x射线显影化合物及其制备方法、x射线显影栓塞材料及其制备方法
技术领域
1.本技术涉及x射线下可视化的栓塞技术领域,且特别涉及一种x射线显影化合物及其制备方法、x射线显影栓塞材料及其制备方法。
背景技术:
2.自经导管动脉栓塞术(transcatheter arterial embolization,简称tae)的概念提出40多年来,选择合适的栓塞材料是该技术发展关键的一环。由于碘原子对x射线的强吸收,含碘化合物,尤其是含碘有机化合物,可在x射线检测下人体内显影。因此,可以在栓塞材料上连接含碘的有机化合物,从而使栓塞材料具有x射线显影的功能。
技术实现要素:
3.针对现有技术的不足,本技术实施例的目的包括提供一种x射线显影化合物及其制备方法、x射线显影栓塞材料及其制备方法,基于一类新的显影化合物,较为容易的使栓塞材料具有x射线显影功能。
4.第一方面,本技术实施例提供了一种x射线显影化合物,化合物的结构式如下:
[0005][0006]
其中,x为由至少一个碘取代的c5-12芳基或c5-12杂芳基;
[0007]
y不存在;或者,y为c1-6亚烷基、c2-6亚烯基、c2-6亚炔基、c1-6亚烷氧基和c1-6烷氧基亚烷基的一种。
[0008]
在上述技术方案中,通过酰胺键直接或间接将咪唑基团或苯并咪唑基团与碘取代的芳基或杂芳基连接起来,形成x射线显影化合物,该显影化合物中的酰胺键处的c-n键容易发生断裂,以便将断裂处的c与栓塞材料键接(断裂和键接可以一步反应),以便使栓塞材料具有x射线显影功能。
[0009]
在本技术的部分实施例中,x为由至少一个碘取代的六元芳基或六元杂芳基;y不存在或y为亚甲基或y为亚烷氧基。
[0010]
在上述技术方案中,可以使酰胺键中的c-n键更加容易断裂,使具有x射线显影功能的栓塞材料的制备更加方便。
[0011]
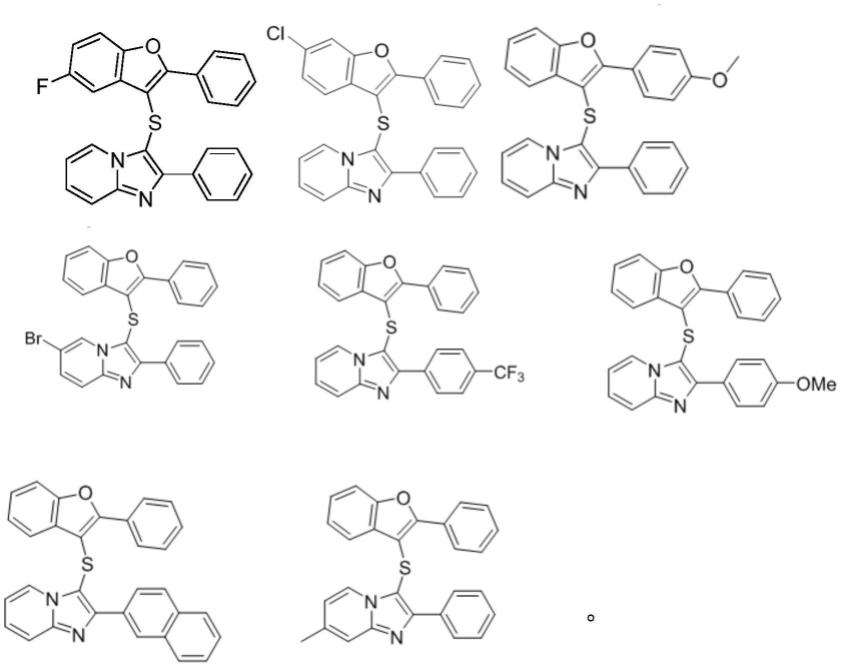
在本技术的部分实施例中,x射线显影化合物的结构式为:
[0012][0013]
第二方面,本技术提供一种上述x射线显影化合物的制备方法,x射线显影化合物为化合物ⅰ,制备方法包括:
[0014]
反应式一:
[0015][0016]
或,反应式二:
[0017][0018]
或,反应式三:
[0019][0020]
或,x射线显影化合物为化合物ⅱ,制备方法包括:
[0021]
反应式四:
[0022][0023]
其中,反应式一、反应式二和反应式三中的化合物ⅲ中的x和y,与化合物ⅰ中的x和y一致;反应式四中的化合物ⅲ中的x和y,与化合物ⅱ中的x和y一致。
[0024]
反应式一或反应式四中,通过氯化亚砜作为中间反应物,将含有羧酸基的碘取代的芳基或杂芳基与咪唑或苯并咪唑缩合,制备较为方便。反应式二或反应式三中,通过n,n'-羰基二咪唑(cdi)或2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(hatu)作为缩合剂,将含有羧酸基的碘取代的芳基或杂芳基与缩合剂缩合,制备较为方便。
[0025]
第三方面,本技术提供一种x射线显影栓塞材料,在栓塞材料上通过酯基连接-y-x
基团,其中,x为由至少一个碘取代的c5-12芳基或c5-12杂芳基;y不存在;或者,y为c1-6亚烷基、c2-6亚烯基、c2-6亚炔基、c1-6亚烷氧基和c1-6烷氧基亚烷基的一种。
[0026]
在上述技术方案中,该x射线显影栓塞材料通过酯基将栓塞材料与显影基团(碘取代的c5-12芳基或c5-12杂芳基)连接起来,栓塞材料具有显影功能,且稳定性较好。
[0027]
在本技术的部分实施例中,x射线显影栓塞材料的结构式为:
[0028]
其中m和n为正整数。
[0029]
该x射线显影栓塞材料可以是固体栓塞剂或液体栓塞剂,其均能够实现显影功能。
[0030]
在本技术的部分实施例中,x为由至少一个碘取代的六元芳基或六元杂芳基;y不存在或y为亚甲基或y为亚烷氧基。
[0031]
第四方面,本技术提供一种x射线显影栓塞材料的制备方法,包括:在1,8-二氮杂双环[5.4.0]十一碳-7-烯(dbu)的催化作用下,使第一方面任一项提供的x射线显影化合物的酰胺键与栓塞材料上的至少部分羟基反应,形成酯基连接。
[0032]
在上述技术方案中,可以使x射线显影栓塞材料的制备更加简单,以便使栓塞材料具有x射线显影功能,且通过酯基连接显影分子和栓塞材料,可以使x射线显影栓塞材料的稳定性较好。
[0033]
在本技术的部分实施例中,栓塞材料为固体栓塞剂,制备方法包括:将聚乙烯醇微球、x射线显影化合物、1,8-二氮杂双环[5.4.0]十一碳-7-烯与溶剂混合形成混合液,将混合液于40~90℃反应24~48h。
[0034]
在上述条件下,以1,8-二氮杂双环[5.4.0]十一碳-7-烯作为催化剂,可以使x射线显影化合物中的c-n键处断裂,并将断裂处的c与聚乙烯醇的至少部分羟基处脱氢键连接,该条件较为温和,容易达到,且最终得到的显影微球的稳定性较好。
[0035]
在本技术的部分实施例中,栓塞材料为液体栓塞剂,制备方法包括:将乙烯乙烯醇共聚物(evoh)、x射线显影化合物、1,8-二氮杂双环[5.4.0]十一碳-7-烯与溶剂混合形成混合液,将混合液于40~90℃反应24~48h;将反应物溶于溶剂中,得到液体栓塞剂。
