1.本发明涉及结构物及其制造方法、使用了该结构物的吸附体、以及生物粒子的纯化方法。
背景技术:
2.病毒、病毒样颗粒或病毒载体等生物粒子在基因治疗、疫苗、检查诊断、血液净化等医疗领域中是非常重要的分子。另外,其去除在预防及防止感染、确保医药品在病毒学上的安全性方面是非常重要的。
3.近年来,这些医疗领域是备受关注的领域,除了高效地去除生物粒子的技术以外,还迫切需要从细胞培养液、血液等生物试样中高效地回收生物粒子的方法。
4.通常,高纯度的病毒的纯化采用如下方法:首先,通过过滤从产生病毒的转化细胞的培养液、鸡卵的绒毛膜尿囊液中去除细胞、细胞片等固体物质,然后通过色谱法、超速离心法等进行纯化。
5.亲和色谱法是使用将与对象分子特异性结合的抗体等配体固定于珠基材而得到的载体将对象物与除此以外的杂质进行分离的方法。从稳定性、生产效率的观点考虑,已知将低分子化抗体用于配体的例子(专利文献1等)。另外,在现有的病毒纯化方法中,还已知使用了固定化有低分子化抗体的珠载体的方法(非专利文献1等)、市售品(ge公司的capto(注册商标)avb、thermo scientific公司的poros(注册商标)captureselect(注册商标)aavx等)。
6.然而,在现有的使用了珠载体的色谱法中,由于珠载体昂贵且难以制备,而且珠基材的孔径小,因此,在高效地回收病毒等生物粒子方面仍存在课题。
7.因此,廉价且溶出工序的生物粒子回收率高、清洗工序的生物粒子残留率低的结构物及其制造方法、使用其的吸附体、以及生物粒子的纯化方法完全未知,迫切需要提供这些技术。
8.现有技术文献
9.专利文献
10.专利文献1:国际公开第01/44301号小册子
11.非专利文献
12.非专利文献1:mol.ther.methods clin.dev.2018 9:33
技术实现要素:
13.为了解决现有的上述各问题,本发明以实现以下目的作为课题。即,本发明的目的在于提供廉价且溶出工序的生物粒子回收率高
く
、清洗工序的生物粒子残留率低的结构物及其制造方法、使用了该结构物的吸附体、以及生物粒子的纯化方法。此外,就生物粒子的去除而言,作为它们的高效去除手段,本发明也提供有效的方法。
14.解决课题的方法
15.本发明人等为了实现上述目的而进行了深入研究,结果发现,通过具有水不溶性纤维和连接于上述水不溶性纤维的低分子化抗体的结构物,能够提供廉价且溶出工序的生物粒子回收率高、清洗工序的生物粒子残留率低的结构物,通过采用以包括将水不溶性纤维与低分子化抗体连接的工序为特征的结构物的制造方法,能够提供廉价且溶出工序的生物粒子回收率高、清洗工序的生物粒子残留率低的结构物的制造方法。
16.本发明基于本发明人等的上述见解,上述解决课题的方法如下所述。即:
17.<1>一种结构物,其具有水不溶性纤维、和连接于上述水不溶性纤维的低分子化抗体。
18.<2>一种结构物的制造方法,该方法包括:将水不溶性纤维与低分子化抗体进行连接的工序。
19.<3>一种吸附体,其具有上述<1>所述的结构物。
20.<4>一种生物粒子的纯化方法,该方法包括:使用上述<1>所述的结构物、或上述<3>所述的吸附体。
21.发明的效果
22.根据本发明,可以解决现有的上述各问题而实现上述目的,能够提供廉价且溶出工序的生物粒子回收率高、清洗工序的生物粒子残留率低的结构物及其制造方法、使用了该结构物的吸附体、以及生物粒子的纯化方法。
附图说明
23.图1是示出使用孔径测定装置(permporometer)算出的平均孔径与通气时间的平方根的倒数的相关性的图。
24.图2是示出载体7的aav吸附、解吸的色谱图的图。
25.图3是示出poros(注册商标)captureselect(注册商标)aavx的aav吸附、解吸的色谱图的图。
具体实施方式
26.(结构物)
27.上述结构物具有水不溶性纤维、和连接于上述水不溶性纤维的低分子化抗体,可以进一步具有其它要素。
28.即,对于上述结构物而言,上述水不溶性纤维与上述低分子化抗体可以直接连接在一起,上述水不溶性纤维与上述低分子化抗体也可以经由其它要素而连接在一起。
[0029]-水不溶性纤维-[0030]
上述水不溶性纤维可以用作上述结构物的基材。
[0031]
作为上述水不溶性纤维的厚度的下限值,没有特别限制,可以根据目的而适当选择,从制造的稳定性的观点考虑,优选为0.08mm以上、更优选为0.10mm以上、进一步优选为0.12mm以上。
[0032]
作为上述水不溶性纤维的厚度的上限值,没有特别限制,可以根据目的而适当选择,从单位面积重量的均匀性的观点考虑,优选为0.50mm以下、更优选为0.40mm以下、进一
步优选为0.30mm以下。
[0033]
作为上述水不溶性纤维的单位面积重量的下限值,没有特别限制,可以根据目的而适当选择,从制造的稳定性的观点考虑,优选为5g/m2以上、更优选为10g/m2以上。
[0034]
作为上述水不溶性纤维的单位面积重量的上限值,没有特别限制,可以根据目的而适当选择,从水不溶性纤维的均匀性的观点考虑,优选为100g/m2以下、更优选为90g/m2以下、进一步优选为80g/m2以下、特别优选为70g/m2以下。
[0035]
作为上述水不溶性纤维的堆积密度的下限值,没有特别限制,可以根据目的而适当选择,从装入设备后的水不溶性纤维结构的变化少的观点考虑,优选为50kg/m3以上、更优选为60kg/m3以上、进一步优选为70kg/m3以上。
[0036]
作为上述水不溶性纤维的堆积密度的上限值,没有特别限制,可以根据目的而适当选择,从通液性能的观点考虑,优选为400kg/m3以下、更优选为350kg/m3以下、进一步优选为300kg/m3以下。
[0037]
需要说明的是,上述堆积密度是指对每1m3上述水不溶性纤维的重量进行测定而得到的值。
[0038]
作为上述水不溶性纤维的形状,没有特别限制,可以根据目的而适当选择,可以列举例如:圆形、四边形、三角形、捆包形(bale shape)等。
[0039]
上述水不溶性纤维的表面可以通过接枝聚合、聚合物涂层、碱、酸等试剂处理、等离子体处理等进行改性。
[0040]
作为上述接枝聚合,没有特别限制,可以根据目的而适当选择,可以列举例如:进行电子束照射的接枝聚合法等。
[0041]
进行上述电子束照射的接枝聚合法有如下方法:预照射法,预先对水不溶性纤维照射电子束,产生自由基后,对该产生了自由基的水不溶性纤维赋予自由基聚合性化合物,通过随后的后聚合促进接枝聚合性化合物的聚合;同时照射法,对水不溶性纤维赋予自由基聚合性化合物后,对其照射电子束而产生自由基,通过随后的后聚合促进接枝聚合化合物的聚合。在本发明中,可以采用上述预照射法和同时照射法的任一种。
[0042]
在上述预照射法及同时照射法的任一种的情况下,对水不溶性纤维赋予自由基聚合性化合物后,在直至后聚合结束为止的期间,优选在水不溶性纤维的表面预先层压膜。由此,可以防止自由基聚合性化合物的挥散,均匀地引发接枝聚合,且通过阻隔空气而抑制空气中的氧所导致的自由基失活。另外,在同时照射法的情况下,电子束照射时水不溶性纤维表面被膜密封,水不溶性纤维和自由基聚合性化合物与空气中的氧阻隔,因此不容易发生因空气中的氧导致的水不溶性纤维的氧化。
[0043]
作为上述膜,使用具有0.01~0.20mm厚度的高分子膜,其根据使用的电子束的透射力而具有适当的厚度。
[0044]
作为上述膜的材质,没有特别限制,可以根据目的而适当选择,可以列举例如:聚对苯二甲酸乙二醇酯等聚酯类、聚烯烃类等,其中,优选为聚对苯二甲酸乙二醇酯。特别是在同时照射法的情况下,在电子束照射中聚合物自由基的生成效率低、且氧透过性低的聚对苯二甲酸乙二醇酯膜是合适的。
[0045]
需要说明的是,上述聚酯类是指聚酯及其衍生物,上述聚烯烃类是指聚烯烃及其衍生物。
[0046]
在上述预照射法中,首先,对水不溶性纤维照射电子束,生成引起聚合反应的自由基(聚合物自由基等)。对于电子束照射时的气体氛围温度而言,低温度时自由基的生成效率提高,可以为通常的室温。
[0047]
作为预照射法时的上述电子束的照射条件,没有特别限制,可以根据目的而适当选择,优选在将照射气体氛围的氧浓度设定为300ppm以下的状态下在加速电压100~2000千伏(以下简称为“kv”)、更优选为120~300kv及电流1~100ma的范围根据水不溶性纤维的厚度、目标接枝率等而适当确定。
