和氯化钠溶液20ml
×
3萃取,柱层析纯化,得到固体(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(3-(6-碘-2-甲基-4-氧代喹唑啉)-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯,收率为56.9%,熔点为110.9-111.6℃。
[0114]
实施例23:(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(3-(7-氯-2-甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯的合成(化合物编号为a23),包括以下步骤:
[0115]
(1)同实施例6的第(1)步
[0116]
(2)将7-氯-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),3-氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到7-氯-3-(3-羟基苯基)-2-甲基喹唑啉-4(3h)-酮。
[0117]
(3)将7-氯-3-(3-羟基苯基)-2-甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应结束。将反应体系用氢氧化钠水20ml
×
3溶液,饱和氯化钠溶液20ml
×
3萃取,柱层析纯化,得到固体(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(3-(7-氯-2-甲基-4-氧代喹唑啉-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯,收率为74.4%,熔点为105.3-106.7℃。
[0118]
实施例24:(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(3-(8-氟-2-甲基-4-氧代喹唑啉)-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯的合成(化合物编号为a24),包括以下步骤:
[0119]
(1)同实施例11的第(1)步
[0120]
(2)将8-氟-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),3-氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到8-氟-3-(3-羟基苯基)-2-甲基喹唑啉-4(3h)-酮。
[0121]
(3)将8-氟-3-(3-羟基苯基)-2-甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应结束。将反应体系用氢氧化钠水20ml
×
3溶液,饱和氯化钠溶液20ml
×
3萃取,柱层析纯化,得到固体(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(3-(8-氟-2-甲基-4-氧代喹唑啉)-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯,收率为45.0%,熔点为125.9-126.7℃。
[0122]
实施例25:(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(3-(5-氟-2-甲基-4-氧代喹唑啉)-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯的合成(化合物编号为a25),包括以下步骤:
[0123]
(1)同实施例10的第(1)步
[0124]
(2)将5-氟-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),3-氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将
体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到5-氟-3-(3-羟基苯基)-2-甲基喹唑啉-4(3h)-酮。
[0125]
(3)将5-氟-3-(3-羟基苯基)-2-甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应结束。将反应体系用氢氧化钠水20ml
×
3溶液,饱和氯化钠溶液20ml
×
3萃取,柱层析纯化,得到固体(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(3-(5-氟-2-甲基-4-氧代喹唑啉)-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯,收率为30.0%,熔点为112.9-113.5℃。