[0036]
在上述条件下,以1,8-二氮杂双环[5.4.0]十一碳-7-烯作为催化剂,可以使x射线显影化合物中的c-n键处断裂,并将断裂处的c与乙烯乙烯醇共聚物的至少部分羟基处脱氢键连接,该条件较为温和,容易达到,且最终得到的显影液体栓塞剂的稳定性较好。
附图说明
[0037]
为了更清楚地说明本技术实施例的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本技术的某些实施例,因此不应被看作是对
范围的限定,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图。
[0038]
图1为本技术实施例1提供的目标产物i-1的核磁氢谱图;
[0039]
图2为实施例一提供的显影微球的光学图片;
[0040]
图3为实施例二提供的显影微球的光学图片;
[0041]
图4为实施例三提供的显影微球的光学图片;
[0042]
图5为实施例四提供的显影微球的光学图片;
[0043]
图6为实施例五提供的显影微球的光学图片;
[0044]
图7为实施例六提供的显影微球的光学图片;
[0045]
图8为实施例七提供的显影微球的光学图片。
具体实施方式
[0046]
为使本技术实施例的目的、技术方案和优点更加清楚,下面对本技术的技术方案进行清楚、完整地描述。
[0047]
x射线显影化合物
[0048]
本技术中,x射线显影化合物的结构式如下:
[0049][0050]
其中,x为由至少一个碘取代的c5-12芳基或c5-12杂芳基;
[0051]
y不存在;或者,y为c1-6亚烷基、c2-6亚烯基、c2-6亚炔基、c1-6亚烷氧基和c1-6烷氧基亚烷基的一种。
[0052]
在上述技术方案中,通过酰胺键直接或间接将咪唑基团或苯并咪唑基团与碘取代的芳基或杂芳基连接起来,形成x射线显影化合物,该显影化合物中的酰胺键处的c-n键容易发生断裂,以便将断裂处的c与栓塞材料键接(断裂和键接可以一步反应),以便使栓塞材料具有x射线显影功能。
[0053]
在一些实施例中,x为由至少一个碘取代的六元芳基或六元杂芳基;y不存在或y为亚甲基或y为亚烷氧基。可以使酰胺键中的c-n键更加容易断裂,使具有x射线显影功能的栓塞材料的制备更加方便。
[0054]
例如:x为例如:x为
等。
[0055]
在另一些实施例中,x还可以是至少一个碘取代的五元芳基或五元杂芳基;或,x还可以是至少一个碘取代的七元芳基或七元杂芳基;或,x还可以是至少一个碘取代的八元芳基或八元杂芳基;或,x还可以是至少一个碘取代的九元芳基或九元杂芳基;或,x还可以是至少一个碘取代的十元芳基或十元杂芳基;或,x还可以是至少一个碘取代的十一元芳基或十一元杂芳基;或,x还可以是至少一个碘取代的十二元芳基或十二元杂芳基。
[0056]
在另一些实施例中,y还可以是c2亚烷基、c3亚烷基、c4亚烷基、c5亚烷基、c6亚烷基、c2亚烯基、c3亚烯基、c4亚烯基、c5亚烯基、c6亚烯基、c2亚炔基、c3亚炔基、c4亚炔基、c5亚炔基、c6亚炔基、c2亚烷氧基、c3亚烷氧基、c4亚烷氧基、c5亚烷氧基、c6亚烷氧基、c1烷氧基亚烷基、c2烷氧基亚烷基、c3烷氧基亚烷基、c4烷氧基亚烷基、c5烷氧基亚烷基或c6烷氧基亚烷基的一种。
[0057]
在一些实施例中,x射线显影化合物的结构式为:
[0058][0059]
x射线显影化合物的制备方法
[0060]
上面介绍了x射线显影化合物的结构式以后,下面对x射线显影化合物的制备方法进行具体介绍,分别对化合物ⅰ和化合物ⅱ的制备方法进行介绍如下:
[0061]
x射线显影化合物为化合物ⅰ,化合物ⅰ制备方法可以有如下三种:
[0062]
第一种方式,其反应式一如下:
[0063]
化合物iii中的x和y满足:x为由至少一个碘取代的c5-12芳基或c5-12杂芳基;y不存在;或者,y为c1-6亚烷基、c2-6亚烯基、c2-6亚炔基、c1-6亚烷氧基和c1-6烷氧基亚烷基的一种。其中,目标产物化合物ⅰ的x和y的选择与反应物化合物iii的x和y的选择一致。
[0064]
可选地,在绝氧环境中,向化合物iii溶液中添加氯化亚砜(socl2),加热回流浓缩后,得到酰氯固体;将酰氯固体溶解在二氯甲烷(ch2cl2、dcm)中形成酰氯溶液;在绝氧环境中,将含有咪唑(化合物iv)、二氯甲烷(ch2cl2、dcm)和三乙胺(c6h
15
n,et3n)的溶液降温到0~5℃条件下,然后添加酰氯溶液,添加完成以后,在室温条件下反应得到化合物i。
偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(hatu)和无水二甲基甲酰胺(dmf),搅拌溶解后,再缓慢加入三乙胺(et3n),保持室温反应。tlc监控原料反应完全后,向反应液中加入水淬灭反应,然后加入乙酸乙酯(ea)萃取分层。留有机相,有机相用水洗涤三次,用无水硫酸钠干燥后,浓缩有机相,浓缩到一定程度后(留有一定量的ea)析出大量固体,降温到-5~0℃,搅拌析出固体,然后抽滤,再用少量冷的ea淋洗滤饼,滤饼在45℃下真空干燥2h后,得到较为纯净的化合物i,为白色固体。
[0074]
x射线显影化合物为化合物ⅱ,化合物ⅱ的制备方法中,反应式四如下:
[0075]
化合物iii中的x和y满足:x为由至少一个碘取代的c5-12芳基或c5-12杂芳基;y不存在;或者,y为c1-6亚烷基、c2-6亚烯基、c2-6亚炔基、c1-6亚烷氧基和c1-6烷氧基亚烷基的一种。其中,目标产物化合物ⅱ的x和y的选择与反应物化合物iii的x和y的选择一致。
[0076]
可选地,在绝氧环境中,向化合物iii溶液中添加氯化亚砜(socl2),加热回流浓缩后,得到酰氯固体;将酰氯固体溶解在二氯甲烷(dcm)中形成酰氯溶液;在绝氧环境中,将含有苯并咪唑(化合物v)、二氯甲烷(dcm)和三乙胺(et3n)的溶液降温到0~5℃条件下,然后添加酰氯溶液,添加完成以后,在室温条件下反应得到化合物ⅱ。
[0077]
例如:氮气保护下,在单口反应瓶中依次加入化合物iii、无水四氢呋喃(1,4-环氧丁烷,thf)搅拌溶解后,再缓慢加入氯化亚砜(socl2),升温到70℃让其回流反应3h后,tlc监控(取少量反应液用甲醇淬灭)原料反应完全后,把反应液直接浓缩至干得到灰白色酰氯固体。将酰氯固体溶解在无水二氯甲烷(ch2cl2、dcm)中形成酰氯溶液。在另一个三口烧瓶中,氮气保护下,依次加入苯并咪唑(化合物v)、无水二氯甲烷(ch2cl2、dcm)、三乙胺(c6h
15
n,et3n),然后降温到0~5℃下,缓慢滴加酰氯溶液,加完后,移到室温让其反应1.5~3h后,tlc监控原料反应完后,向反应液中加入水淬灭反应,然后萃取分层,留有机相,有机相浓缩至干后,再加入乙酸乙酯让其溶液后,再用水洗涤三次后,留有机相,用无水硫酸钠干燥后,浓缩有机相,浓缩到一定程度后析出大量固体,降温到-5~0℃,搅拌析出固体,然后抽滤,再用少量冷的乙酸乙酯淋洗滤饼,滤饼再在45℃下真空干燥2h后,即得到较为纯净的化合物i,为白色固体。
[0078]
反应式一或反应式四中,通过氯化亚砜作为中间反应物,将含有羧酸基的碘取代的芳基或杂芳基与咪唑或苯并咪唑缩合,制备较为方便。反应式二或反应式三中,通过n,n'-羰基二咪唑或2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯作为缩合剂,将含有羧酸基的碘取代的芳基或杂芳基与缩合剂缩合,制备较为方便。
[0079]
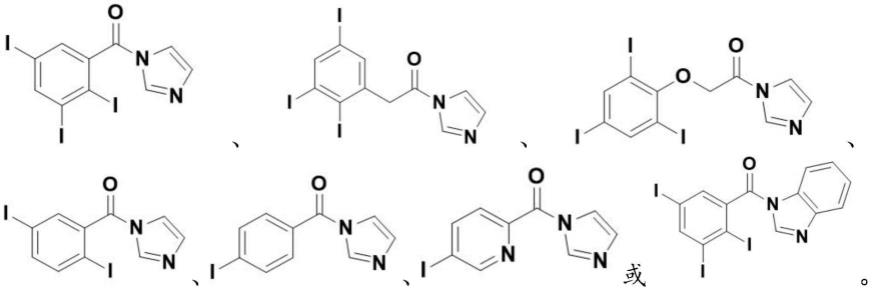
在一些实施例中,化合物ⅲ的结构式为:
[0080][0081]
经过上述四个反应式中的任意一种反应方式,可以得到的结构式如下的任意一种x射线显影化合物:
[0082][0083]
其中,化合物ⅲ的结构式中的x和y的选择与x射线显影化合物的x和y的选择是一一对应关系,在进行反应的时候,化合物ⅲ中x和y的选择与对应得到的x射线显影化合物的x和y的选择不发生变化。
[0084]
x射线显影栓塞材料
[0085]
本技术中,x射线显影栓塞材料是在栓塞材料上通过酯基连接-y-x基团,其中,x为由至少一个碘取代的c5-12芳基或c5-12杂芳基;y不存在;或者,y为c1-6亚烷基、c2-6亚烯基、c2-6亚炔基、c1-6亚烷氧基和c1-6烷氧基亚烷基的一种。
[0086]
该x射线显影栓塞材料通过酯基将栓塞材料与显影基团(碘取代的c5-12芳基或c5-12杂芳基)连接起来,栓塞材料具有显影功能,且稳定性较好。
[0087]
在本技术的部分实施例中,x射线显影栓塞材料的结构式为:
[0088]
其中m和n为正整数。
[0089]
该x射线显影栓塞材料可以是固体栓塞剂或液体栓塞剂,其均能够实现显影功能。
[0090]
在一些实施例中,x为由至少一个碘取代的六元芳基或六元杂芳基;y不存在或y为
亚甲基或y为亚烷氧基。
[0091]
在另一些实施例中,x还可以是至少一个碘取代的五元芳基或五元杂芳基;或,x还可以是至少一个碘取代的七元芳基或七元杂芳基;或,x还可以是至少一个碘取代的八元芳基或八元杂芳基;或,x还可以是至少一个碘取代的九元芳基或九元杂芳基;或,x还可以是至少一个碘取代的十元芳基或十元杂芳基;或,x还可以是至少一个碘取代的十一元芳基或十一元杂芳基;或,x还可以是至少一个碘取代的十二元芳基或十二元杂芳基。
[0092]
在另一些实施例中,y还可以是c2亚烷基、c3亚烷基、c4亚烷基、c5亚烷基、c6亚烷基、c2亚烯基、c3亚烯基、c4亚烯基、c5亚烯基、c6亚烯基、c2亚炔基、c3亚炔基、c4亚炔基、c5亚炔基、c6亚炔基、c2亚烷氧基、c3亚烷氧基、c4亚烷氧基、c5亚烷氧基、c6亚烷氧基、c1烷氧基亚烷基、c2烷氧基亚烷基、c3烷氧基亚烷基、c4烷氧基亚烷基、c5烷氧基亚烷基或c6烷氧基亚烷基的一种。
[0093]
在一些实施例中,x射线显影栓塞材料的结构式为:
[0094][0095]
x射线显影栓塞材料的制备方法
[0096]
本技术中,通过上述的x射线显影化合物进行x射线显影栓塞材料的制备,具体可以是:在1,8-二氮杂双环[5.4.0]十一碳-7-烯的催化作用下,使第一方面任一项提供的x射线显影化合物的酰胺键与栓塞材料上的至少部分羟基反应,形成酯基连接。可以使x射线显影栓塞材料的制备更加简单,以便使栓塞材料具有x射线显影功能,且通过酯基连接显影分子和栓塞材料,可以使x射线显影栓塞材料的稳定性较好。
[0097]
