1.本发明涉及化合物纯化/精制技术领域,具体涉及一种去氧孕烯的精制方法。
背景技术:
2.去氧孕烯,化学名称:(17α)-13-乙基-11-亚甲基-18,19-双失碳孕甾-4-烯-20-炔基-17-醇,cas:54024-22-5,是一种强效孕激素,其特点是没有雄激素和雌激素活性,具有明显的排卵抑制作用,临床广泛用作避孕药,其效果可靠,周期控制好,不降低血浆高密度脂蛋白(hdl),有利于脂质代谢,无增加体重,雄性症等副作用。去氧孕烯是目前市场上口服避孕药的重要品种。但是由于去氧孕烯制备成本高,产品价格昂贵,因此寻找最终产品的高效精制方法对于去氧孕烯的规模化生产具有重要价值。
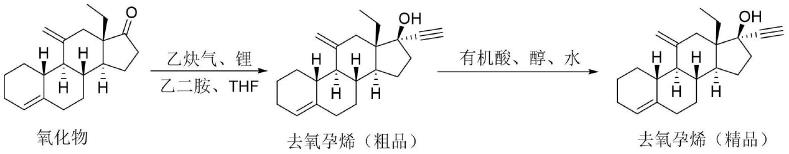
3.去氧孕烯的结构式:
[0004][0005]
虽然去氧孕烯有多种合成方法,但最后一步反应通常采用氧化物的炔基化,再经后处理和精制得到去氧孕烯产品。如美国专利us3927046公开的方法中最后一步反应如式一所示:
[0006][0007]
反应为氧化物在乙炔钾/thf溶液中炔基化,经柱色谱纯化、然后在甲醇中重结晶得到产物去氧孕烯,摩尔收率为78%,未报道产物纯度。
[0008]
2009年公开的专利cn 101445542a中,最后一步反应为氧化物在乙炔气体/thf/koh/无水丙酮体系中炔基化,经活性炭吸附之后,以正己烷为溶剂重结晶得到产物去氧孕烯,摩尔收率为77%,同样未报道产物纯度。
[0009]
在2021年公开的专利cn112225771a中,最后一步反应为氧化物在乙炔气体/金属锂/乙二胺/thf体系中炔基化得到去氧孕烯粗品,粗品收率85%-90%,粗品纯度》98%。继而经活性炭吸附处理、正己烷/异丙醚重结晶得到去氧孕烯精制产品,精制收率80%,精制品纯度》99%,亦即,最后一步合成去氧孕烯粗品及产品精制的总收率为68%-72%。
[0010]
除目前专利公开的方法外,也有文献报道去氧孕烯的合成,例如chem.eur.j.2008,14,1541-1551报道的方法中最后一步反应为氧化物在乙炔气体/金属锂/乙二胺/thf体系中炔基化,经柱色谱分离得到油状的去氧孕烯,收率83%。然后采用正
戊烷重结晶,重结晶收率和产物纯度未说明。
[0011]
按照已有文献报道的精制方法,主要通过低极性溶剂如正己烷或正己烷/异丙醚对去氧孕烯进行精制处理,由于产品和杂质的极性都很小,所以采用文献方法精制过程中造成产品的损失比较大。且现有去氧孕烯制备工艺中,通过柱层析或在溶剂中添加活性炭或硅胶等吸附剂吸附,然后进行重结晶,操作繁琐,同样也会造成产品损失,收率降低。
技术实现要素:
[0012]
为了解决现有技术中存在的问题,本发明提出了一种去氧孕烯的精制方法,将以13-乙基-11-亚甲基-18,19-双失碳雌甾-4-烯-17-酮(简称:氧化物)为原料经一步炔基化反应制备得到的去氧孕烯粗品采用有机酸、醇和水按照一定比例作为混合溶剂进行重结晶,无需柱层析或使用活性炭等吸附剂处理,从而简化操作、提高去氧孕烯的精制效率,去氧孕烯产品的纯度可达99.9%。并且,采用本发明方法可以严格控制去氧孕烯产品中的单个杂质含量,其中杂质i含量《0.1%,且总杂质含量(以下简称“总杂”)《0.1%。本发明通过提高重结晶收率(》90%),可以有效降低成本。
[0013][0014]
本发明提供一种去氧孕烯的精制方法,将以13-乙基-11-亚甲基-18,19-双失碳雌甾-4-烯-17-酮(简称:氧化物)为原料经一步炔基化反应制备得到的去氧孕烯粗品,采用有机酸、醇和水作为混合溶剂进行重结晶。
[0015]
所述去氧孕烯粗品中,杂质主要包括以下杂质i、ii、iii的一种或几种:
[0016]
