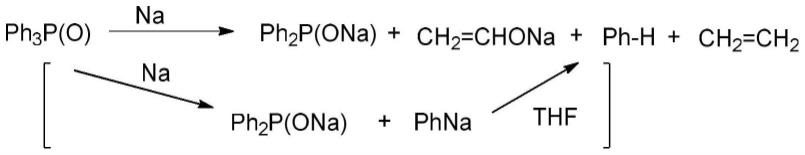
1.本发明涉及有机合成技术领域,更具体地说,涉及一种膦基化合物的制备方法及其应用。
背景技术:
2.磷化合物在工业生产中有各种用途。三苯基氧化膦ph3p(o)是维生素制造过程中使用wittig(维蒂希)反应产生的大量副产的工业废弃物,由于三苯基氧膦没有好的用途,大量的三苯基氧膦,通常作为固废焚烧处理,这会造成磷资源的浪费及环境的污染。
3.现有资料中记载,ph3p(o)与钠的反应虽然可以在低温液氨中进行生成有利用价值的ph2pona(1:esteban r.n.bornancini等人、 j.org.chem.1990,55,2332-2326;2:marek stankevic等人、tetrahedron 67(2011)8671-8678),但是,液氨的毒性高,容易迅速气化,反应操作繁杂;另外,这个条件下也会因过度还原等产生大量的副产物。还有资料中记载, ph3p(o)可以和nah-lii反应生成使ph3p(o)还原而产生ph2pona,但是nah昂贵,而且这个反应过程必须同时使用lii,此工艺繁杂(ciputra tejo等人、 chem.commun.,2018,54,1782-1785)。
[0004][0005]
phna会和溶剂反应生成rona等。rona和ph2pona难分离,只能混合物用。
[0006]
还有文献杂志中记载,在thf等极性醚溶剂里,ph3p(o)和na反应可生成 ph2pona。但是,该反应使用块状钠,会伴随许多副产物(zhang,j.et al j. org.chem.2020,85,14166-14173)。还有的资料中记载通过使用微米钠,可以高选择性可得到ph2pona,从而达到高收率生产各种膦氧化合物(zhang, j.-q.et al communications chemistry| (2020)3:1|https://doi.org/10.1038/s42004-019-0249-6|www.nature.com /commschem),但是,该反应中会生成的phna,会消耗溶剂,同时,生成的杂质rona可溶解在溶剂里面,不好将ph2pona从溶剂中分离出来,ph2pona与 rona无法有效的分离。
[0007]
截至目前为止,上述文献中记载,ph3po和na反应会产生ph2pona,均只是被假设提出,没有得到过纯的ph2pona。ph2pona的结构也从来没有被确凿无疑地证明过,如表征一个化合物的构造不可缺少的氢谱和碳谱,至今为止 ph2pona从来没有被报道过。
[0008]
再者,ph3po和na反应会产生的副产rona和ph2pona一起溶解在溶液里面,由于rona和ph2pona具有类似的反应性,还会形成竞争反应,还会影响 ph2pona参与反应的利用率。如想利用ph3po和na反应的反应产物ph2pona与 me3sicl反应,制备ph2posime3时,rona也会和me3sic反应生成rosime3,这不但会造成原料浪费,还使得产物ph2posime3的后续分离纯化步骤复杂。
[0009][0010]
rona和ph2pona会形成竞争反应,生成副产物rosime3[0011]
通过上述分析发现现有在thf等极性醚溶剂里,ph3p(o)和na反应可生成 ph2pona的制备方法中存在如下缺陷:
[0012]
1.使用块状金属钠时,会导致ph2pona选择性低,还会伴随产生很多副产物;
[0013]
2.ph3p(o)与金属钠反应时,thf溶剂还会遭破坏;
[0014]
3.ph3p(o)与金属钠反应产生的副产物和推测反应产物ph2pona均可溶在溶剂中,无法得到纯的ph2pona,更无法去验证这个ph2pona的用途及应用价值。