在一些实施例中,x射线显影化合物为化合物i,咪唑基脱除,并与栓塞材料的至少部分羟基处的o键连接,形成x射线显影栓塞材料;在另一些实施例中,x射线显影化合物为化合物ⅱ,苯并咪唑基脱除,并与栓塞材料的至少部分羟基处的o键连接,形成x射线显影栓塞材料。
[0098]
在一些实施例中,栓塞材料为固体栓塞剂,制备方法包括:将聚乙烯醇微球、x射线显影化合物、1,8-二氮杂双环[5.4.0]十一碳-7-烯(dbu)与溶剂混合形成混合液,将混合液
于40~90℃反应24~48h。在上述条件下,以1,8-二氮杂双环[5.4.0]十一碳-7-烯作为催化剂,可以使x射线显影化合物中的c-n键处断裂,并将断裂处的c与聚乙烯醇的至少部分羟基处脱氢键连接,该条件较为温和,容易实现,且最终得到的显影微球的稳定性较好。
[0099]
例如:将聚乙烯醇微球、x射线显影化合物(化合物i或ii)与溶剂(二甲基亚砜(dmso)或n,n-二甲基甲酰胺(dmf))混合后加热溶胀,然后加入1,8-二氮杂双环[5.4.0]十一碳-7-烯(dbu),于40~90℃反应24~48h。反应后将微球依次用dmso、10%碳酸氢钠和水洗涤,除去残留的显影剂和酸。可选地,化合物i或ii的质量为聚乙烯醇微球质量的1.5-3倍;1,8-二氮杂双环[5.4.0]十一碳-7-烯的物质的量为化合物i或ii的物质的量的0.5-2倍。
[0100]
其中,聚乙烯醇微球的制备方法为:将聚乙烯醇与n-(2,2-二甲氧基乙基)-2-丙烯酰胺进行酸催化反应,使得聚乙烯醇上接枝n-(2,2-二甲氧基乙基)-2-丙烯酰胺,得到大分子聚乙烯醇单体;将大分子聚乙烯醇单体与带磺酸基的水溶性单体、引发剂和水配制成水相加入油相中,形成油包水的反相悬浮聚合体系;将反相悬浮聚合体系升温至反应温度,在搅拌条件下,加入催化剂进行反应;从反应后的体系中分离出微球,再净化、干燥。在其他实施例中,聚乙烯醇微球也可以是现有任意方法制备得到的材料,或购买得到。
[0101]
使用聚乙烯醇微球与x射线显影化合物来制备x射线显影栓塞材料的反应式五如下:
[0102][0103]
其中,反应式五中示出的是:聚乙烯微球上的多个羟基发生反应,并连接了x射线显影化合物。
[0104]
在本技术的部分实施例中,栓塞材料为液体栓塞剂,制备方法包括:将乙烯乙烯醇共聚物、x射线显影化合物、1,8-二氮杂双环[5.4.0]十一碳-7-烯与溶剂混合形成混合液,将混合液于40~90℃反应24~48h;将反应物溶于溶剂中,得到液体栓塞剂。在上述条件下,以1,8-二氮杂双环[5.4.0]十一碳-7-烯作为催化剂,可以使x射线显影化合物中的c-n键处断裂,并将断裂处的c与乙烯乙烯醇共聚物的至少部分羟基处脱氢键连接,该条件较为温和,容易实现,且最终得到的显影液体栓塞剂的稳定性较好。
[0105]
例如:将乙烯乙烯醇共聚物、x射线显影化合物(化合物i或ii)与溶剂(二甲基亚砜(dmso)或n,n-二甲基甲酰胺(dmf))混合后加热溶胀,然后加入1,8-二氮杂双环[5.4.0]十一碳-7-烯(dbu),于40~90℃反应24~48h。反应结束后冷却至室温,将聚合物溶液倒入200-300ml甲醇中,沉淀析出聚合物固体。过滤,聚合物固体用150ml dmso于80℃加热溶解后,倒入200-300ml甲醇中,沉淀析出聚合物固体。重复此操作3次。将此聚合物于80℃再次溶于100ml dmso或n-甲基吡咯烷酮,冷却至室温,制得具有x射线显影功能的液体栓塞剂。可选地,化合物i或ii的质量为乙烯乙烯醇共聚物质量的1.5-3倍;1,8-二氮杂双环[5.4.0]十一碳-7-烯的物质的量为化合物i或ii的物质的量的0.5-2倍。
[0106]
使用乙烯乙烯醇共聚物与x射线显影化合物来制备x射线显影栓塞材料的反应式六如下:
[0107][0108]
其中,反应式六中示出的是:乙烯乙烯醇共聚物的单体结构上的羟基发生反应,并连接了x射线显影化合物,其仅仅是一个示例,是为了解释说明x射线显影液体栓塞剂的制备机理,在实际反应中,一般是一部分羟基上均会连接x射线显影化合物,而不是所有羟基上连接。
[0109]
基于上述的x射线显影化合物i或ii,制备得到的x射线显影的栓塞微球或液体栓塞剂,均稳定性较好,且具有x射线显影功能。
[0110]
为使本技术实施例的目的、技术方案和优点更加清楚,下面将对本技术实施例中的技术方案进行清楚、完整地描述。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
[0111]
实施例1:化合物i-1的合成
[0112][0113]
氮气保护下,在100ml单口反应瓶中依次加入化合物iii-1(5g,1.0eq.)、无水thf(50ml)搅拌溶解后,再缓慢加入氯化亚砜(10eq.),升温到70℃让其回流反应3h后,tlc监控(取少量反应液用甲醇淬灭)原料反应完全后,把反应液直接浓缩至干得到灰白色酰氯固体。无水dcm溶解酰氯固体得到酰氯溶液。
[0114]