[0048]
另外,上述电子束的照射剂量可以考虑目标接枝率及照射所引起的水不溶性纤维的物性降低而适当确定,通常,10~300千戈瑞(以下简称为“kgy”)左右是适当的,优选为10~200kgy。照射剂量低于10kgy时,不会生成足够的接枝聚合量所需要的自由基,超过300kgy时,水不溶性纤维由具有放射线耐性的高分子原材料形成的情况下会发生因主链的切断所导致的物性降低,因此不优选。
[0049]
需要说明的是,预照射法时的照射气体氛围优选为氮气等非活性气体氛围,可以为空气氛围。但是,在空气氛围中,水不溶性纤维可能由于空气中的氧而被氧化。
[0050]
接着,对上述电子束照射后的水不溶性纤维赋予自由基聚合性化合物。例如,在通过通气氮气而去除了溶解氧的自由基聚合性化合物溶液的槽中浸渍、或在上述自由基聚合性化合物溶液的槽中浸渍并通过,由此,使其停留给定时间,对水不溶性纤维充分赋予自由基聚合性化合物。
[0051]
需要说明的是,本发明中的“浸渍”是指水不溶性纤维与自由基聚合性化合物溶液接触。因此,作为对水不溶性纤维赋予自由基聚合性化合物的方法,可以使用各种涂敷方法。其中,浸渍涂布、缺角轮直接涂布、缺角轮反向涂布、吻涂、凹版涂布等能够效率良好地进行涂敷,因此是优选的。
[0052]
将赋予了自由基聚合性化合物的水不溶性纤维从上述溶液槽取出。此时,优选在水不溶性纤维的表面层压膜。例如,在水不溶性纤维为片状或纤维状的情况下,将赋予了自由基聚合性化合物溶液的水不溶性纤维夹在2片膜之间并使其密合。通过使膜与赋予了自由基聚合性化合物溶液的水不溶性纤维密合,能够将自由基聚合性化合物溶液的赋予率控制为一定水平,且能够对水不溶性纤维均匀地赋予自由基聚合性化合物。另外,通过在层压膜而密闭的空间内使水不溶性纤维与自由基聚合性化合物进行反应,接枝反应进一步受到促进。
[0053]
需要说明的是,本发明中的“层压”是指使水不溶性纤维与膜接触。
[0054]
将从上述溶液槽取出的水不溶性纤维在给定温度的后聚合槽中停留给定的时间,促进自由基聚合性化合物的接枝聚合(后聚合)。此时的后聚合温度为0℃~130℃、更优选为40℃~70℃。由此,水不溶性纤维与自由基聚合性化合物的接枝聚合反应受到促进。然后,通过清洗、干燥,可以得到接枝化纤维。需要说明的是,后聚合气体氛围优选为氮气等非活性气体氛围,在层压有膜的状态下进行后聚合时,可以为空气氛围。
[0055]
另外,在上述同时照射法的情况下,通过在利用通气氮气而去除了溶解氧的自由基聚合性化合物溶液的槽中浸渍、或在上述槽中浸渍通过,使其停留给定时间,对水不溶性纤维充分赋予自由基聚合性化合物。然后,从上述溶液槽中取出,照射电子束。优选在从溶液槽中取出时,在水不溶性纤维的表面层压膜,在该状态下照射电子束。
[0056]
同时照射法时的加速电压可以根据高分子原材料的种类、赋予了自由基聚合性化合物溶液的水不溶性纤维及层压的膜的合计厚度、目标接枝率而适当确定,通常,加速电压100~2000kv左右是适当的。另外,电子束的照射剂量可以与上述预处理方法的情况相同。电子束照射时的气体氛围优选为氮、氦等非活性气体氛围,在水不溶性纤维的表面层压有膜的情况下,由于照射气体氛围对接枝聚合没有影响,因此,考虑到经济性,空气中照射是适当的。
[0057]
经电子束照射的水不溶性纤维与上述预照射法的情况同样地进行自由基聚合性化合物的后聚合。然后,通过清洗、干燥,可以得到接枝化纤维。需要说明的是,在同时照射法中,后聚合气体氛围优选为氮气等非活性气体氛围,在层压了膜的状态下进行后聚合的情况下,可以为空气氛围。
[0058]
另外,本发明中使用的自由基聚合性化合物为与通过电子束照射而在水不溶性纤维生成的聚合物自由基成键的化合物。具体而言,可以列举:丙烯酸、甲基丙烯酸、衣康酸、甲基丙烯基磺酸、苯乙烯磺酸等具有酸性基团的不饱和化合物及它们的酯;丙烯酸酰胺、甲基丙烯酸酰胺等不饱和羧酸酰胺、末端具有缩水甘油基、羟基的不饱和化合物;乙烯基膦酸酯等不饱和有机磷酸酯、季铵盐、叔胺盐等具有碱性的甲基丙烯酸酯、氟丙烯酸酯、丙烯腈等,但并不限定于此。这些化合物可以单独使用,或者混合2种以上使用。通过使用2种以上的自由基聚合性化合物,可以得到接枝链由至少2种以上自由基聚合性化合物的共聚物形成的复合接枝化纤维。
[0059]
在这些自由基聚合性化合物当中,在本发明中,从接枝率的观点考虑,优选使用丙烯酸类单体。进一步,从与具有氨基、羟基、硫醇基等的配体的反应性的观点考虑,优选为分子末端具有羧基、环氧基的丙烯酸类单体,更优选为选自丙烯酸、甲基丙烯酸及甲基丙烯酸缩水甘油酯(以下简称为“gma”)中的至少1种。
[0060]
需要说明的是,上述丙烯酸类是指丙烯酸及其衍生物。
[0061]
上述的自由基聚合性化合物可以是以水、低级醇这样的有机溶剂或它们的混合溶液作为溶剂的稀释溶液。该稀释溶液的自由基聚合性化合物的浓度根据希望的接枝率而变化,可以以1~70容量%制备。另外,在使用易于产生均聚物的自由基聚合性化合物的情况下,可以通过在自由基聚合性化合物的稀释溶液中添加铜、铁的金属盐来抑制均聚物的生成。
[0062]
作为上述溶液中的上述自由基聚合性化合物的浓度的下限值,没有特别限制,可以根据目的而适当选择,优选为1重量%以上、更优选为2.5重量%以上、进一步优选为5重量%以上、特别优选为10重量%以上。
[0063]
作为上述溶液中的上述自由基聚合性化合物的浓度的上限值,没有特别限制,可以根据目的而适当选择,优选为70重量%以下、更优选为60重量%以下、进一步优选为50重量%以下、特别优选为40重量%以下。
[0064]
另外,在上述自由基聚合性化合物溶液包含溶剂时,接枝率提高。在该情况下,溶剂中的乳化剂的浓度优选调整为0.1~5重量%的范围。
[0065]
作为上述乳化剂,没有特别限制,可以根据目的而适当选择,例如,优选为聚山梨酯,作为上述聚山梨酯,可以举出聚山梨酯20、60、65、80等,其中,更优选为亲水性高的聚山梨酯20。
[0066]
需要说明的是,自由基聚合性化合物溶液优选预先通过吹入氮气等非活性气体而去除溶解氧。
[0067]
作为上述接枝聚合反应的接枝率,没有特别限制,可以根据目的而适当选择,优选为50%以上。
[0068]
本发明中,上述“接枝率”是根据接枝反应前的水不溶性纤维干燥重量(w1)和接枝反应后的接枝化纤维干燥重量(w2)如下所述计算出的值。
[0069]
接枝率=〔(w2-w1)/w1〕
×
100(%)
[0070]
作为上述水不溶性纤维的材料,没有特别限制,可以根据目的而适当选择,可以列举例如:聚烯烃类、聚丙烯、聚丙烯马来酸酐、改性聚丙烯、聚乙烯、纤维素、再生纤维素、乙酸纤维素、二乙酸纤维素、三乙酸纤维素、乙基纤维素、乙酸纤维素、聚对苯二甲酸乙二醇酯(pet)、聚对苯二甲酸丁二醇酯(pbt)、丙烯酸树脂、聚碳酸酯、聚酯类、聚丙烯腈、聚酰胺、聚苯乙烯、溴化聚苯乙烯、聚(甲基)丙烯酸烷基酯、聚氯乙烯、聚氯丁烯、聚氨酯、聚乙烯醇、聚乙酸乙烯酯、聚砜、聚醚砜、聚丁二烯、丁二烯-丙烯腈共聚物、苯乙烯-丁二烯共聚物、乙烯-乙烯醇共聚物、芳族聚酰胺、玻璃、尼龙、人造丝等。这些材料可以单独使用1种,也可以组合使用2种以上。其中,从电子束接枝聚合的反应性良好的观点考虑,优选为聚烯烃类或纤维素类,更优选为聚烯烃类,进一步优选为聚丙烯。
[0071]
需要说明的是,上述聚烯烃类是指聚烯烃及其衍生物,上述聚酯类是指聚酯及其衍生物,上述纤维素类是指纤维素及其衍生物。
[0072]
作为上述水不溶性纤维的平均纤维直径的下限值,没有特别限制,可以根据目的而适当选择,从具有良好的拉伸强度的观点、或生产性的观点考虑,优选为0.3μm以上、更优选为0.4μm以上、进一步优选为0.5μm以上。
[0073]
作为上述水不溶性纤维的平均纤维直径的上限值,没有特别限制,可以根据目的而适当选择,从纯化性能高的观点考虑,优选为15μm以下、更优选为10μm以下、进一步优选为5μm以下、特别优选为3μm以下。平均纤维直径超过15μm,则纯化性能低,由此不优选。
[0074]
作为上述水不溶性纤维的平均孔径的下限值,没有特别限制,可以根据目的而适当选择,从具有良好的通液性能的观点、或生产性的观点考虑,优选为1.0μm以上、更优选为大于1.0μm、进一步优选为1.5μm以上、更进一步优选为2.0μm以上、特别优选为2.5μm以上、最优选为3.0μm以上。