[0126]
实施例26:(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(3-(2,8-二甲基-4-氧代喹唑啉)-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯的合成(化合物编号为a26),包括以下步骤:
[0127]
(1)同实施例8的第(1)步
[0128]
(2)将2,8-二甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),3-氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到3-(3-羟基苯基)-2,8-二甲基喹唑啉-4(3h)-酮。
[0129]
(3)将3-(3-羟基苯基)-2,8-二甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应结束。将反应体系用氢氧化钠水20ml
×
3溶液,饱和氯化钠溶液20ml
×
3萃取,柱层析纯化,得到固体(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(3-(2,8-二甲基-4-氧代喹唑啉)-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯,收率为52.1%,熔点为116.1-117.8℃。
[0130]
实施例27:(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(3-(7-氟-2-甲基-4-氧代喹唑啉)-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯的合成(化合物编号为a27),包括以下步骤:
[0131]
(1)同实施例15的第(1)步
[0132]
(2)将7-氟-2-甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),3-氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到7-氟-3-(3-羟基苯基)-2-甲基喹唑啉-4(3h)-酮。
[0133]
(3)将7-氟-3-(3-羟基苯基)-2-甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应结束。将反应体系用氢氧化钠水20ml
×
3溶液,饱和氯化钠溶液20ml
×
3萃取,柱层析纯化,得到固体(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(3-(7-氟-2-甲基-4-氧代喹唑啉)-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯,收率为58.5%,熔点为133.2-134.9℃。
[0134]
实施例28:(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(3-(2,5-二甲基-4-氧代喹唑啉)-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯的合成(化合物编号为a28),包括以下步骤:
[0135]
(1)将2-氨基-6-甲基-苯甲酸(10.0mmol),以原乙酸三乙酯50ml为溶剂,加入到
100ml三口烧瓶中回流反应4-5h,待反应结束,冷却至室温后,过滤收集固体,得到中间体2,5-二甲基-4h-苯并[d][1,3]噁嗪-4-酮。
[0136]
(2)将2,5-二甲基-4h-苯并[d][1,3]噁嗪-4-酮(1.0mmol),3-氨基苯酚(1.1mmol),以冰乙酸30ml为溶剂,加热回流4-6h,经tlc跟踪监测反应进程,反应完毕后,将体系倒入带有磁力搅拌器的冰水中,充分搅拌。待温度将至室温后,过滤收集固体,得到3-(3-羟基苯基)-2,5-二甲基喹唑啉-4(3h)-酮。
[0137]
(3)将3-(3-羟基苯基)-2,5-二甲基喹唑啉-4(3h)-酮(1.0mmol),乙酰溴-α-d-葡萄糖(3.0mmol),以二氯甲烷30ml为溶剂,加热至35℃,经tlc跟踪监测反应进程,待反应结束。将反应体系用氢氧化钠水20ml
×
3溶液,饱和氯化钠溶液20ml
×
3萃取,柱层析纯化,得到固体(2s,3s,4r,5s,6s)-2-(乙酰氧基甲基)-6-(3-(2,5-二甲基-4-氧代喹唑啉)-3(4h)-基)苯氧基)四氢-2h-吡喃-3,4,5-三乙酸三酯,收率为25.3%,熔点为142.1-143.8℃。