在另一个三口烧瓶中,氮气保护下,依次加入化合物iv(2.0eq.)、无水dcm(50ml)、三乙胺(3.0eq.),然后降温到0~5℃下,缓慢滴加上面制备好的酰氯溶液。加完后,移到室温让其反应1.5h后,tlc监控原料反应完后,向反应液中加入水淬灭反应,然后萃取分层,留有机相,有机相浓缩至干后,再加入乙酸乙酯让其溶液后,再用水洗涤三次后,留有机相,用无水硫酸钠干燥后,浓缩有机相,浓缩到一定程度后析出大量固体,降温到-5~0℃,搅拌析出固体,然后抽滤,再用少量冷的乙酸乙酯淋洗滤饼,滤饼再在45℃下真空干燥2hr后,即得到目标产物i-1(5g,91%),为白色固体(纯度98%),熔点255~257℃。
[0115]
将得到的目标产物i-1经hplc(高效液相色谱),lcms(液相质谱)和1h-nmr(核磁氢谱)表征。方法如下:
[0116]
hplc:测试仪器为安捷伦1260型高效液相色谱仪;色谱柱为十八烷基键合硅胶柱;流动相为0.1%磷酸水溶液和甲醇;检测器为vwd检测器;取0.1mg左右样品于样品瓶中,加dmso溶解后直接进样检测;根据积分面积确定产品纯度。
[0117]
lcms:测试仪器为安捷伦1260-6125型高效液相色谱质谱联用仪;色谱柱为十八烷
基键合硅胶柱;流动相为0.1%甲酸水溶液和甲醇;检测器为质谱检测器;取0.1mg左右样品于样品瓶中,加dmso溶解后直接进样检测,识别主要离子峰。
[0118]1h-nmr:测试仪器为bruker 400m核磁测试仪;采用探头为常温氢谱探头;取20-30mg样品加入核磁管中,加入0.3-0.5ml氘代dmso溶解后测试。得到的目标产物i-1的核磁氢谱如图1,1h nmr(400mhz,dmso-d6)δ8.45(d,j=1.9hz,1h),8.11(s,1h),8.01(d,j=1.9hz,1h),7.61(s,1h),7.15-7.14(m,1h);ms(esi):m/z 551(m h
)。
[0119]
实施例2:化合物i-1的合成
[0120][0121]
氮气保护下,在500ml单口反应瓶中依次加入化合物iii-1(30g,1.0eq.)、cdi(11.68g,1.2eq.)和无水thf(300ml),搅拌溶解后,再缓慢加入三乙胺(3.0eq.),保持室温反应。经tlc点板监控原料反应完全后,向反应液中加入水淬灭反应,然后加入ea萃取分层。留有机相,用水洗涤三次后,无水硫酸钠干燥,浓缩有机相。浓缩到一定程度后(留有一定量的ea)析出大量固体,降温到-5~0℃,搅拌析出固体,然后抽滤,再用少量冷的ea淋洗滤饼,滤饼在45℃下真空干燥2h后,得到目标产物i-1(30.4g,92%),为白色固体(纯度99%),熔点255~257℃。检测方法同实施例1,得到结果为:1h nmr(400mhz,dmso-d6)δ8.45(d,j=1.9hz,1h),8.11(s,1h),8.01(d,j=1.9hz,1h),7.61(s,1h),7.15-7.14(m,1h);ms(esi):m/z 551(m h
)。
[0122]
实施例3:化合物i-1的合成
[0123][0124]
氮气保护下,在单口反应瓶中依次加入化合物iii-1(5g,1.0eq.)、化合物iv(2.0eq.)、hatu(1.5eq.)和无水dmf(50ml),搅拌溶解后,再缓慢加入三乙胺(3.0eq.),保持室温反应。tlc监控原料反应完全后,向反应液中加入水淬灭反应,然后加入ea萃取分层。留有机相,有机相用水洗涤三次,用无水硫酸钠干燥后,浓缩有机相,浓缩到一定程度后(留有一定量的ea)析出大量固体,降温到-5~0℃,搅拌析出固体,然后抽滤,再用少量冷的ea淋洗滤饼,滤饼在45℃下真空干燥2h后,得到目标产物i-1(4.3g,78%),为白色固体(纯度98%),熔点255~257℃。检测方法同实施例1,得到结果为:1h nmr(400mhz,dmso-d6)δ8.45(d,j=1.9hz,1h),8.11(s,1h),8.01(d,j=1.9hz,1h),7.61(s,1h),7.15-7.14(m,1h);ms(esi):m/z 551(m h
)。
[0125]
实施例4:化合物ii-1的合成
[0126][0127]
氮气保护下,在100ml单口反应瓶中依次加入化合物iii-1(5g,1.0eq.)、无水thf(50ml)搅拌溶解后,再缓慢加入氯化亚砜(10eq.),升温到70℃让其回流反应3hr后,tlc监控(取少量反应液用甲醇淬灭)原料反应完全后,把反应液直接浓缩至干得到灰白色酰氯固体;同时在另一个三口烧瓶中,氮气保护下,依次加入化合物v(2.0eq.)、无水dcm(50ml)、三乙胺(3.0eq.),然后降温到0~5℃下,缓慢滴加上面制备好的酰氯溶液(用无水dcm溶解制备好的酰氯固体)。加完后,移到室温让其反应1.5h后,tlc监控原料反应完后,向反应液中加入水淬灭反应,然后萃取分层,留有机相,有机相浓缩至干后,再加入乙酸乙酯让其溶液后,再用水洗涤三次后,留有机相,用无水硫酸钠干燥后,浓缩有机相,浓缩到一定程度后析出大量固体,降温到-5~0℃,搅拌析出固体,然后抽滤,再用少量冷的乙酸乙酯淋洗滤饼,滤饼再在45℃下真空干燥2h后,即得到目标产物ii-1(5g,83%),为白色固体(纯度98%),熔点278~279℃。检测方法同实施例1,得到结果为:1h nmr(400mhz,dmso-d6)δ8.46(d,j=1.9hz,1h),8.13(s,1h),8.02(d,j=1.9hz,1h),8.00-7.98(m,1h),7.61-7.63(m,1h),7.33-7.31(m,1h),7.15-7.13(m,1h);ms(esi):m/z 601(m h