杂质i:(5α,17α)-13-乙基-11-亚甲基-18,19-双失碳孕甾-3-烯-20-炔基-17-醇(去氧孕烯
△3异构体)
[0017][0018]
杂质ii:13-乙基-11-亚甲基-18,19-双失碳雌甾-4-烯-17-酮
[0019][0020]
杂质iii:(17α)-13-乙基-17-羟基-11-亚甲基-18,19-双失碳孕甾-4-烯-20-炔基-3-酮
[0021][0022]
由于杂质i和去氧孕烯的极性相似,所以杂质i最难去除。
[0023]
本发明方法是付出了创造性劳动及大量试验后提出的,以13-乙基-11-亚甲基-18,19-双失碳雌甾-4-烯-17-酮(简称:氧化物)为原料经一步炔基化反应制备得到的去氧孕烯粗品,采用有机酸、醇和水作为混合溶剂进行重结晶,获得了高收率、高纯度的去氧孕烯。与现有技术中采用正己烷、正戊烷、正己烷/异丙醚、丙酮等溶剂进行重结晶的方法作比较,本发明的精制方法收率显著提高、且操作简便。并且无需柱层析或通过活性炭吸附即可去除粘稠的杂质。以原料氧化物计,获得的粗品精制后的总收率显著提高。
[0024]
进一步地,在本发明的去氧孕烯精制方法中,采用的有机酸为较为常见的有机酸,如包括但不限于甲酸,乙酸,丙酸,丁酸等有机酸,优选甲酸和乙酸。
[0025]
进一步地,在本发明的去氧孕烯精制方法中,采用的醇为较为常见的有机醇,如包括但不限于甲醇,乙醇,异丙醇,正丁醇等有机醇,优选异丙醇。
[0026]
进一步地,在本发明的去氧孕烯精制方法中,第一次重结晶时有机酸、醇和水混合溶剂用量非限制性地优选为底物(即去氧孕烯粗品)的4当量、5当量、6当量,7当量、8当量;最优选为底物的7.5当量。第二次重结晶时有机酸、醇和水混合溶剂用量非限制性地优选为底物(即第一次重结晶后的去氧孕烯固体)的4当量、5当量、6当量、7当量、8当量;最优选为底物的7.5当量。具体地,经第一次重结晶,得到的去氧孕烯纯度由粗品的94%-96%提升至98%以上;经第二次重结晶,得到的去氧孕烯纯度提高至99.9%及以上。
[0027]
本发明中,术语“当量”指有机酸、醇和水混合溶剂质量与底物的质量比。
[0028]
进一步,在本发明的去氧孕烯的精制方法中,采用有机酸、醇和水混合溶剂中有机酸、醇和水的质量比例为1-20:0.5-5:0.5-5,优选区间为3-5:0.5-2:0.5-2。
[0029]
具体的,在本发明的去氧孕烯的精制方法中,采用有机酸、醇和水的混合溶剂对去氧孕烯进行精制,具有更好的精制效果。
[0030]
更为具体地,所述精制方法包括:
[0031]
第一次精制:将去氧孕烯粗品(纯度为94%-96%),有机酸、醇和水加入到设有回流冷凝装置的加热容器中,加热至溶清,之后降至室温,在10℃-15℃水浴中析晶1-2h,过滤,得到第一次精制品,纯度为98.0%-98.9%。
[0032]
第二次精制:将第一次精制得到的去氧孕烯,有机酸、醇和水加入到设有回流冷凝装置的加热容器中,加热至溶清,之后降至室温,在10℃-15℃水浴中析晶1-2h,过滤,将滤饼在40℃-45℃下真空干燥1-8h,得到第二次精制品,纯度为99.9%-99.99%。
[0033]
具体地,经第一次重结晶,纯度为94%-96%的去氧孕烯粗品纯度提升至98%以上;经第二次重结晶,去氧孕烯纯度可高达99.9%,本发明第一次、第二次的重结晶处理方法相同。若继续经过第三次重结晶,得到的去氧孕烯纯品的纯度依旧维持99.9%,未见明显变化,所以本发明两次重结晶即可确保去氧孕烯的纯度。
[0034]
进一步,所述第一次精制和第二次精制中的加热至溶清,优选在60℃-120℃条件下加热。
[0035]
本发明创新提出了一种不需要添加活性炭等吸附剂就可以去除粘稠杂质的去氧孕烯精制方法。