[0015]
4.通过现有的ph3p(o)与金属钠制备方法所得的反应产物去制备 ph2posime
3,
很难得到纯净的ph2posime
3,
同时若想通过ph3p(o)与金属钠反应产物去制备ph2posime
3,
常常需要加入过量的me3sicl才行,这会造成原料浪费。
[0016]
上述技术问题,一直是我们亟待解决的技术难题。
技术实现要素:
[0017]
1.要解决的技术问题
[0018]
针对现有技术中存在的问题,本发明的目的在于提供一种膦基化合物的制备方法,它可弥补以前使用r3po与钠反应的制备方法制备出r1r2pona所带来的各种缺陷。
[0019]
2.技术方案
[0020]
为解决上述问题,本发明采用如下的技术方案。
[0021]
一种膦基化合物的制备方法,所述方法包括如下步骤:第一步:在惰性气体或氮气气氛下,将膦氧类化合物r3po与质子化合物进行混合,获得混合物;
[0022]
r3po为r1r2r3po,r1、r2和r3分别独立选自芳香族或脂肪族取代基,r1、 r2和r3中至少一个为芳香族取代基,r1、r2和r3可相同也可不同;
[0023]
所述质子化合物选自水、醇、有机酸或无机酸;
[0024]
所述膦氧类化合物r3po与质子化合物的摩尔比为0.2-3∶1;
[0025]
第二步:向所述第一步中所获得的混合物中加入金属钠,膦氧类化合物 r3po、质子化合物和金属钠进行充分反应生成naoh或质子化合物钠盐、r2pona 以及r系有机物,r2pona结构中含有两个r,r1、r2和r3中的任意两个r为r2pona 中的两个r,剩余一个r为r系有机物中的r;
[0026]
第三步:反应结束后,分离反应生成的naoh或质子化合物钠盐、r2pona 以及r系有机物,得r2pona。
[0027]
进一步的,所述第一步中的膦氧类化合物r3po与质子化合物进行混合后,混合物中的膦氧类化合物r3po与质子化合物反应生成络合物,络合物生成后,再进行第二步。
[0028]
进一步的,所述第一步中的膦氧类化合物r3po与质子化合物进行混合后,混合物中的膦氧类化合物r3po与质子化合物不反应生成络合物,直接进入第二步。
[0029]
进一步的,所述第二步中,在膦氧类化合物r3po、质子化合物和金属钠进行反应前,可向膦氧类化合物r3po与质子化合物形成的混合物中加入第一溶剂,所述第一溶剂为烷烃、芳烃或有机醚中的一种或任意多种混合。
[0030]
进一步的,所述第二步中,在膦氧类化合物r3po、质子化合物和金属钠进行反应前,不向膦氧类化合物r3po与质子化合物形成的混合物中加入第一溶剂,所述第一溶剂为烷烃、芳烃或有机醚中的一种或任意多种混合。
[0031]
进一步的,所述第一溶剂为非醚类有机溶剂时,所述第三步中,反应结束后,向反应体系中加入第二溶剂将生成的r1r2pona进行溶解,然后进行过滤去除反应生成的naoh或质子化合物钠盐,最后减压旋蒸去除第一溶剂、第二溶剂和r系有机物得固体r2pona;所述第二溶剂为r2pona可溶但质子化合物钠盐不溶的有机醚。
[0032]
也就是说,若膦氧类化合物r3po、质子化合物和金属钠在无第一溶剂或在r2pona不溶、质子化合物钠盐也不溶的第一溶剂条件下,所述第三步中,反应结束后,向反应体系中加入第二溶剂将生成的r1r2pona进行溶解,然后进行过滤去除反应生成的naoh或质子化合物钠盐,最后减压旋蒸去除第一溶剂、第二溶剂和r系有机物得固体r2pona;所述第二溶剂为r2pona可溶但质子化合物钠盐不溶的有机醚。
[0033]
所述第二溶剂可为thf等有机醚。
[0034]
进一步的,所述第一溶剂为r2pona可溶但质子化合物钠盐不溶的有机醚时,所述第三步中,反应结束后,直接进行过滤去除反应生成的naoh或质子化合物钠盐,得有机相,最后对有机相进行减压旋蒸去除第一溶剂和r系有机物得固体r2pona。