[0138]
对上述实施例a1-a28合成的含喹唑啉结构的糖苷类衍生物的核磁共振氢谱(1hnmr)、碳谱(
13
cnmr)和高分辨质谱(hrms)数据如表1所示。
[0139]
表1.实施例a1-a28化合物的波谱数据
[0140]
[0141]
[0142]
[0143]
[0144]
[0145]
[0146]
[0147]
[0148]
[0149][0150]
实施例29:目标化合物对猕猴桃溃疡病菌的抑制活性
[0151]
(1)测试方法
[0152]
采用浊度法(yangl.;etal.,2017)测定化合物的杀菌活性。制备浓度为100μg/ml的被测化合物。配制nb培养基(3.0g牛肉提取物,5.0g蛋白胨,1.0g酵母粉,10.0g葡萄糖,1000ml蒸馏水,ph7.0-7.2),分别用接菌环划一小块含有烟草青枯病菌、魔芋软腐病菌的培养基放入两个nb培养基中,塞好塞子,在28℃,180rpm恒温摇床振荡培养到生长对数期(od=0.6-0.8)备用。取40μl的菌液、4ml水-吐温(1%吐温20)、1ml配制好的化合物溶液,将试管28
±
1℃下培养,并以180rpm连续摇动1-3天。通过测量600nm(od600)处的光密度来监测细菌的生长,但含有同样浓度的溶剂和0.1%tween20作为空白对照,叶枯唑作为对照药剂,每处理重复三次。通过以下公式计算药剂对细菌的抑制率:
[0153]
i=(ctur-ttur)/ctur
×
100%
[0154]
其中i为抑制率,ctur代表未经药物处理的试管中细菌生长的校正的浊度值(空白对照),ttur代表经化合物处理的试管中细菌生长的校正的浊度值。
[0155]
(2)生物测试结果
[0156]
表2目标化合物在100μg/ml下对猕猴桃溃疡病菌的抑制活性
[0157]
[0158][0159]
采用离体生长速率法,在浓度为100μg/ml,以叶枯唑为对照药剂测试了目标化合物的抑菌活性,从表2生物活性测定结果可以看出含喹唑啉结构的糖苷类衍生物对猕猴桃溃疡病菌具有较好的抑制活性,其中a23的活性最好,抑制率为90.8%,优于对照药剂叶枯唑。实施例30:目标化合物抗黄瓜花叶病毒治疗、钝化和保护活性
[0160]
(1)测试方法
[0161]
a.病毒提纯
[0162]
采用gooding方法(gooding;etal.1967),选取接种3周以上,cmv系统侵染寄主心叶烟(nicotianaglutinosal.)植株上部叶片,在磷酸缓冲液中匀浆,双层纱布过滤,1000rpm离心,经2次聚乙二醇处理,再离心,沉淀用磷酸缓冲液悬浮,即得到cmv的粗提液。整个试验在4℃下进行。用紫外分光光度计测定260nm波长的吸光度值,根据公式计算病毒浓度。
[0163]
病毒浓度(mg/ml)=(a260
×
稀释倍数)/e0.1%1cm260nm
[0164]
其中e表示消光系数,即波长260nm时,浓度为0.1%(1mg/ml)的悬浮液,在光程为lcm时的光吸收(光密度)值。tmv的e0.1%1cm260nm是3.1。
[0165]
b、药剂对cmv侵染的活性治疗作用:选长势一致的心叶烟,先用毛笔蘸取病毒汁液,全叶接种病毒,接种后用清水冲洗。待叶片干后,在右半叶涂施药剂,左半叶涂施对应剂量的溶剂作对照。随后在光照培养箱中保湿培养,控制温度23
±
1℃,光照10000lux,3-4d后观察并记录产生枯斑的数目。每药剂处理设3株,每株3~4片叶。按上述方法每药剂进行3次重复,按下列公式计算抑制率。
[0166]
c、药剂对cmv侵染的活体保护作用
[0167]
药剂对cmv侵染的活体保护作用:选长势一致的心叶烟,先用毛笔在右半叶涂施药剂,左半叶涂施对应剂量的溶剂作对照,待叶片干后,笔蘸取病毒汁液,全叶接种病毒,接种后用清水冲洗。随后在光照培养箱中保湿培养,控制温度23
±
1℃,光照10000lux,3-4d后观察并记录产生枯斑的数目。每药剂处理设3株,每株3~4片叶。按上述方法每药剂进行3次重复,按下列公式计算抑制率。
[0168]
d、药剂对cmv侵染的活体钝化作用
[0169]
药剂对cmv侵染的活体钝化作用:选长势一致的心叶烟,向全叶撒匀金刚砂,将化合物与等体积的病毒汁液混合钝化30分钟,用排笔人工摩擦接种于撒有金刚砂的适龄笕色
黎右半叶,对应剂量的溶剂与病毒汁液混合接种于撒有金刚砂的适龄笕色黎左半叶,3-4d后观察并记录产生枯斑的数目。每药剂处理设3株,每株3~4片叶。按上述方法每药剂进行3次重复,按下列公式计算抑制率。
[0170]
y=(c-a)/c
×
100%
[0171]
其中:y为化合物对烟草花叶病毒的抑制率;c为对照组(左半叶)枯斑个数,a为对照组(右半叶)枯斑个数。
[0172]
(2)生物测试结果
[0173]
表3目标化合物对黄瓜花叶病毒的治疗、保护、钝化活性