)。
[0128]
实施例5:化合物i-2的合成
[0129][0130]
氮气保护下,在100ml单口反应瓶中依次加入化合物iii-2(5g,1.0eq.)、cdi(1.2eq.)和无水thf(50ml),搅拌溶解后,再缓慢加入三乙胺(3.0eq.),保持室温反应。经tlc点板监控原料反应完全后,向反应液中加入水淬灭反应,然后加入ea萃取分层。留有机相,用水洗涤三次后,无水硫酸钠干燥,浓缩有机相。浓缩到一定程度后(留有一定量的ea)析出大量固体,降温到-5~0℃,搅拌析出固体,然后抽滤,再用少量冷的ea淋洗滤饼,滤饼在45℃下真空干燥2h后,得到目标产物i-2(5.2g,94%),为白色固体(纯度98%),熔点230~232℃。检测方法同实施例1,得到结果为:1h nmr(400mhz,dmso-d6)δ8.40(d,j=1.9hz,1h),8.00(s,1h),7.95(d,j=1.9hz,1h),7.61(s,1h),7.15-7.14(m,1h),4.23(s,2h);ms(esi):m/z 565(m h
)。
[0131]
实施例6:化合物i-3的合成
[0132][0133]
氮气保护下,在100ml单口反应瓶中依次加入化合物iii-3(5g,1.0eq.)、cdi
(1.2eq.)和无水thf(50ml),搅拌溶解后,再缓慢加入三乙胺(3.0eq.),保持室温反应。经tlc点板监控原料反应完全后,向反应液中加入水淬灭反应,然后加入ea萃取分层。留有机相,用水洗涤三次后,无水硫酸钠干燥,浓缩有机相。浓缩到一定程度后(留有一定量的ea)析出大量固体,降温到-5~0℃,搅拌析出固体,然后抽滤,再用少量冷的ea淋洗滤饼,滤饼在45℃下真空干燥2h后,得到目标产物i-3(4.9g,90%),为白色固体(纯度98%),熔点210~211℃。检测方法同实施例1,得到结果为:1h nmr(400mhz,dmso-d6)δ8.45(s,2h),8.01(s,1h),7.61(s,1h),7.15-7.14(m,1h),4.57(s,2h);ms(esi):m/z 581(m h
)。
[0134]
实施例7:化合物i-4的合成
[0135][0136]
氮气保护下,在100ml单口反应瓶中依次加入化合物iii-4(5g,1.0eq.)、cdi(1.2eq.)和无水thf(50ml),搅拌溶解后,再缓慢加入三乙胺(3.0eq.),保持室温反应。经tlc点板监控原料反应完全后,向反应液中加入水淬灭反应,然后加入ea萃取分层。留有机相,用水洗涤三次后,无水硫酸钠干燥,浓缩有机相。浓缩到一定程度后(留有一定量的ea)析出大量固体,降温到-5~0℃,搅拌析出固体,然后抽滤,再用少量冷的ea淋洗滤饼,滤饼在45℃下真空干燥2h后,得到目标产物i-4(5.3g,93%),为白色固体(纯度98%),熔点250~251℃。检测方法同实施例1,得到结果为:1h nmr(400mhz,dmso-d6)δ8.17(d,j=0.7hz,1h),8.10(s,1h),8.03(dd,j=5.6,0.7hz,1h),7.94(d,j=5.6hz,1h),7.60-7.59(m,1h),7.16-7.15(m,1h);ms(esi):m/z 425(m h
)。
[0137]
实施例8:化合物i-5的合成
[0138][0139]
氮气保护下,在100ml单口反应瓶中依次加入化合物iii-5(5g,1.0eq.)、cdi(1.2eq.)和无水thf(50ml),搅拌溶解后,再缓慢加入三乙胺(3.0eq.),保持室温反应。经tlc点板监控原料反应完全后,向反应液中加入水淬灭反应,然后加入ea萃取分层。留有机相,用水洗涤三次后,无水硫酸钠干燥,浓缩有机相。浓缩到一定程度后(留有一定量的ea)析出大量固体,降温到-5~0℃,搅拌析出固体,然后抽滤,再用少量冷的ea淋洗滤饼,滤饼在45℃下真空干燥2h后,得到目标产物i-5(5.5g,91%),为白色固体(纯度98%),熔点253~254℃。检测方法同实施例1,得到结果为:1h nmr(400mhz,dmso-d6)δ7.85(s,1h),7.91(d,j=6.3hz,2h),7.74(d,j=6.3hz,2h),7.61-7.59(m,1h),7.18-7.17(m,1h);ms(esi):m/z 299(m h
)。
[0140]
实施例9:化合物i-6的合成
[0141][0142]
氮气保护下,在100ml单口反应瓶中依次加入化合物iii-6(5g,1.0eq.)、cdi(1.2eq.)和无水thf(50ml),搅拌溶解后,再缓慢加入三乙胺(3.0eq.),保持室温反应。经tlc点板监控原料反应完全后,向反应液中加入水淬灭反应,然后加入ea萃取分层。留有机相,用水洗涤三次后,无水硫酸钠干燥,浓缩有机相。浓缩到一定程度后(留有一定量的ea)析出大量固体,降温到-5~0℃,搅拌析出固体,然后抽滤,再用少量冷的ea淋洗滤饼,滤饼在45℃下真空干燥2h后,得到目标产物i-6(4.4g,73%),为白色固体(纯度98%),熔点270~271℃。检测方法同实施例1,得到结果为:1h nmr(400mhz,dmso-d6)δ8.37(d,j=0.5hz,1h),8.13(s,1h),8.14(dd,j=4.4,0.5hz,1h),7.99(d,j=4.4hz,1h),7.61-7.59(m,1h),7.26-7.25(m,1h);ms(esi):m/z 300(m h