[0075]
作为上述水不溶性纤维的平均孔径的上限值,没有特别限制,可以根据目的而适当选择,从纯化性能高的观点考虑,优选为50μm以下、更优选为40μm以下、进一步优选为35μm以下、特别优选为30μm以下。
[0076]
上述水不溶性纤维的平均孔径如以下所述进行测定。
[0077]
对于上述水不溶性纤维的平均孔径而言,将使用孔径测定装置(porous materials inc.(pmi公司)制、capillary flow porometer cfp-1200aexlcshj)测得的平均流量孔径(mean flow pore)作为平均孔径。上述平均孔径的测定、计算使用测定软件(autofilling perm-porometer v6.73.90)及分析软件(caprep v6.71.51)。
[0078]
将待测定的水不溶性纤维冲裁成直径26mm的圆形,在galwick(美国注册商标)渗透液(pmi公司制、表面张力15.9dynes/cm)中浸渍后,放入干燥器中用真空泵减压1分钟左右,去除空气,填充渗透液。将该水不溶性纤维设置于装置附带的试样台(φ26),对于在渗
透液中浸渍后的样品,使空气的流量逐渐增加至200000cc/m,然后,在干燥状态下将空气的流量逐渐减少至0kpa的压力。绘制此时的压力,通过泡点法计算出平均流量孔径(平均孔径)。测定条件如下所述。
[0079]
maxflow 200000(cc/m)
[0080]
bublflow 2.00(cc/m)
[0081]
f/pt 200(oldbubltime)
[0082]
minbpress 0(kpa)
[0083]
zerotime 1.0(sec)
[0084]
v2incr 5(cts*3)
[0085]
preginc 0.3
[0086]
pulsedelay 2
[0087]
maxpres 1378.9466(kpa)
[0088]
pulsewidth 0.2(sec)
[0089]
mineqtime 30(sec)
[0090]
presslew 10(cts*3)
[0091]
flowslew 50(cts*3)
[0092]
equiter 5(0.1sec)
[0093]
aveiter 20(0.1sec)
[0094]
maxpdif 0.69(kpa)
[0095]
maxfdif 50(cc/m)
[0096]
作为上述水不溶性纤维的gurley透气度(通过300ml空气总量所需要的时间(秒)),没有特别限制,可以根据目的而适当选择,优选为60秒钟以下、更优选为30秒钟以下、进一步优选为10秒钟以下、更进一步优选为8秒钟以下、特别优选为5秒钟以下、最优选为2.5秒钟以下。
[0097]
上述gurley透气度是通过以下方式得到的值:依据jis l1096:2010(8.26.2b法(gurley型法)),将水不溶性纤维设置于面积642mm2的圆形的通气面,使用高度254mm、外径76.2mm、内径74mm、质量567g的圆筒,使空气300ml通过水不溶性纤维,测定300ml空气总量通过所需要的时间(通气时间:秒)。此时,根据孔径,将1~30片水不溶性纤维重合,每个水平各测定3次,计算出每1片的平均值。
[0098]
作为上述水不溶性纤维,没有特别限制,可以根据目的而适当选择,可以为无纺布、织布或编物,从简化制造工序的观点考虑,优选为无纺布。
[0099]
作为上述无纺布的制造方法,没有特别限制,可以根据目的而适当选择,可以列举例如:湿式法、干式法、熔喷法、静电纺丝法、闪蒸纺丝法、抄造法、纺粘法、热熔粘合法、化学粘合法、针刺法、水刺法(水流缠结法)、缝编法、蒸汽喷射法等。其中,从可得到极细纤维的观点考虑,优选为熔喷法、静电纺丝法、闪蒸纺丝法、抄造法等。
[0100]
作为上述熔喷法,没有特别限制,可以根据目的而适当选择,可以举出例如,利用高温/高速的空气流将挤出机中熔融的热塑性树脂从熔喷模头以丝状喷出,在输送机上聚集拉伸成纤维状的树脂,由此,纤维彼此抱合并发生熔粘,得到无粘合剂的自粘接型极细纤维的无纺布方法等。此时,可以通过调整树脂粘度、熔融温度、喷出量、热风温度、风压、dcd
(纺丝喷嘴和表面至输送机的距离)等来控制上述无纺布的纤维直径、单位面积重量、纤维取向、纤维分散性。进一步,可以通过热压加工、拉幅机加工等进行无纺布的厚度、平均孔径的控制。
[0101]-低分子化抗体-[0102]“抗体”是着眼于免疫球蛋白的功能的一般的名称。免疫球蛋白是淋巴细胞的b细胞所产生的糖蛋白,具有识别并结合特定的蛋白质等分子的作用。免疫球蛋白具有与该特定的分子(抗原)特异性结合的功能、和与其它生物分子、细胞协同而将具有抗原的因子无毒化/去除的功能。
[0103]
全部免疫球蛋白基本上由相同的分子结构形成,以“y”字型的4根链结构(轻链/重链的2根多肽链各2根)作为基本结构。轻链(l链)有λ链和κ链这2种,全部的免疫球蛋白均具有它们中的任何一者。重链(h链)有γ链、μ链、α链、δ链、ε链这样的结构不同的5种,根据该重链的差异,免疫球蛋白的种类(同种型)发生变化。免疫球蛋白g(以下有时简称为“igg”)为单体型的免疫球蛋白,由2根重链(γ链)和2根轻链构成,具有2个部位的抗原结合部位。
[0104]
将位于抗体的“y”字的下半部分的竖直部分的部位称为fc区域,将上半部分的“v”字部分称为fab区域。fc区域具有引起抗体与抗原结合后的反应的效应器功能,fab区域具有与抗原结合的功能。重链的fab区域和fc区域通过铰链区连接在一起,木瓜中包含的蛋白质分解酶木瓜蛋白酶将该铰链区分解而切断成2个fab区域(片段)和1个fc区域。fab区域中,接近“y”字前端的部分(结构域)在氨基酸序列中可观察到丰富多彩的变化,以便能够与多种多样的抗原结合,也被称为可变区(v区域)。将轻链的可变区称为vl区域,将重链的可变区称为vh区域。v区域以外的fab区域和fc区域是变化较少的区域,被称为恒定区(c区域)。将轻链的恒定区称为cl区域,将重链的恒定区称为ch区域,ch区域可进一步分为ch1~ch3这3种。重链的fab区域由vh区域和ch1形成,重链的fc区域由ch2和ch3形成。铰链区位于ch1与ch2之间。
[0105]
在骆驼科动物来源抗体、鲨鱼等鱼类来源抗体当中存在重链抗体,所述重链抗体是不存在轻链而仅由重链构成的抗体。骆驼科动物来源重链抗体与具有轻链的通常的igg抗体(igg1)相区别,被称为igg2、igg3。另一方面,鱼类来源的重链抗体被称为ignar(new antigen receptor)。
[0106]
作为本发明的低分子化抗体,只要是具有与抗原的结合能力的全长抗体(whole atibody、例如whole igg等)的一部分缺失的抗体片段即可,没有特别限定。
[0107]
本发明的低分子化抗体没有特别限制,可以根据目的而适当选择,优选不具有ch2结构域及ch3结构域。
[0108]
本发明的低分子化抗体优选包含重链可变区(vh)及轻链可变区(vl)中的任一者或两者。vh或vl的氨基酸序列可以包含添加、缺失和/或置换。此外,只要与抗原结合,也可以使vh及vl中的任一者或两者的一部分缺失。
[0109]
作为上述低分子化抗体,没有特别限制,可以根据目的而适当选择,可以列举例如包含骆驼科动物来源重链抗体的可变区(vhh)、鱼类来源重链抗体的可变区(v-nar)、fab、fab’、f(ab’)2、单链抗体(single chain antibody:scfv)、diabody、triabody、微抗体(minibody)的抗体等。其中,从稳定性及生产效率的观点考虑,优选包含骆驼科动物来源重链抗体的可变区(vhh)及单链抗体(scfv)。
[0110]
作为上述骆驼科动物来源的重链抗体的可变区(vhh),没有特别限制,可以根据目的而适当选择,可以列举例如:由免疫了上述吸附对象的骆驼科动物的血清纯化而得到的可变区、在宿主细胞中使vhh基因表达而制备的可变区、基于氨基酸序列进行化学合成而得到的可变区等。
[0111]
另外,上述vhh可以利用市售品。
[0112]
作为上述骆驼科动物,没有特别限制,可以根据目的而适当选择,可以列举例如:双峰驼、单峰驼、美洲驼、羊驼(alpaca)、骆马(vicuna)、原驼等。