[0036]
具体实施方案中,所述粘稠杂质包括但不限于杂质i、杂质ii或者杂质iii。
[0037]
可选地,本发明所述底物即去氧孕烯粗品的制备方法包括:
[0038]
将氧化物13-乙基-11-亚甲基-18,19-双失碳雌甾-4-烯-17-酮、乙炔锂乙二胺络合物和无水四氢呋喃加入到反应瓶中。在0℃~5℃搅拌下反应3小时,反应完全后加入正己烷稀释反应液,加入水,分出有机层。有机层用硫酸水溶液洗涤,再用水洗涤至中性,无水硫酸钠干燥过滤、洗涤滤饼,有机层浓缩干后得去氧孕烯粗品。
[0039]
本发明涉及到的试剂均为普通市售产品,涉及到的去氧孕烯粗品均为通过经典反应方法,即原料氧化物经一步炔基化反应制备得到的去氧孕烯粗品,涉及到的操作如无特殊说明均为本领域常规操作。本发明所述底物即去氧孕烯粗品亦可为普通市售产品。
[0040]
在符合本领域常识的基础上,上述各优选条件,可以相互组合,得到具体实施方式。
[0041]
本发明有益效果在于:本发明提供一种去氧孕烯的精制方法,通过采用特定用量的有机酸、醇和水作为混合溶剂对由13-乙基-11-亚甲基-18,19-双失碳雌甾-4-烯-17-酮(简称:氧化物)为原料经一步炔基化反应制备得到的去氧孕烯粗品进行两次重结晶,成功实现了有效控制杂质(包括杂质i、ii、iii)含量、保证产品纯度的同时,显著提高了去氧孕烯的收率,并且不需要采用柱层析或者活性炭等吸附剂处理。以原料氧化物计,获得的粗品精制后的总收率保持在90%以上,显著优于本发明背景技术中的精制方法(收率68%-78%)。由于去氧孕烯价格昂贵,最终产品收率的提升对于降低产品成本、提升产品的竞争力具有重要价值。
[0042]
本发明提出的精制方法的构思,是根据产品和杂质的物化特性,通过选用合适的混合溶剂,在保持去氧孕烯高的重结晶收率的前提下,有效去除杂质,通过两次重结晶,使得去氧孕烯的纯度达到99.9%。突破了以往的利用低极性溶剂(相似相溶的原理)进行纯化的方式。
具体实施方式
[0043]
结合以下具体实施例,对发明作进一步的详细说明。实施本发明的过程、条件、实验方法等,除以下专门提及的内容之外,均为本领域的普遍知识和公知常识,本发明没有特别限制内容。需要理解的是以下实施例的给出仅是为了起到说明的目的,并不是用于对本发明的范围进行限制。本领域的技术人员在不背离本发明的宗旨和精神的情况下,可以对本发明进行各种修改和替换。下述实施例中所使用的实验方法如无特殊说明,均为常规方法。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
[0044]
实施例1去氧孕烯粗品制备
[0045]
将91g氧化物13-乙基-11-亚甲基-18,19-双失碳雌甾-4-烯-17-酮、90g乙炔锂乙二胺络合物和600ml无水四氢呋喃加入到反应瓶中。在0℃-5℃搅拌下反应3小时,进行tlc检测。反应完全后加入1800ml正己烷稀释反应液,加入600ml水,分出有机层。有机层用10%硫酸水溶液200ml洗涤,再用水洗涤至中性,无水硫酸钠干燥过滤、洗涤滤饼,有机层浓干得去氧孕烯粗品100g,收率100%,色谱纯度95.174%。
[0046]
实施例2对去氧孕烯粗品(以本发明实施例1制备的去氧孕烯粗品为例)进行精制
[0047]
取本发明实施例1所得去氧孕烯粗品20g,加入甲酸90g、甲醇30g、纯水10g,升温至固体全溶后,降至20℃~25℃再加入纯水20g继续降温至10℃~15℃析晶1~2小时过滤,得去氧孕烯一次精制品湿料24g,色谱纯度98.595%。
[0048]