)。
[0143]
实施例一:显影微球的制备
[0144]
(1)pva干球的制备:将聚乙烯醇与n-(2,2-二甲氧基乙基)-2-丙烯酰胺进行酸催化反应,使得聚乙烯醇上接枝n-(2,2-二甲氧基乙基)-2-丙烯酰胺,得到大分子聚乙烯醇单体;将大分子聚乙烯醇单体与带磺酸基的水溶性单体、引发剂和水配制成水相加入油相中,形成油包水的反相悬浮聚合体系;将反相悬浮聚合体系升温至反应温度,在搅拌条件下,加入催化剂进行反应;从反应后的体系中分离出微球,再净化、干燥。
[0145]
(2)显影微球的制备:
[0146][0147]
反应瓶中依次加入1g上述制备的pva干球、2g化合物i-1(实施例1提供的化合物i-1)和25ml二甲亚砜(dmso),加热溶胀后加入0.5ml dbu,80℃反应48h。反应结束后沥出微球,用dmso和热的碳酸氢钠溶液各洗三次,水洗后得到显影微球。取2.0ml湿球,用丙酮干燥后,105℃下烘干5h至恒重后称量其重量为831.6mg。
[0148]
实施例二:显影微球的制备
[0149]
pva干球的制备方法与实施例一相同,其不同在于:
[0150][0151]
反应瓶中依次加入1g上述制备的pva干球、2g化合物ii-1(实施例4提供的化合物
ii-1)和25ml二甲亚砜(dmso),加热溶胀后加入0.5ml dbu,80℃反应48h。反应结束后沥出微球,用dmso和热的碳酸氢钠溶液各洗三次,水洗后得到显影微球。取2.0ml湿球,用丙酮干燥后,105℃下烘干5h至恒重后称量其重量为861.3mg。
[0152]
实施例三:显影微球的制备
[0153]
pva干球的制备方法与实施例一相同,其不同在于:
[0154][0155]
反应瓶中依次加入1g上述制备的pva干球、2g化合物i-2(实施例5提供的化合物i-2)和25ml二甲亚砜(dmso),加热溶胀后加入0.5ml dbu,80℃反应48h。反应结束后沥出微球,用dmso和热的碳酸氢钠溶液各洗三次,水洗后得到显影微球。取2.0ml湿球,用丙酮干燥后,105℃下烘干5h至恒重后称量其重量为861.0mg。
[0156]
实施例四:显影微球的制备
[0157]
pva干球的制备方法与实施例一相同,其不同在于:
[0158][0159]
反应瓶中依次加入1g上述制备的pva干球、2g化合物i-3(实施例6提供的化合物i-3)和25ml二甲亚砜(dmso),加热溶胀后加入0.5ml dbu,80℃反应48h。反应结束后沥出微球,用dmso和热的碳酸氢钠溶液各洗三次,水洗后得到显影微球。取2.0ml湿球,用丙酮干燥后,105℃下烘干5h至恒重后称量其重量为891.4mg。
[0160]
实施例五:显影微球的制备
[0161]
pva干球的制备方法与实施例一相同,其不同在于:
[0162][0163]
反应瓶中依次加入1g上述制备的pva干球、2g化合物i-4(实施例7提供的化合物i-4)和25ml二甲亚砜(dmso),加热溶胀后加入0.5ml dbu,80℃反应48h。反应结束后沥出微球,用dmso和热的碳酸氢钠溶液各洗三次,水洗后得到显影微球。取2.0ml湿球,用丙酮干燥后,105℃下烘干5h至恒重后称量其重量为830.7mg。
[0164]
实施例六:显影微球的制备
[0165]
pva干球的制备方法与实施例一相同,其不同在于:
[0166][0167]
反应瓶中依次加入1g上述制备的pva干球、2g化合物i-5(实施例8提供的化合物i-5)和25ml二甲亚砜(dmso),加热溶胀后加入0.5ml dbu,80℃反应48h。反应结束后沥出微球,用dmso和热的碳酸氢钠溶液各洗三次,水洗后得到显影微球。取2.0ml湿球,用丙酮干燥后,105℃下烘干5h至恒重后称量其重量为820.6mg。
[0168]
实施例七:显影微球的制备
[0169]
pva干球的制备方法与实施例一相同,其不同在于:
[0170][0171]
反应瓶中依次加入1g上述制备的pva干球、2g化合物i-6(实施例9提供的化合物i-6)和25ml二甲亚砜(dmso),加热溶胀后加入0.5ml dbu,80℃反应48h。反应结束后沥出微球,用dmso和热的碳酸氢钠溶液各洗三次,水洗后得到显影微球。取2.0ml湿球,用丙酮干燥后,105℃下烘干5h至恒重后称量其重量为692.4mg。
[0172]
实施例八:液体栓塞材料的制备
[0173][0174]
反应瓶中依次加入5g乙烯乙烯醇共聚物(evoh)上、10g化合物i-1(实施例1提供的化合物i-1)和125ml二甲亚砜(dmso),加热充分溶解后加入2.5ml dbu,80℃反应48h。反应结束后冷却至室温,将聚合物溶液倒入200-300ml甲醇中,沉淀析出聚合物固体。过滤,聚合物固体用150ml dmso于80℃加热溶解后,倒入200-300ml甲醇中,沉淀析出聚合物固体。重复此操作3次。得到的聚合物固体用丙酮干燥后,105℃下烘干5h至恒重后称量其重量为6.9g。将此聚合物于80℃再次溶于100ml n-甲基吡咯烷酮,冷却至室温,制得具有x射线显影功能的液体栓塞剂。此栓塞材料在体外为流动性良好的液体,注射于人体血管内后沉淀析出固体,原位产生栓塞。