[0113]
作为表达vhh基因的上述宿主细胞,没有特别限制,可以根据目的而适当选择,可以列举例如:大肠杆菌等细菌、酵母等真菌、动物细胞、植物细胞等。
[0114]
作为对上述骆驼科动物免疫上述吸附对象的方法,没有特别限制,可以根据目的而适当选择,可以举出例如国际公开第2020/067418号小册子中记载的方法等。
[0115]
作为在宿主细胞中表达vhh基因而制备的方法,没有特别限制,可以根据目的而适当选择,可以举出例如国际公开第2020/067418号小册子及日本特开2015-119637号公报中记载的方法等。
[0116]
作为上述vhh的市售品,没有特别限制,可以根据目的而适当选择,可以列举例如:thermo scientific公司的captureselect(注册商标)biotin anti-hsa conjugate、captureselect(注册商标)biotin anti-igg-fc(human)conjugate、captureselect(注册商标)biotin anti-aavx conjugate等。
[0117]
上述单链抗体(scfv)可以通过将抗体的vh和vl连接而得到。在scfv中,vh和vl经由连接子(linker)、优选为肽连接子而连接(proc.natl.acad.sci.u.s.a 1988 85:5879)。该肽连接子没有特别限制。例如,可以使用由3~25个残基左右形成的任意的单链肽作为连接子。
[0118]
作为上述scfv,没有特别限制,可以根据目的而适当选择,可以列举例如:在宿主细胞表达scfv基因而制备的scfv、基于氨基酸序列而化学合成的scfv、将序列已知的抗体的重链可变区和轻链可变区通过连接子连接而成的scfv等。
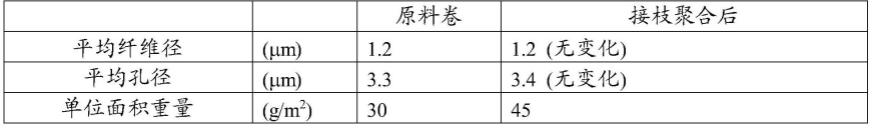
[0119]
另外,上述scfv可以利用市售品。
[0120]
作为表达scfv基因的上述宿主细胞,没有特别限制,可以根据目的而适当选择,可以列举例如:大肠杆菌等细菌、酵母等真菌、动物细胞、植物细胞等。
[0121]
本发明中的低分子化抗体优选包含单域抗体。
[0122]
作为上述单域抗体,没有特别限制,可以根据目的而适当选择,优选包含上述骆驼科动物来源的重链抗体的可变区(vhh)。
[0123]
本发明中的低分子化抗体可以经嵌合化、人源化。
[0124]
作为上述低分子化抗体的分子量,没有特别限制,可以根据目的而适当选择,优选为130000以下、更优选为100000以下、进一步优选为50000以下、特别优选为30000以下、最优选为20000以下。
[0125]
作为上述低分子化抗体的吸附对象,没有特别限制,可以根据目的而适当选择,可以列举例如:病毒、病毒样颗粒、病毒载体、核酸、抗体、酶、激素、以及它们的复合体、代谢产物等。其中,优选为病毒、病毒样颗粒、病毒载体。
[0126]
上述病毒样颗粒主要为构成衣壳的病毒外壳蛋白质的全部或一部分,由于不包含
核酸,因此,没有感染的隐患,另一方面,由于引起免疫反应,因此,可以作为疫苗的有效成分而使用。
[0127]
作为上述病毒、病毒样颗粒、病毒载体,没有特别限制,可以根据目的而适当选择,可以列举例如:腺相关病毒(aav)、腺病毒、肠病毒、细小病毒、乳多空病毒、人乳头瘤病毒、轮状病毒、柯萨奇病毒、札幌病毒、诺如病毒、脊髓灰质炎病毒、埃可病毒、甲型肝炎病毒、戊型肝炎病毒、鼻病毒、星状病毒、圆环病毒、猿猴病毒等无包膜病毒病毒、逆转录病毒、慢病毒、仙台病毒、单纯疱疹病毒等疱疹病毒、牛痘病毒、麻疹病毒、杆状病毒、流感病毒、白血病病毒、辛德比斯病毒等包膜病毒、以及这些病毒的衣壳、或包含它们的基因的病毒载体等。其中,优选为腺相关病毒、或包含腺相关病毒的衣壳、或包含腺相关病毒基因的载体。
[0128]
上述腺相关病毒是衣壳中包含直链状单链dna的属于细小病毒科的病毒。作为上述腺相关病毒的血清型,已知有1型aav(aav1)、2型aav(aav2)、3型aav(aav3)、4型aav(aav4)、5型aav(aav5)、6型aav(aav6)、7型aav(aav7)、8型aav(aav8)、9型aav(aav9)及10型aav(aav10)等。
[0129]
在上述低分子化抗体的吸附对象为上述腺相关病毒的情况下,作为上述低分子化抗体,可以利用国际公开第2018/104528号小册子中记载的公知的vhh。
[0130]
另外,也可以利用thermo scientific公司的captureselect(注册商标)biotin anti-aavx conjugate等市售品。
[0131]
此外,还可以将genetex公司的intact particles[a20]抗体等igg抗体片段化而利用。
[0132]
在从序列已知的抗体制备scfv等低分子化抗体时,可以从公共的数据库获得,例如,可以利用蛋白质数据库中注册的与aav2结合的抗体的重链序列(pdb:3j1s_h)及轻链序列(pdb:3j1s_l)等。
[0133]
‑‑
aav结合容量
‑‑
[0134]
作为上述结构物的aav结合容量,没有特别限制,可以根据目的而适当选择,优选为1.0
×
10
12
vg/ml-column以上、更优选为5.0
×
10
12
vg/ml-column以上、进一步优选为1.0
×
10
13
vg/ml-column以上、特别优选为1.5
×
10
13
vg/ml-column以上。需要说明的是,作为结合容量的计算方法,有基于从负载量减去溶出至流过级分的量而得到的总吸附量的方法、以及基于在吸附后通过溶出工序回收的总量的方法。在本发明中,基于后者的总回收量计算出结合容量。
[0135]
上述结构物的aav结合容量通过以下的方法测定。
[0136]
使用上述结构物通过亲和色谱法对aav粗纯化液进行纯化。
[0137]
制备下述a~f液,在使用前用0.2μm过滤器进行了过滤。
[0138]
a液:tris-hcl(20mmol/l),氯化钠(500mmol/l),ph8.0
[0139]
b液:柠檬酸(100mmol/l),氯化钠(500mmol/l),ph2.1
[0140]
c液:naoh(10mmol/l)
[0141]
d液:磷酸(120mmol/l),乙酸(167mmol/l),苯甲醇(2.2%v/v)
[0142]
e液:20%乙醇
[0143]
f液:用a液稀释aav粗纯化液并调整为ph8.0的溶液(2
×
10
13
vg左右)
[0144]
将柱连接于akta avant 25,将流速设定为0.2ml/min,用纯水清洗,用a液平衡化。
然后,通过f液,使aav保持于柱载体后,用a液清洗,用b液将aav溶出。溶出结束后,按照a液、c液、a液、d液的顺序清洗柱。通过qpcr对各级分中包含的aav量进行定量,根据溶出工序中包含的aav量计算出结合容量。
[0145]
‑‑
配体密度
‑‑
[0146]
作为上述结构物的配体密度(低分子化抗体密度),没有特别限制,可以根据目的而适当选择,优选为0.01mg/ml-column以上且100mg/ml-column以下,更优选为0.1mg/ml-column以上且50mg/ml-column以下,进一步优选为1mg/ml-column以上且10mg/ml-column以下。
[0147]
上述配体密度如下所述进行测定。
[0148]
将配体固定于水不溶性纤维时的滤液回收,测定吸光度,由此计算出固定于水不溶性纤维的配体的配体密度。
[0149]-其它要素-[0150]
作为上述其它要素,没有特别限制,可以根据目的而适当选择,可以举出例如间隔物等。
[0151]
‑‑
间隔物
‑‑
[0152]
作为包含上述间隔物的结构物,没有特别限制,可以根据目的而适当选择,可以举出例如,上述低分子化抗体经由间隔物而连接于上述水不溶性纤维的结构物等。
[0153]
作为上述间隔物所具有的官能团,没有特别限制,可以根据目的而适当选择,可以列举例如:氨基、羟基、环氧基、羧基等。其中,从稳定性、反应性、与配体的连接的容易程度的观点考虑,优选为环氧基。
[0154]
作为上述间隔物,没有特别限制,可以根据目的而适当选择,可以列举例如包含聚合物、单体、二聚物、三聚物、四聚物的间隔物等。其中,优选为包含聚合物的间隔物。上述聚合物可以为共聚物。