去氧孕烯一次精制品湿料24g,加入甲酸90g、甲醇30g、纯水10g,升温至固体全溶后,降至20℃~25℃再加入纯水20g继续降温至10℃~15℃析晶1~2小时过滤,烘干得白色去氧孕烯二次精制品18.02g,收率90.1%(以本发明实施例1中的氧化物计),色谱纯度99.909%。杂质i残留仅为0.05%,杂质ii和iii未检出。
[0049]
hplc检测方法如下:
[0050]
色谱条件:用空间保护的十八烷基硅烷键合硅胶为填充剂的色谱柱(agilent zorbax sb c184.6
×
250mm,5μm,或效能相当的色谱柱;以乙腈-水(73:27)为流动相;检测波长为205nm;柱温为50℃;流速为每分钟1.0ml;进样体积15μl。
[0051]
系统适用性要求:系统适用性溶液的色谱图中,杂质ⅲ、杂质ⅰ和杂质ⅱ峰相对于主成分峰(保留时间约为22min)的相对保留时间分别约为0.25、0.95和1.05。去氧孕烯峰与杂质ⅱ峰之间的峰谷比(峰谷比=hp/hv,其中hp为杂质ⅱ峰在基线上的高度,hv为分离主成分峰与杂质ⅱ峰曲线的最低点在基线上的高度)应不得小于2.0,去氧孕烯峰与杂质ⅰ峰的分离度不得低于1.2。
[0052]
表1.本发明实施例2中两次精制后的产品hplc含量分析结果
[0053]
保留时间(min)5.98813.57514.42521.01122.71523.66525.14238.427峰归属杂质iii///杂质i产品杂质ii/粗品(%)0.0300.0390.2720.2000.88995.1742.3960.669一次精制品(%)/0.0250.0390.0270.32998.5950.6250.210二次精制品(%)////0.05299.909//
[0054]
实施例3对两次精制后的去氧孕烯(以本发明实施例2制备的去氧孕烯为原料)实施第三次精制
[0055]
取本发明实施例2制备的去氧孕烯二次精制品18g,加入甲酸90g、甲醇30g和纯水10g,升温至固体全溶后,降至20℃~25℃再加入纯水20g继续降温至10℃~15℃析晶1~2小时过滤,烘干得白色去氧孕烯三次精制品17.02g,色谱纯度99.921%。
[0056]
表2.本发明实施例3中第三次精制的产品hplc含量分析结果
[0057]
保留时间(min)5.98813.57514.42521.01122.71523.66525.14238.427峰归属杂质iii///杂质i产品杂质ii/粗品(%)0.0300.0390.2720.2000.88995.1742.3960.669二次精制品(%)////0.05299.909//三次精制品(%)////0.04399.921//
[0058]
实施例4对去氧孕烯粗品(以本发明实施例1制备的去氧孕烯粗品为例)进行精制,以乙酸、异丙醇和水为混合溶剂进行重结晶。
[0059]
取本发明实施例1所得去氧孕烯粗品20g,加入乙酸90g、异丙醇30g、纯水10g,升温至固体全溶后,降至20℃~25℃再加入纯水20g继续降温至10℃~15℃析晶1~2小时过滤,得去氧孕烯一次精制品湿料24.2g,色谱纯度98.621%。
[0060]
去氧孕烯一次精制品湿料24.2g,加入乙酸90g、异丙醇30g、纯水10g,升温至固体全溶后,降至20℃~25℃再加入纯水20g继续降温至10℃~15℃析晶1~2小时过滤,烘干得白色去氧孕烯二次精制品18.10g,收率90.5%(以本发明实施例1中的氧化物计),色谱纯度99.91%。杂质i残留仅为0.05%,杂质ii和iii未检出。
[0061]
表3.本发明实施例4中两次精制后的产品hplc含量分析结果
[0062]