[0174][0175][0176]
采用半叶枯斑法,浓度为500μg/ml,以宁南霉素为对照药剂测试了目标化合物的抗tmv活性,从表3生物活性测定结果可以看出含喹唑啉结构的糖苷类衍生物对cmv均具有中等到优秀的抑制活性,其中a23在治疗、保护、钝化方面,均优于对照药剂宁南霉素。
[0177]
实施例31:目标化合物抗烟草花叶病毒治疗、钝化和保护活性
[0178]
(1)测试方法
[0179]
a.病毒提纯
[0180]
采用gooding方法(gooding;etal.1967),选取接种3周以上,tmv系统侵染寄主心叶烟(nicotianaglutinosal.)植株上部叶片,在磷酸缓冲液中匀浆,双层纱布过滤,1000rpm离心,经2次聚乙二醇处理,再离心,沉淀用磷酸缓冲液悬浮,即得到tmv的粗提液。整个试验在4℃下进行。用紫外分光光度计测定260nm波长的吸光度值,根据公式计算病毒浓度。
[0181]
病毒浓度(mg/ml)=(a260
×
稀释倍数)/e0.1%1cm260nm
[0182]
其中e表示消光系数,即波长260nm时,浓度为0.1%(1mg/ml)的悬浮液,在光程为lcm时的光吸收(光密度)值。tmv的e0.1%1cm260nm是3.1。
[0183]
b、药剂对tmv侵染的活性治疗作用:选长势一致的心叶烟,先用毛笔蘸取病毒汁液,全叶接种病毒,接种后用清水冲洗。待叶片干后,在右半叶涂施药剂,左半叶涂施对应剂量的溶剂作对照。随后在光照培养箱中保湿培养,控制温度23
±
1℃,光照10000lux,3-4d后观察并记录产生枯斑的数目。每药剂处理设3株,每株3~4片叶。按上述方法每药剂进行3次重复,按下列公式计算抑制率。
[0184]
c、药剂对tmv侵染的活体保护作用
[0185]
药剂对tmv侵染的活体保护作用:选长势一致的心叶烟,先用毛笔在右半叶涂施药剂,左半叶涂施对应剂量的溶剂作对照,待叶片干后,笔蘸取病毒汁液,全叶接种病毒,接种后用清水冲洗。随后在光照培养箱中保湿培养,控制温度23
±
1℃,光照10000lux,3-4d后观察并记录产生枯斑的数目。每药剂处理设3株,每株3~4片叶。按上述方法每药剂进行3次重复,按下列公式计算抑制率。
[0186]
d、药剂对tmv侵染的活体钝化作用
[0187]
药剂对tmv侵染的活体钝化作用:选长势一致的心叶烟,向全叶撒匀金刚砂,将化合物与等体积的病毒汁液混合钝化30分钟,用排笔人工摩擦接种于撒有金刚砂的适龄笕色黎右半叶,对应剂量的溶剂与病毒汁液混合接种于撒有金刚砂的适龄笕色黎左半叶,3-4d后观察并记录产生枯斑的数目。每药剂处理设3株,每株3~4片叶。按上述方法每药剂进行3次重复,按下列公式计算抑制率。
[0188]
y=(c-a)/c
×
100%
[0189]
其中:y为化合物对烟草花叶病毒的抑制率;c为对照组(左半叶)枯斑个数,a为对照组(右半叶)枯斑个数。
[0190]
(2)生物测试结果
[0191]
表4目标化合物对烟草花叶病毒的治疗、保护、钝化活性
[0192][0193][0194]
采用半叶枯斑法,浓度为500μg/ml,以宁南霉素为对照药剂测试了目标化合物的抗tmv活性,从表4生物活性测定结果可以看出含喹唑啉结构的糖苷类衍生物对tmv均具有中等到优秀的抑制活性,其中a23在治疗、保护、钝化方面,均优于对照药剂宁南霉素。
[0195]
实施例32:目标化合物a23组合物的制备
[0196]
下述所用的部分制剂通过市场购买的制剂进行浓度调节制备得到。以下实施例是对本发明的进一步说明,但本发明不局限于本实施例中的比例、制剂类型和用途。下面实施例中,目标化合物a23和松脂酸铜的可湿性粉剂制备复配组合物,各组合物中,目标化合物a23和松脂酸铜比例按照质量配比计算。根据需要制备如下目标化合物a23和松脂酸铜的制剂。
[0197]
组合物1:a23:松脂酸铜可湿性粉剂=1:1
[0198]
组合物2:a23:松脂酸铜可湿性粉剂=1:2
[0199]
组合物3:a23:松脂酸铜可湿性粉剂=2:1
[0200]
实施例33:目标化合物a23的组合物的抗猕猴桃溃疡病活性
[0201]
按照实施例29中提到的抑菌活性测试方法对目标化合物a23与松脂酸铜的组合物进行抗猕猴桃溃疡病菌活性测试。
[0202]
表5药物对猕猴桃溃疡病菌的抑制活性
[0203][0204]
采用离体生长速率法,在浓度为100μg/ml,测试了组合物的抗猕猴桃溃疡病菌活性,从表5生物活性测定结果可以看出复配组合物的抗猕猴桃溃疡病菌活性相对比复配前的化合物有所提高,组合物3(a23:松脂酸铜可湿性粉剂=2:1)对猕猴桃溃疡病菌的抑制活性达100.0%。因此,a23与松脂酸铜的复配组合物对猕猴桃溃疡病有协同增效的效果。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。