[0155]
作为上述聚合物,没有特别限制,可以根据目的而适当选择,可以列举例如包含亲水性聚合物、疏水性聚合物的聚合物等。其中,从能够进行水性溶剂中的操作的观点、抑制蛋白质与间隔物的非特异性疏水性作用的观点考虑,优选为包含亲水性聚合物的聚合物。
[0156]
作为上述亲水性聚合物,没有特别限制,可以根据目的而适当选择,可以举出例如,包含多胺、多糖类的聚合物等。
[0157]
作为上述多胺,没有特别限制,可以根据目的而适当选择,可以列举例如:聚乙烯亚胺、聚烯丙胺、聚乙烯胺、聚赖氨酸、乙二胺、1,2-丙二胺、1,6-六亚甲基二胺、哌嗪、2,5-二甲基哌嗪、异佛尔酮二胺、4,4
’‑
二环己基甲烷二胺、1,4-环己烷二胺等二胺类;二亚乙基三胺、二亚丙基三胺、三亚乙基四胺等多胺类;肼、n,n
’‑
二甲基肼、1,6-六亚甲基双肼等肼类;丁二酸二酰肼、己二酸二酰肼、戊二酸二酰肼、癸二酸二酰肼、间苯二甲酸二酰肼等二酰肼类等。这些多胺、可以单独使用1种,也可以组合使用2种以上。其中,从能够容易获得分子量不同的分子的观点考虑,优选为包含聚乙烯亚胺或聚烯丙胺的多胺。
[0158]
上述多糖类,没有特别限制,可以根据目的而适当选择,可以列举例如:壳聚糖、几丁质、纤维素、琼脂糖、卡拉胶、肝素、透明质酸、果胶、木葡聚糖、葡甘聚糖、淀粉、糖原等。这些多糖类可以单独使用1种,也可以组合使用2种以上。其中,从包含氨基的观点考虑,优选为壳聚糖。
[0159]
作为上述聚合物的摩尔质量的下限值,没有特别限制,可以根据目的而适当选择,从生物粒子的吸附性的观点考虑,优选为500g/mol以上、更优选为5000g/mol以上、进一步优选为10000g/mol以上、特别优选为60000g/mol以上。
[0160]
作为上述聚合物的摩尔质量的上限值,没有特别限制,可以根据目的而适当选择,从生物粒子的吸附性的观点考虑,优选为2000000g/mol以下、更优选为1000000g/mol以下、进一步优选为500000g/mol以下、特别优选为200000g/mol以下。
[0161]
上述聚合物可以是具有侧链的聚合物,也可以是线状聚合物,从生物粒子的吸附性的观点考虑,优选为具有侧链的聚合物。
[0162]
(结构物的制造方法)
[0163]
上述结构物的制造方法包括将水不溶性纤维与低分子化抗体进行连接的工序,可以进一步包括其它工序。
[0164]
上述水不溶性纤维及上述低分子化抗体如上所述。
[0165]
作为上述水不溶性纤维与上述低分子化抗体的连接,没有特别限制,可以根据目的而适当选择,例如,除了将上述低分子化抗体加入上述水不溶性纤维并颠倒混合的方法、向上述水不溶性纤维加入其它要素颠倒混合并进一步加入低分子化抗体颠倒混合的方法等。
[0166]
作为上述颠倒混合的时间,没有特别限制,可以根据目的而适当选择,优选为1小时以上至48小时以下、更优选为2小时以上至48小时以下、进一步优选为3小时以上至24小时以下、特别优选为8小时以上至24小时以下。
[0167]
作为包含上述间隔物的结构物的制造方法,没有特别限制,可以根据目的而适当选择,可以举出例如,将间隔物的一端连接于水不溶性纤维、并将另一端连接于低分子化抗体的工序等。
[0168]
上述间隔物、上述水不溶性纤维及上述低分子化抗体如上所述。
[0169]
这里,间隔物的一端与水不溶性纤维的连接和间隔物的另一端与低分子化抗体的连接的顺序没有限定,从易于控制用于连接的反应的观点考虑,优选在间隔物的一端连接于水不溶性纤维之后进行间隔物的另一端与低分子化抗体的连接。
[0170]
作为上述间隔物的一端与水不溶性纤维的连接,没有特别限制,可以根据目的而适当选择,可以列举例如,向水不溶性纤维中加入间隔物溶液并颠倒混合的方法等。
[0171]
作为上述颠倒混合的时间,没有特别限制,可以根据目的而适当选择,优选为1小时以上至48小时以下、更优选为2小时以上至48小时以下、进一步优选为3小时以上至24小时以下、特别优选为8小时以上且24小时以下。
[0172]
作为上述间隔物的另一端与低分子化抗体的连接,没有特别限制,可以根据目的而适当选择,可以列举例如,向水不溶性纤维加入低分子化抗体并颠倒混合的方法等。
[0173]
作为上述颠倒混合的时间,没有特别限制,可以根据目的而适当选择,优选为1小时以上至48小时以下、更优选为2小时以上至48小时以下、进一步优选为3小时以上至24小时以下、特别优选为8小时以上且24小时以下。
[0174]
本发明的结构物可以根据目的而进行加工,其形状没有特别限定。例如,可以选择平膜状、中空丝状、褶皱状、卷状、螺旋状、管状等形状。这些经加工的结构物可以以单体使用,也可以层叠,还可以串联及并联地连接而使用。
[0175]
用于填充上述结构物的设备的形状没有特别限定,可以选择圆盘状、圆筒状、板状等。从实现均匀的通液的观点考虑,优选为圆盘状及圆筒状。
[0176]
加工后的结构物例如可以作为用于生物粒子的纯化的柱、以生物粒子的吸附及去除为目的的过滤器及口罩等而使用。作为过滤器,例如,可以作为血液制剂等生物医药品的制造工序中的病毒去除、hepa(high efficiency particulate air、高效空气过滤器)等的过滤器而使用。
[0177]
(吸附体)
[0178]
上述吸附体具有上述的结构物,可以进一步具有其它的构成要素。
[0179]
上述吸附体的吸附对象如上述的低分子化抗体的吸附对象所述。
[0180]
(生物粒子的纯化方法)
[0181]
上述生物粒子的纯化方法包括通过使用了上述的结构物或上述的吸附体的亲和色谱纯化法进行分离的工序,可以进一步包括其它工序。
[0182]
上述生物粒子的纯化方法的纯化对象如上述的低分子化抗体的吸附对象所述。
[0183]
作为上述纯化,没有特别限制,可以根据目的而适当选择,可以列举例如,使用了填充有上述结构物或上述吸附体的柱的纯化等。
[0184]
作为上述柱的材质,没有特别限制,可以根据目的而适当选择,可以列举例如,玻璃、聚丙烯、丙烯酸等树脂、不锈钢等金属等。
[0185]
上述生物粒子的纯化法基本上可以通过与一般公知的使用了包含蛋白质配体的亲和柱的纯化法相同的步骤来实现(可以举出例如,作为市售品而存在的蛋白质a柱等。journal of chromatography a 2007 1160:44-55)。
[0186]
即,在将含有对象生物粒子的缓冲液调整为中性后,使该溶液通过填充有本发明的结构物或吸附体的亲和柱,吸附对象生物粒子。
[0187]
接着,使纯的缓冲液适量通过亲和柱,清洗柱内部。此时,期望的生物粒子被吸附于柱内的本发明的结构物或吸附体。
[0188]
接着,使调整为适当的ph的酸性或碱性的缓冲液(有时包含促进从该结构物或该吸附体的解离的物质)通过柱,将期望的生物粒子溶出,由此实现高纯度的纯化。
[0189]
通过使不会将配体(低分子化抗体)、载体的基材(水不溶性纤维)的功能完全损害的程度的适当的强酸性或强碱性的纯缓冲液(有时为包含适当的改性剂或有机溶剂的溶液)通过本发明的结构物或吸附体并进行清洗,可以进行再利用。
[0190]-其它工序-[0191]
作为上述其它工序,没有特别限制,可以根据目的而适当选择,可以列举例如,在通过上述亲和色谱纯化法进行分离的工序之前或在通过上述亲和色谱纯化法进行分离的工序之后的离子交换色谱纯化工序、超离心纯化工序等其它的纯化工序等。
[0192]
上述离子交换色谱纯化工序是基于与带有电荷的离子交换配体的静电相互作用而将混合物分离的工序。
[0193]
另外,上述超离心纯化工序通过将混合物离心而分离为分子量接近的各物质的工序。
[0194]
与上述其它的纯化方法相比,由于亲和色谱纯化法利用对对象生物粒子具有特异性结合能力的配体,因此具有易于从培养液等包含大量杂质的混合物中以高纯度分离目标
生物粒子的优点。
[0195]-生物粒子在流通液(ft)中的溶出率、溶出工序的生物粒子回收率、以及清洗工序的生物粒子残留率-[0196]
作为上述生物粒子的纯化方法中的生物粒子在流通液(ft)中的溶出率,没有特别限制,可以根据目的而适当选择,优选低于100%、更优选为50%以下、进一步优选为20%以下、特别优选为0%。
[0197]