[0175]
实施例九:液体栓塞材料的制备
[0176][0177]
反应瓶中依次加入5g乙烯乙烯醇共聚物(evoh)上、10g化合物i-2(实施例5提供的化合物i-2)和125ml二甲亚砜(dmso),加热充分溶解后加入2.5ml dbu,80℃反应48h。反应结束后冷却至室温,将聚合物溶液倒入200-300ml甲醇中,沉淀析出聚合物固体。过滤,聚合物固体用150ml dmso于80℃加热溶解后,倒入200-300ml甲醇中,沉淀析出聚合物固体。重复此操作3次。得到的聚合物固体用丙酮干燥后,105℃下烘干5h至恒重后称量其重量为7.2g。将此聚合物于80℃再次溶于100ml dmso,冷却至室温,制得具有x射线显影功能的液体栓塞剂。此栓塞材料在体外为流动性良好的液体,注射于人体血管内后沉淀析出固体,原位产生栓塞。
[0178]
对比例一:显影微球的制备
[0179]
pva干球的制备方法与实施例一相同,其不同在于:
[0180][0181]
反应瓶中依次加入1g上述制备的pva干球、2g化合物x(实施例9提供的化合物i-6)和25ml二甲亚砜(dmso),加热溶胀后加入0.5ml dbu,80℃反应48h。反应结束后沥出微球,用dmso和热的碳酸氢钠溶液各洗三次,水洗后得到显影微球。取2.0ml湿球,用丙酮干燥后,105℃下烘干5h至恒重后称量其重量为473.4mg。
[0182]
图2为实施例一提供的显影微球的光学图片;图3为实施例二提供的显影微球的光学图片;图4为实施例三提供的显影微球的光学图片;图5为实施例四提供的显影微球的光学图片;图6为实施例五提供的显影微球的光学图片;图7为实施例六提供的显影微球的光学图片;图8为实施例七提供的显影微球的光学图片。从图2~图8可以看出,本技术实施例一~实施例七制备得到的产品为微球结构。
[0183]
检测实施例一~实施例九以及对比例一提供的显影栓塞材料的性能进行检测,测试结果如表1,其测试方法如下:
[0184]
取2.0ml湿球,用丙酮干燥后,105℃下烘干5h至恒重后称量其重量。取部分干球测量其干球碘含量。
[0185]
称取约5mg样品于无尘灰纸上,按照《中国药典》第二部操作,吸收液为:10ml 5g/l
氢氧化钠与10ml 1%维生素c,急速通氧1min,用表面皿覆盖瓶口,静置10min,点燃包有样品的滤纸尾部,迅速将瓶塞插入燃烧瓶中,按紧瓶塞,并用少量水封闭瓶口,燃烧完毕后(应无灰色、黑色碎片或颗粒,若燃烧后留有应无灰色、黑色碎片或颗粒,表示供试品燃烧不完全,遇此情况应重新取样燃烧),立即充分振摇,使生成的烟雾完全吸入吸收液中,放置15min,开启瓶塞,用少量水冲洗瓶塞及铂丝,合并洗液及吸收液,同时同法做空白实验。加入3滴署红指示剂,用0.01mol/l硝酸银标准滴定液滴定,记录消耗体积,平行操作两次。
[0186]
计算:
[0187]
x=(c
×v×
127
×m湿球
)/(m
×v湿球
)
×
100%
[0188]
x:为样品中碘含量;
[0189]
c:为硝酸银浓度,mol/l;
[0190]
v:为硝酸银消耗体积,ml;
[0191]
m:为样品质量,mg;
[0192]
m(湿球):为湿球质量,mg;
[0193]
v(湿球):为湿球体积,ml;
[0194]
127:为碘的相对分子质量。
[0195]
湿球碘含量计算公式如下:
[0196]
湿球碘含量=(微球干重
×
干球碘含量)/湿球体积
[0197]
hu值测试方法:
[0198]
将各实施例显影微球进行micro ct测试,对根据上面方法制备的显影微球样品的不透射线性进行评估。在nunc冻存管小瓶中将珠粒悬浮于0.5%琼脂糖凝胶,使用装备有钨阳极的微型ct成像系统扫描器,利用微计算机断层扫描技术(微型ct成像系统),对显影微球的不透射线性进行测试。然后将图像分割从孔隙结构中分离出聚合物以便报告聚合物辐射密度。然后,使用在同一天得到的水标准品计算以hu为单位的辐射密度。
[0199]
稳定性测试方法:
[0200]
取2ml上述制备的显影微球,加入20ml 10%的碳酸氢钠溶液,煮沸后继续加热30min,清水洗净后用丙酮干燥,105℃下烘干5h至恒重后称量其重量,测干球碘含量并计算其湿球碘含量。比较碳酸氢钠溶液处理前后的湿球碘含量,数值无明显变化或减少较少为稳定性较好;反之则稳定性较差。
[0201]
表1显影栓塞材料的性能
[0202][0203]
从表1可以看出,本技术提供的x射线显影化合物可以使栓塞材料具有显影功能,且可以使得到的栓塞材料的稳定性较高。
[0204]
以上所描述的实施例是本技术一部分实施例,而不是全部的实施例。本技术的实施例的详细描述并非旨在限制要求保护的本技术的范围,而是仅仅表示本技术的选定实施例。基于本技术中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本技术保护的范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。