[0035]
进一步的,所述第一溶剂选自正己烷、正庚烷、聚乙烯、聚丙烯、苯、甲苯、二甲苯、三甲苯、thf、二氧杂环乙烷、meoch2ch2ome中的一种或任意多种混合。
[0036]
进一步的,所述第一溶剂选自四氢呋喃、甲苯和二氧杂环乙烷中的一种或任意多种混合。
[0037]
进一步的,r1、r2和r3分别独立选自c
1-c
20
的直链或支链烷基或链烯基、 c
3-c
20
环烷基、c
7-c
20
的芳烷基、c
6-c
20
的芳基或c
2-c
20
的杂环基。
[0038]
进一步的,r1、r2和r3独立选自带c
1-c6长链或支链烷基的苯基、含n、o、 s原子的杂环芳烃以及c
1-c
20
长链或支链烷基。
[0039]
进一步的,金属钠与膦氧类化合物r3po的摩尔比为0.1-5∶1。
[0040]
进一步的,金属钠与膦氧类化合物r3po的摩尔比为1-2∶1。
[0041]
进一步的,所述第一溶剂与膦氧类化合物r3po的重量比为0.5-20∶1。
[0042]
进一步的,所述第一溶剂与膦氧类化合物r3po的重量比为2-5∶1。
[0043]
进一步的,所述膦氧类化合物r3po、质子化合物和金属钠进行反应的反应温度为0℃-170℃。
[0044]
进一步的,所述膦氧类化合物r3po、质子化合物和金属钠进行反应的反应温度为60℃-150℃。
[0045]
进一步的,所述质子化合物的结构式为x-oh或y-h。
[0046]
进一步的,质子化合物选自x-oh结构时,所述第二步中,膦氧类化合物 r3po、质子化合物和金属钠进行充分反应生成naoh、质子化合物钠盐rx和
[0047]
[0048]
r2pona,反应式为:r3为r1r2r3,r1、r2、r3分别独立选自芳香族或脂肪族取代基。
[0049][0050]
上述制备过程为:r3na x-oh
→
naoh r3x, r1r2r3po与金属钠反应生成r1r2pona和r3na,r3na不与第一溶剂发生反应,r3na 与反应体系中的质子化合物x-oh反应生成r3x和naoh;
[0051]
当将膦氧类化合物r3po与质子化合物x-oh混合获得的混合物在无第一溶剂或第一溶剂是非醚类有机溶剂时,在反应结束后,分离之前,可向反应体系中加入第二溶剂将生成的r1r2pona进行溶解;第二溶剂为有机醚;r1r2pona 易溶于第二溶剂,naoh不溶于有机醚以及第一溶剂,然后进行过滤去除反应生成的白色沉淀naoh,最后减压旋蒸去除第一溶剂、第二溶剂和生成的r系有机物r3x得固体r1r2pona。
[0052]
当将膦氧类化合物r3po与质子化合物x-oh混合获得的混合物是在第一溶剂的条件下进行,第一溶剂为有机醚,r1r2pona溶于有机醚,naoh不溶于有机醚,反应结束后,进行过滤去除反应生成的白色沉淀naoh得有机相,最后对有机相进行减压旋蒸去除第一溶剂和生成r系有机物r3x得固体r1r2pona, r1r2pona为r2pona。
[0053]
进一步的,若质子化合物选自y-h结构时,所述第二步中,膦氧类化合物r3po、质子化合物和金属钠进行充分反应生成质子化合物钠盐nay、rh和
[0054][0055]
r2pona,反应式为:r3为r1r2r3,r1、r2、r3分别独立选自芳香族或脂肪族取代基。
[0056][0057]
上述制备过程为:r3na y-h
→
nay r3h, r1r2r3po与金属钠反应生成r1r2pona和r3na,r3na不与反应体系中的第一溶剂发生反应,其与反应体系中的质子化合物y-h反应生成nay和r系有机物r3h;
[0058]