作为上述生物粒子的纯化方法中的溶出工序的生物粒子回收率,没有特别限制,可以根据目的而适当选择,优选为5%以上、更优选为10%以上、进一步优选为50%以上、特别优选为70%以上。
[0198]
作为上述生物粒子的纯化方法中的清洗工序的生物粒子残留率,没有特别限制,可以根据目的而适当选择,优选为30%以下、更优选为20%以下、进一步优选为10%以下、特别优选为5%以下。
[0199]
生物粒子在流通液(ft)中的溶出率、溶出工序的生物粒子回收率、以及清洗工序的生物粒子残留率通过以下的方法进行测定。
[0200]
通过基于无纺布载体的色谱法对aav前处理液进行纯化。
[0201]
制备下述a~f液,在使用前用0.2μm过滤器进行过滤。
[0202]
a液:tris-hcl(20mmol/l),氯化钠(500mmol/l),ph8.0
[0203]
b液:柠檬酸(100mmol/l),氯化钠(500mmol/l),ph2.1
[0204]
c液:naoh(10mmol/l)
[0205]
d液:磷酸(120mmol/l),乙酸(167mmol/l),苯甲醇(2.2%v/v)
[0206]
e液:20%乙醇
[0207]
f液:用a液稀释aav前处理液并调整为ph8.0的溶液(1
×
10
12
vg左右)
[0208]
将填充有载体的柱连接于akta avant 25,将流速设定为0.2ml/min,用纯水清洗,用a液平衡化。然后,通过f液,使aav保持于柱载体后,用a液清洗,用b液将aav溶出。溶出结束后,按照a液、c液、a液、d液的顺序清洗柱。
[0209]
通过使用了quantstudio3实时pcr系统(thermo fisher scientific公司制)的定量pcr(qpcr)对各级分中包含的aav量进行定量,计算出溶出峰的aav量相对于全部回收液中包含的aav量的值作为回收率。另外,计算出流通液(ft)溶液中的aav量相对于通液的f液的总aav量的值作为溶出率,计算出碱清洗工序的aav量相对于全部回收液中包含的aav量的值作为残留率。
[0210]
实施例
[0211]
以下,对本发明的实施例进行说明,但本发明并不受这些实施例的任何限定。
[0212]
<制造例1:水不溶性纤维(无纺布1)的制作>
[0213]
以聚丙烯作为材料,通过熔喷法制作了平均纤维直径1.19μm、单位面积重量30g/m2、厚度0.17mm的无纺布。对于自由基聚合性化合物溶液,将gma/聚山梨酯20/水以重量%计为10/2/88的方式进行混合,通入氮气,由此去除溶解氧。在加速电压250kv、照射剂量50kgy的条件下、氮气氛围中、以室温对制成的无纺布进行电子束照射。将电子束照射后的无纺布浸渍于自由基聚合性化合物溶液,在反应槽中以50℃加热40分钟,促进聚合反应。然后,用常温的水清洗无纺布后,进行干燥,得到了接枝率50%的水不溶性纤维(无纺布1)。
[0214]
如下所述,测定无纺布的平均纤维直径及平均孔径,对于接枝聚合前后的无纺布,总结于表1。对于接枝聚合后的结果,也示于表2。
[0215]
无纺布的平均纤维直径基于扫描电子显微镜观察而求出。从得到的电子显微镜图像中测量随机选择的100根以上纤维的直径。将100个以上的测量值的数平均值作为平均纤维直径。另外,接枝率是测定接枝反应前的无纺布(w1)和接枝反应后的无纺布重量(w2)并如下所述计算出的值。
[0216]
接枝率=〔(w2-w1)/w1〕
×
100(%)
[0217]
对于无纺布的平均孔径而言,将使用孔径测定装置(porous materials inc.(pmi公司)制、capillary flow porometer cfp-1200aexlcshj)测得的平均流量孔径作为平均孔径。平均孔径的测定在计算时使用了测定软件(autofilling perm-porometer v6.73.90)及分析软件(caprep v6.71.51)。
[0218]
将待测定的无纺布冲裁成直径26mm的圆形,在galwick(美国注册商标)渗透液(pmi公司制、表面张力15.9dynes/cm)中浸渍后,放入干燥器中用真空泵减压1分钟左右,去除空气,填充了渗透液。将该无纺布设置于装置附带的试样台(φ26),对于在渗透液中浸渍后的样品,使空气的流量逐渐增加至200000cc/m,然后,在干燥状态下将空气的流量逐渐减少至0kpa的压力。绘制此时的压力,通过泡点法计算出平均流量孔径(平均孔径)。如下所述示出测定条件。
[0219]
maxflow 200000(cc/m)
[0220]
bublflow 2.00(cc/m)
[0221]
f/pt 200(oldbubltime)
[0222]
minbpress 0(kpa)
[0223]
zerotime 1.0(sec)
[0224]
v2incr 5(cts*3)
[0225]
preginc 0.3
[0226]
pulsedelay 2
[0227]
maxpres 1378.9466(kpa)
[0228]
pulsewidth 0.2(sec)
[0229]
mineqtime 30(sec)
[0230]
presslew 10(cts*3)
[0231]
flowslew 50(cts*3)
[0232]
equiter 5(0.1sec)
[0233]
aveiter 20(0.1sec)
[0234]
maxpdif 0.69(kpa)
[0235]
maxfdif 50(cc/m)
[0236]
[表1]
[0237]
[0238][0239]
[表2]
[0240]
ꢀꢀ
无纺布1无纺布2无纺布3无纺布4无纺布5无纺布6平均纤维径(μm)1.20.60.60.63.010平均孔径(μm)3.43.73.53.510.932.8单位面积重量(g/m2)452618174550厚度(mm)0.20.170.110.130.280.26
[0241]
<制造例2:水不溶性纤维(无纺布2)的制作>
[0242]
以聚丙烯作为材料,制作了平均纤维直径0.56μm、单位面积重量15g/m2、厚度0.11mm的无纺布。使用将gma/聚山梨酯20/水以重量%计为8/2/90的方式进行混合而得到的自由基聚合性化合物溶液,与制造例1同样地进行电子束接枝聚合,得到了接枝率70%的水不溶性纤维(无纺布2)。
[0243]
与制造例1同样地测定无纺布的平均纤维直径及平均孔径,示于表2。
[0244]
<制造例3:水不溶性纤维(无纺布3)的制作>
[0245]
以聚丙烯作为材料,制作了平均纤维直径0.56μm、单位面积重量15g/m2、厚度0.11mm的无纺布。使用将gma/聚山梨酯20/水以重量%计为2.4/2/95.6的方式进行混合而得到的自由基聚合性化合物溶液,与制造例1同样地进行电子束接枝聚合,得到了接枝率20%的水不溶性纤维(无纺布3)。
[0246]
与制造例1同样地测定无纺布的平均纤维直径及平均孔径,示于表2。
[0247]
<制造例4:水不溶性纤维(无纺布4)的制作>
[0248]
以聚丙烯作为材料,制作了平均纤维直径0.56μm、单位面积重量15g/m2、厚度0.11mm的无纺布。对于自由基聚合性化合物溶液,将丙烯酸(aa)/neoethanol以重量%计为20/80的方式进行混合,通入氮气,由此去除溶解氧。在加速电压200kv、照射剂量10kgy的条件下、氮气氛围中、以室温对制成的无纺布进行电子束照射。将电子束照射后的无纺布浸渍于自由基聚合性化合物溶液,在反应槽中以50℃加热30分钟,促进聚合反应。然后,用常温的水清洗无纺布后,进行干燥,得到了接枝率12%的水不溶性纤维(无纺布4)。
[0249]
与制造例1同样地测定无纺布的平均纤维直径及平均孔径,示于表2。
[0250]
<制造例5:水不溶性纤维(无纺布5)的制作>
[0251]
以聚丙烯作为材料,通过熔喷法制作了平均纤维直径3μm、单位面积重量30g/m2、厚度0.28mm的无纺布。使用将gma/聚山梨酯20/水以重量%计为10/2/88的方式进行混合而得到的自由基聚合性化合物溶液,与制造例1同样地进行电子束接枝聚合,得到了接枝率50%的水不溶性纤维(无纺布5)。
[0252]
与制造例1同样地测定无纺布的平均纤维直径及平均孔径,示于表2。
[0253]
<水不溶性纤维(无纺布6)>
[0254]