保留时间(min)5.98813.57514.42521.01122.71523.66525.14238.427峰归属杂质iii///杂质i产品杂质ii/粗品(%)0.0300.0390.2720.2000.88995.1742.3960.669一次精制品(%)/0.0310.0370.1250.23198.6210.5110.314二次精制品(%)////0.04999.913//
[0063]
实施例5采用不同比例有机酸、醇和水混合溶剂进行重结晶,以乙酸、异丙醇和水为混合溶剂进行重结晶。
[0064]
取本发明实施例1所得去氧孕烯粗品20g,加入乙酸60g、异丙醇20g、纯水5g,升温至固体全溶后,降至20℃~25℃再加入纯水15g继续降温至10℃~15℃析晶1~2小时过滤,得去氧孕烯一次精制品湿料24.5g,色谱纯度97.757%。
[0065]
去氧孕烯一次精制品湿料24.5g,加入乙酸60g、异丙醇20g、纯水5g,升温至固体全溶后,降至20℃~25℃再加入纯水15g继续降温至10℃~15℃析晶1~2小时过滤,得去氧孕烯二次精制品湿料23.2g,色谱纯度99.208%。
[0066]
去氧孕烯二次精制品湿料23.2g,加入乙酸60g、异丙醇20g、纯水5g,升温至固体全溶后,降至20℃~25℃再加入纯水15g继续降温至10℃~15℃析晶1~2小时过滤,烘干得白色去氧孕烯三次精制品18.3g,收率91.5%(以本发明实施例1中的氧化物计),色谱纯度99.847%。杂质i残留仅为0.05%,杂质ii和iii未检出。
[0067]
表4.本发明实施例5采用不同比例有机酸、醇和水混合溶剂进行重结晶所得产品的hplc含量分析结果
[0068][0069][0070]
表4数据显示,采用乙酸、异丙醇和水的混合溶剂(三者的当量比例为12:4:1)进行三次重结晶,第一次精制后,产品纯度从95.174%提升至97.757%;第二次精制后,产品纯度提高至99.208%;第三次精制后,产品纯度提高至99.847%,表明有机酸、醇和水的不同当量配比对精制结果有影响(同本发明实施例4进行比较)。
[0071]
实施例6对比试验:采用文献方法中的正己烷/异丙醚为溶剂对去氧孕烯粗品进行
精制
[0072]
取本发明实施例1所得去氧孕烯粗品20g,加入正己烷220g,活性炭2.2g,升温至40℃~50℃搅拌1-2小时过滤、正己烷漂洗滤饼,滤液合并减压浓缩干溶剂后再加入正己烷60g和异丙醚10g,升温至固体全溶后,降至0℃~5℃析晶1~2小时过滤,固体再加入60g正己烷,升温至固体全溶后,降至0℃~5℃析晶1~2小时过滤,烘干得去氧孕烯精制纯品14g,收率70%,色谱纯度99.2%。
[0073]
表5.本发明实施例6对比实验中以正己烷/异丙醚为溶剂精制所得产品的hplc含量分析结果
[0074]
保留时间(min)5.98813.57514.42521.01122.71523.66525.14238.427峰归属杂质iii///杂质i产品杂质ii/粗品(%)0.0300.0390.2720.2000.88995.1742.3960.669精制品(%)0.0100.0250.0290.0270.21199.200.3130.075
[0075]
表5数据显示,采用文献中常用的正己烷/异丙醚为溶剂对去氧孕烯粗品进行重结晶,不但精制后的产品纯度只有99.20%,而且重结晶收率也仅为70%,此精制方案损失大,杂质去除效果欠佳,仍有少量杂质残留在产品中,尤其以杂质i(0.21%)和杂质ii(0.31%)残留较为明显。
[0076]
本发明的保护内容不局限于以上实施例。在不背离发明构思的精神和范围下,本领域技术人员能够想到的变化和优点都被包括在本发明中,并且以所附的权利要求书为保护范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。