当将膦氧类化合物r3po与质子化合物y-h混合获得的混合物在无第一溶剂或第一溶剂是非醚类有机溶剂时,在反应结束后,分离之前,可向反应体系中加入第二溶剂将生成的r1r2pona进行溶解;第二溶剂为有机醚;质子化合物钠盐nay不溶于有机醚,然后进行过滤去除反应生成的质子化合物钠盐 nay得有机相,最后对有机相进行减压旋蒸去除第一溶剂、第二溶剂和生成的 r系有机物r3x得固体r1r2pona,r1r2pona为r2pona。
[0059]
当将膦氧类化合物r3po与质子化合物y-h混合获得的混合物在第一溶剂的条件下进行,第一溶剂为有机醚,r1r2pona溶于有机醚,质子化合物钠盐nay不溶于有机醚,反应结束后,进行过滤去除反应生成的质子化合物钠盐 nay得有机相,最后对有机相进行减压旋蒸去除第一溶剂、第二溶剂和生成的 r系有机物r3x得固体r1r2pona,r1r2pona为r2pona。
[0060]
进一步的,所述质子化合物是水、h2o、meoh、etoh、meco2h、hcl和phsh 中的一种或任意多种混合。
[0061]
进一步的,所述质子化合物为水。
[0062]
进一步的,所述r3po与质子化合物的摩尔比为0.5-1.5∶1。
[0063]
进一步的,r1、r2和r3均选自苯基。
[0064]
进一步的,ph2pona的中文名称为二苯基次磷酸钠。
[0065]
ph2pona的结构式为:其在空气下或遇水会发生反应,其需在惰性气体或氮气气氛下进行保存。
[0066]
钠在ph3po与质子化合物的混合物体系中,在有无第一溶剂的条件下进行反应,分离纯化可制备出纯度为99%以上的ph2pona,制备出的纯净的ph2pona 可用于制备其他膦氧化合物,并可获得一个较好的收率。
[0067]
由本发明提供的制备方法制备的ph2pona纯度要远优于背景技术中记载,例如ph2pona制备过程中,质子化合物为水,第一溶剂选为thf溶剂;在惰性气体或氮气气氛下,ph3po先与金属钠进行反应生成ph2pona和phna;生成的 phna活性较活泼,phna与水反应生成苯和naoh,naoh不溶于thf溶剂,ph2pona 溶于thf溶剂中,在惰性气体或氮气气氛下进行过滤,去除反应生成的白色沉淀naoh,最后减压旋蒸去除thf溶剂和苯后,得纯净的固体ph2pona;
[0068]
ph3po na=ph2pona phna,phna h2o=phh naoh。
[0069]
背景技术中记载在thf等极性醚溶剂里,ph3p(o)和na反应可生成 ph2pona,该过程会产生较多的副产物,这些副产物与产物ph2pona会溶解在溶剂中,不易将ph2pona与制备过程中产生的副产物进行分离,最后无法获得较纯的ph2pona;通过对比可以发现,本发明提供的制备方法制备的ph2pona 纯度要远优于背景技术中记载。
[0070][0071]
由一种膦基化合物的制备方法制备的r2pona作为中间体在制备其他膦氧基化合物的应用。
[0072]
通过本发明提供的膦基化合物的制备方法制备的纯净的r1r2pona可作为制备其他膦氧基化合物的中间体;
[0073]
r1r2pona通过与r4z反应制备r1r2por4;
[0074]
r1r2pona r4z
→
r1r2por4 naz;
[0075]
r4z为芳香族或脂肪族化合物,r1r2pona可与r4z反应制备生成r1r2por4和naz,r1r2por4溶于有机溶剂,naz不溶于有机溶剂,利用这一特性,反应结束后,可向反应体系中加入有机溶剂,过滤将naz分离,后利用减压旋蒸将有机溶剂去除,水洗,即得到r1r2por4。
[0076]
r1r2pona通过与z
1-r
5-z2反应制备r1r2por5por1r2;
[0077]