对于纤维素无纺布(以下记载为无纺布6,505swjd,futamura chemical公司制),与制造例1同样地测定无纺布的平均纤维直径及平均孔径,示于表2。
[0255]
对于制造例2、3及5中制作的无纺布2、3及5、以及无纺布6,使用gurley式密度仪(toyoseiki公司制)测定了gurley透气度。
[0256]
对于上述gurley透气度,依据jis l1096:2010(8.26.2b法(gurley型法)),将水不溶性纤维设置于面积642mm2的圆形的通气面,使用高度254mm、外径76.2mm、内径74mm、质量567g的圆筒,使空气300ml通过各无纺布,测定了300ml空气总量通过所需要的时间(通气时间:秒)。此时,将无纺布2、3及5以5片、10片、15片重合的水平分别各测定3次,将无纺布6以5片、10片、15片、30片重合的水平分别各测定3次,计算出每1片的平均值。将结果示于表3。
[0257]
可以认为,上述通气时间与孔的面积、即平均孔径的平方成反比。因此,取通气时间的平方根的倒数,并比较它们与用孔径测定装置计算出的平均孔径的相关性。将结果示于表3及图1。
[0258]
[表3]
[0259][0260]
作为孔径更小的基材,准备了平均孔径0.2μm的再生纤维素膜(cytiva公司制、产品编号10410314、以下记载为膜1)、平均孔径0.45μm的再生纤维素膜(cytiva公司制、产品编号10410214、以下记载为膜2)、以及平均孔径1μm的再生纤维素膜(cytiva公司制、产品编号10410014、以下记载为膜3)。其中,对于以足以进行透气度测定的尺寸市售的膜1及膜3,与无纺布同样地测定透气度,比较了与平均孔径的相关性。此时,将膜1、3均以1片、3片、5片重合的水平分别各测定3次,计算出每1片的平均值。将结果示于表3及图1。
[0261]
根据表3及图1的结果可以确认,通过孔径测定装置测得的无纺布的平均孔径及膜的平均孔径与通气时间的平方根的倒数良好地相关,不取决于基材的种类、材质。
[0262]
<制造例6:盘的制作>
[0263]
使用冲头,将上述制造例1~5中制作的无纺布1~5及无纺布6冲裁成直径5mm的圆形,制作了用于填充于柱的水不溶性纤维盘(无纺布盘)。
[0264]
通过同样的方法将平均孔径0.2μm的再生纤维素膜(cytiva公司制、产品编号10410314、膜1)、平均孔径0.45μm的再生纤维素膜(cytiva公司制、产品编号10410214、膜2)、以及平均孔径1μm的再生纤维素膜(cytiva公司制、产品编号10410014、膜3)冲裁成直径5mm的圆形,制作了用于填充于柱的基材盘(膜盘)。
[0265]
<制造例7:抗aav-scfv抗体的制备>
[0266]
以与蛋白质数据库中注册的aav2结合的抗体的重链序列(pdb:3j1s_h)及轻链序列(pdb:3j1s_l)作为基础,设计了序列表的序列号1所示的与aav2结合的单链抗体(以下称为抗aav-scfv抗体)。
[0267]
设计在该抗aav-scfv抗体中添加有信号序列(pelb)的大肠杆菌表达用的基因(序列号2),委托制备了将基因合成的序列导入pet-22b( )(merck公司制)载体的ncoi位点、noti位点的质粒(genscript公司)。
[0268]
将基因合成的质粒转化至e.coli bl21(de3)(merck公司制),使用包含卡那霉素
的magic media(thermofisher公司制)在坂口烧瓶中进行培养,生产了抗aav-scfv抗体。
[0269]
使用蛋白l载体kaneka kancap l(株式会社钟化制)从得到的培养液进行亲和纯化,通过离心超滤膜进行缓冲液更换,得到了抗aav-scfv抗体的纯化品。
[0270]
<制造例8:抗aav-vhh抗体的制备1>
[0271]
以国际专利公报wo2018/104528中记载的抗aav-vhh抗体的氨基酸序列(国际专利公报wo2018/104528的序列表的序列号59)作为基础,设计了大肠杆菌表达用的抗aav-vhh抗体(序列号3)。
[0272]
设计在该抗aav-vhh抗体中添加有信号序列(pelb)的大肠杆菌表达用的基因(序列号4),委托制备了将基因合成的序列导入pet-28b( )(merck公司制)载体的xbai、bpu1102i位点的质粒(genscript公司)。
[0273]
将基因合成的质粒转化至e.coli bl21(de3)(merck公司制),使用包含卡那霉素的magic media(thermofisher公司制)在坂口烧瓶中进行培养,生产了抗aav-vhh抗体。
[0274]
使用阳离子交换载体cellufine max s(jnc制)从得到的培养液进行纯化,通过离心超滤膜进行缓冲液更换,得到了抗aav-vhh抗体的纯化品(以下记载为vhh1)。
[0275]
<制造例9:抗aav-vhh抗体的制备2>
[0276]
通过国际公开第2020/067418号小册子的实施例6中记载的方法实施了免疫实验。通过日本特开2015-119637号公报的实施例1中记载的方法从免疫后的羊驼的血液中获取抗体基因群,制备了抗aav-vhh抗体的噬菌体文库。从噬菌体文库筛选抗aav-vhh抗体使用aav的空颗粒(empty particle)作为抗原,通过日本特开2015-119637号公报的实施例1中记载的生物淘选(biopanning)的方法而实施。aav的空颗粒通过国际公开第2020/067418号小册子的实施例3中记载的方法制备。
[0277]
将生物淘选后的经噬菌体感染的大肠杆菌进行连续稀释,在包含100μg/ml氨苄青霉素和2%葡萄糖的2yt琼脂培养基(1.6%胰蛋白胨、1.0%酵母提取物、2.0%琼脂)中进行培养。将单菌落在包含100μg/ml氨苄青霉素和2%葡萄糖的2yt培养基3ml中于37℃下培养过夜。向包含100μg/ml氨苄青霉素的2yt培养基5ml中加入培养过夜的培养液30μl,以37℃、300rpm培养1小时。在培养后,分取培养液3ml,以moi=5感染辅助噬菌体(m13ko7(thermo fisher scientific公司制):1.0
×
1013/ml)。在37℃下静置30分钟后,以37℃、300rpm振荡30分钟。向该培养液中添加50μg/ml的卡那霉素,以37℃、300rpm培养过夜。在培养后,将培养液以4℃、4000rpm离心30分钟,用0.2μm的过滤器(sartorius公司制)过滤上清,去除大肠杆菌,获得了显示出抗aav-vhh抗体的噬菌体。
[0278]
其中,将对aav2、aav5、aav1及6具有亲和性的抗aav-vhh抗体分别称为vhh配体2、3、4。
[0279]
将该噬菌体溶液作为抗体溶液,通过国际公开第2020/067418号小册子中记载的方法评价对aav的结合活性,挑选出结合活性高的克隆。使结合活性高的噬菌体克隆感染e.coli mv1184(takara bio公司制),培育了在细胞周质表达抗aav-vhh抗体的大肠杆菌。使用包含100μg/ml氨苄青霉素的tb培养基(1.2%胰蛋白胨、2.4%酵母提取物、0.8%甘油、0.94%磷酸氢二钾、0.22%磷酸二氢钾)在37℃下将该大肠杆菌培养1小时后,添加异丙基-β-硫代半乳糖苷,使其最终浓度达到1mm,在37℃下培养过夜。
[0280]
通过使用了akta avant 25(ge healthcare公司)的阳离子交换色谱从得到的vhh
表达培养液中纯化了抗aav-vhh抗体。样品负载于用25mm磷酸钠缓冲液(ph6.0)平衡化后的cellufine max s-r 100ml(jnc公司),通过平衡化所使用的缓冲液和包含1m nacl的磷酸钠缓冲液(ph6.0)的线性梯度进行了溶出。用离心超滤单元omega膜1k(pall公司)将经纯化的抗aav-vhh抗体浓缩,得到了纯化vhh抗体(vhh配体2、3、4)。
[0281]
<实施例1:载体1的制作>
[0282]
<实施例1-1:聚乙烯亚胺(pei)间隔物向无纺布1的固定化>
[0283]
对于制造例1中得到的无纺布1,加入pei溶液(alfa aesar公司制、mw:70000、侧链),以37℃颠倒混合16小时。在反应后,将无纺布转移至玻璃过滤器,用纯水清洗,得到了固定化有pei间隔物的无纺布1。