z1r5z2为芳香族或脂肪族化合物,r1r2pona可与z1r5z2反应制备生成 r1r2por5por1r2、naz1和naz2,naz1和naz2不溶于有机溶剂,r1r2por5por1r2溶于有机溶剂,利用这一特性,过滤将naz1和naz2分离出去,后利用减压旋蒸将有机溶剂去除,得到r1r2por5por1r2。
[0078]
z、z1和z2均为卤素基团。
[0079]
r1r2pona通过与h2o反应制备r1r2poh,
[0080]
r1r2pona h2o
→
r1r2poh naoh;
[0081]
r1r2pona可与h2o反应制备生成r1r2poh和h2o,r1r2poh溶于有机溶剂,naoh 不溶于有机溶剂,利用这一特性,反应结束后,可向反应体系中加入有机溶剂,过滤将naoh分离,后利用减压旋蒸将有机溶剂去除,即得到r1r2poh。
[0082]
3.有益效果
[0083]
相比于现有技术,本发明的优点在于:
[0084]
(1)通过本发明开发了一种通过使金属钠与r3po与质子化合物的混合物体系反应,r3po与金属钠反应生成得r3na可与质子化合物迅速反应生成的 naoh或质子化合物钠盐、r2pona以及r系有机物,r系有机物易于旋蒸去除,反应结束后,分离naoh或质子化合物钠盐、r2pona以及r系有机物,利用naoh 或质子化合物钠盐不溶于有机醚溶剂,r2pona以及r系有机物溶于有机醚溶剂这一特征,最后分离,得到的r1r2pona较为纯净。
[0085]
(2)通过本发明开发的将钠与r3po反应制备纯净的膦基化合物r1r2pona 的制备方法,可以更高效的利用制备出的纯净的r1r2pona去制造一系列的磷化合物,减少因之前制备的r1r2pona不纯,导致需消耗一倍以上的r1r2pona 的摩尔倍数原料与r1r2pona反应制备其他磷化合物,进而减少由r1r2pona作为原料制备其他磷化合物的成本。
[0086]
(3)本发明开发的制备方法,适用范围广、工艺简单、成本低廉、安全可控,易于工业化放大生产。
[0087]
(4)通过本发明提供的制备方法,可弥补以前使用r3po与钠反应的制备方法制备出r1r2pona所带来的各种缺陷。
附图说明
[0088]
图1为本发明的ph2pona的结构式;
[0089]
图2为本发明的ph2pona的产品图;
[0090]
图3为本发明的ph2pona的h谱图;
[0091]
图4为本发明的ph2pona的c谱图;
[0092]
图5为本发明的ph2pona的p谱图。
具体实施方式
[0093]
下面以三苯基氧膦为例制备ph2pona对本发明进行进一步描述,但本发明的保护范围并不仅限于此:
[0094][0095]
实施例1ph2pona的制造,分离及精制
[0096]
在氮气氮气下,将10mmol三苯基氧膦、10mmol水,thf 20ml混合搅拌30分钟后,室温下,把金属钠20mmol切成小块,加入混合溶液中;室温下搅拌一夜,得含白色沉淀的深橙色溶液;然后在氮气气氛下,过滤除去白色沉淀naoh,后减压除去溶剂等挥发物,得淡黄色固体ph2pona,收率92%.
[0097]
以下核磁及元素分析表征,证实所得化合物为纯ph2pona。这是世界上第一次成功得到的纯ph2pona。
[0098]
二苯基次磷酸钠(sodium diphenylphosphinite,ph2ona).淡黄色固体,融点355℃(分解)。1h nmr(400mhz,d
8-thf):δ7.47-7.44(m,4h), 7.18-7.14(m,4h),7.07-7.03(m,2h);
13
c{1h}nmr(100mhz,d
8-thf):δ155.1(d,j
p-c
=40.0hz),128.1(d,j
p-c
=20.0hz),127.6(d,j
p-c
= 4.4hz),126.2.
31
p nmr(162mhz,d
8-thf):δ91.2.elemental analysis, calculated for c12h10naop:c,64.29;h,4.50.found:c,64.56;h,4.68.
[0099]
实施例2
[0100]
与实施例1同样条件下,将水换成meoh实施反应,得到95%收率(
31
p nmr 收率)的ph2pona。
[0101]
实施例3
[0102]
与实施例1同样条件下,将水换成乙醇etoh实施反应,得到97%收率 (
31
p nmr收率)的ph2pona。
[0103]
实施例4
[0104]
与实施例1同样条件下,将水换成异丙醇i-proh实施反应,得到93%收率(
31
p nmr收率)的ph2pona。
[0105]
实施例5
[0106]
与实施例1同样条件下,将水换成叔丁醇t-buoh实施反应,得到91%收率(
31
p nmr收率)的ph2pona。
[0107]
实施例6
[0108]
与实施例1同样条件下,将水换成醋酸ch3co2h实施反应,得到90%收率(
31
p nmr收率)的ph2pona。
[0109]
实施例7
[0110]
与实施例1同样条件下,将thf换成meoch2ch2ome实施反应,得到98%收率(
31
p nmr收率)的ph2pona。
[0111]
实施例8
[0112]
与实施例1同样条件下,将thf换成二氧杂环乙烷dioxane实施反应,得到93%收率(
31
p nmr收率)的ph2pona。
[0113]
实施例9
[0114]
与实施例1同样条件下,将thf换成甲苯实施反应,得到90%收率(
31
p nmr收率)的ph2pona。
[0115]
通过实施例1-9可以发现,r3po可与不同的质子化合物在无溶剂或有第一溶剂的条件下与金属钠可进行反应制备r1r2pona,制备出的r1r2pona易于从反应体系中分离出来。
[0116]
实施例10是由本发明提供的一种膦基化合物的制备方法制备的ph2pona 去制备ph2p(o)h的实施例。
[0117]
实施例10
[0118]
与实施例1同样条件下,实施反应后,向混合反应液里面加饱和氯化氨溶液10ml,后用醋酸乙酯萃取,减压除去挥发物,得到98%收率的ph2p(o)h。
[0119]
实施例11-13是由本发明提供的一种膦基化合物的制备方法制备的 r1r2pona与z1r5z2芳香族或脂肪族化合物去制备r1r2por5por1r2的实施例。
[0120]
实施例11
[0121]
与实施例1同样条件下,实施反应后,向混合反应液里面加clch2ch2cl15 ml,搅拌5小时后,减压除去挥发物,水洗,得95%收率的 ph2p(o)ch2ch2p(o)ph2。
[0122]
实施例12
[0123]
与实施例1同样条件下,实施反应后,向混合反应液里面加clch2ch2cl 20 ml,搅拌5小时后,减压除去挥发物,水洗,得95%收率的 ph2p(o)ch2ch2p(o)ph2。
[0124]
实施例13
[0125]
与实施例1同样条件下,实施反应后,向混合反应液里面加 1,4-clch2c6h4ch2cl 10ml,搅拌5小时后,减压除去挥发物,水洗,得91%收率的ph2p(o)ch2c6h4ch2p(o)ph2。
[0126]
实施例14是由本发明提供的一种膦基化合物的制备方法制备的r1r2pona 与r4z芳香族或脂肪族化合物去制备r1r2por4的实施例。
[0127]
实施例14
[0128]
与实施例1同样条件下,反应结束后,滤过除去naoh后,向反应液里面加1当量tmscl 10mmol,搅拌5小时后,减压除去挥发物,得98%收率的油状ph2potms。
[0129]
除上述实施例外,本发明还可以有其他实施方式。凡采用等同替换或等效变换形成的技术方案,均落在本发明要求的保护范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。