[0284]
<实施例1-2:对于固定化有pei间隔物的无纺布的官能团化>
[0285]
对于实施例1-1中得到的固定化有pei间隔物的无纺布1,加入超纯水4ml及2n氢氧化钠水溶液0.8ml,颠倒混合30分钟。加入1,4-丁二醇二缩水甘油醚(nagase chemtex公司制)4ml,在37℃下颠倒混合4小时。在反应后,将无纺布转移至玻璃过滤器,用纯水清洗,得到了环氧化无纺布1。
[0286]
<实施例1-3:scfv配体1向环氧化无纺布1的固定化>
[0287]
对于实施例1-2中得到的环氧化无纺布1,加入包含0.5mol/l nacl的0.2mol/l碳酸盐缓冲液(ph10.0)。向其中添加1.7mg量的制造例7中制备的纯化scfv抗体(scfv配体1),使得液量达到5ml。在37℃下颠倒混合30分钟后,缓慢加入无水硫酸钠0.497g,使其溶解,进一步在37℃下颠倒混合16小时。在反应后,将无纺布转移至玻璃过滤器,用上述碳酸盐缓冲液清洗。回收此时的滤液,测定吸光度,从而计算出固定化于无纺布的scfv配体的配体密度。计算出的配体密度为0.4mg/ml-column(表4)。接着,为了封闭清洗后的无纺布中残留的环氧基,加入包含0.1mol/l的nacl及10%v/v硫代甘油的0.2mol/l磷酸缓冲液(ph8.0),在25℃下颠倒混合16小时。在反应后,将无纺布转移至玻璃过滤器,用纯水及20%乙醇清洗,得到了scfv1固定化无纺布1(载体1)。
[0288]
[表4]
[0289]
[0290][0291]
<实施例2:载体2的制作>
[0292]
将scfv配体1替换为vhh配体1,并将配体固定化时的缓冲液替换为包含0.5mol/l的nacl、1.2mol/l胍盐酸盐的0.2mol/l碳酸盐缓冲液(ph10.0),除此以外,通过与实施例1相同的方法进行间隔物固定化、官能团化、vhh1固定化,得到了vhh1固定化无纺布1(载体2)。
[0293]
与实施例1同样地测定配体密度,将测定结果示于表4。
[0294]
<比较例1:载体3的制作>
[0295]
<比较例1-1:珠1的官能团化>
[0296]
称量poros(注册商标)50oh hydroxyl activated resin(以下记载为珠1,thermo scientific公司制孔径50-1000nm(公称值))5ml-gel,得到了通过与实施例1-2相同的方法对其导入了环氧基的珠1(环氧化珠1)。
[0297]
与实施例1同样地测定配体密度,将测定结果示于表4。
[0298]
<比较例1-2:vhh2向环氧化珠1的固定化>
[0299]
对于比较例1-2中得到的环氧化珠1,加入包含0.5mol/l nacl的0.2mol/l碳酸盐缓冲液(ph10.0)。对其添加5mg量的制造例9中制备的纯化vhh抗体配体2,使得液量达到5ml。在37℃下颠倒混合30分钟后,缓慢加入无水硫酸钠0.497g并使其溶解,进一步在37℃下颠倒混合16小时。在反应后,将珠转移至玻璃过滤器,用上述碳酸盐缓冲液进行清洗。回收此时的滤液并测定吸光度,从而计算出vhh配体的配体密度。接着,为了封闭清洗后的珠中残留的环氧基,加入包含0.1mol/l的nacl及10%v/v硫代甘油的0.2mol/l磷酸缓冲液(ph8.0),在25℃下颠倒混合16小时。在反应后,将珠转移至玻璃过滤器,用纯水及20%乙醇清洗,得到了vhh2固定化珠1(载体3)。
[0300]
与实施例1同样地测定配体密度,将测定结果示于表4。
[0301]
<比较例2:载体4的制作>
[0302]
将珠1替换为再生纤维素膜rc58(膜1),除此以外,通过与比较例1相同的方法进行官能团化及vhh2的固定化,得到了vhh2固定化膜1(载体4)。
[0303]
与实施例1同样地测定配体密度,将测定结果示于表4。
[0304]
<比较例3:载体5的制作>
[0305]
将珠1替换为再生纤维素膜rc55(膜2),除此以外,通过与比较例1相同的方法进行官能团化及vhh2的固定化,得到了vhh2固定化膜2(载体5)。
[0306]
与实施例1同样地测定配体密度,将测定结果示于表4。
[0307]
<比较例4:载体6的制作>
[0308]
将珠1替换为再生纤维素膜rc60(膜3),除此以外,通过与比较例1相同的方法进行
官能团化及vhh2的固定化,得到了vhh2固定化膜3(载体6)。
[0309]
与实施例1同样地测定配体密度,将测定结果示于表4。
[0310]
<实施例3:载体7的制作>
[0311]
将scfv配体1替换为vhh配体2,除此以外,通过与实施例1相同的方法进行间隔物固定化、官能团化、vhh2固定化,得到了vhh2固定化无纺布1(载体7)。
[0312]
与实施例1同样地测定配体密度,将测定结果示于表4。
[0313]
<实施例4:载体8的制作>
[0314]
将无纺布1替换为无纺布2,除此以外,通过与实施例1相同的方法进行间隔物固定化、官能团化、vhh2固定化,得到了vhh2固定化无纺布2(载体8)。
[0315]
与实施例1同样地测定配体密度,将测定结果示于表4。
[0316]
<实施例5:载体9的制作>
[0317]
将无纺布1替换为无纺布3,除此以外,通过与实施例1相同的方法进行间隔物固定化、官能团化、vhh2固定化,得到了vhh2固定化无纺布3(载体9)。
[0318]
与实施例1同样地测定配体密度,将测定结果示于表4。
[0319]
<实施例6:载体10的制作>
[0320]
向无纺布4加入20%乙醇水溶液。对其添加5mg量的制造例9中制备的纯化vhh抗体配体2、以及0.2mg量的4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉氯化物(fujifilm wako pure chemical公司制),使得液量达到5ml,在37℃下颠倒混合4小时。在反应后,将无纺布转移至玻璃过滤器,用纯水清洗,得到了载体10。回收此时的滤液并测定吸光度,从而计算出vhh配体的配体密度。
[0321]
与实施例1同样地测定配体密度,将测定结果示于表4。
[0322]
<实施例7:载体11的制作>
[0323]
将无纺布1替换为无纺布5,除此以外,通过与实施例1相同的方法进行间隔物固定化、官能团化、vhh2固定化,得到了vhh2固定化无纺布5(载体11)。
[0324]
与实施例1同样地测定配体密度,将测定结果示于表4。
[0325]
<实施例8:载体12的制作>
[0326]
将无纺布4替换为无纺布6,除此以外,通过与实施例6相同的方法进行官能团化及vhh2的固定化,得到了vhh2固定化无纺布6(载体12)。
[0327]
与实施例1同样地测定配体密度,将测定结果示于表4。
[0328]
<比较例5:载体13的制作>
[0329]
将vhh配体2替换为vhh配体3,除此以外,通过与比较例1相同的方法进行官能团化及vhh3的固定化,得到了vhh3固定化珠1(载体13)。
[0330]
与实施例1同样地测定配体密度,将测定结果示于表4。
[0331]
<实施例9:载体14的制作>
[0332]
将scfv配体1替换为vhh配体3,除此以外,通过与实施例1相同的方法进行间隔物固定化、官能团化、vhh3固定化,得到了vhh3固定化无纺布1(载体14)。
[0333]
与实施例1同样地测定配体密度,将测定结果示于表4。
[0334]
<实施例10:载体15的制作>
[0335]
将scfv配体1替换为vhh配体4,除此以外,通过与实施例1相同的方法进行间隔物
固定化、官能团化、vhh4固定化,得到了vhh4固定化无纺布1(载体15)。
[0336]
与实施例1同样地测定配体密度,将测定结果示于表4。
[0337]
<制造例10:无纺布载体向柱的填充>
[0338]
将实施例3~10中制备的载体7~12、14、15分别1片1片地重合,以0.2ml-column量填充于tricorn(注册商标)5/20柱。
[0339]
<制造例11:膜载体向柱的填充>
[0340]
将比较例2~4中制备的载体4~6分别1片1片地重合,将0.2ml-column量填充于tricorn(注册商标)5/20柱。
[0341]
<制造例12:珠载体向柱的填充>
[0342]
分别以湿润体积计称量0.2ml-gel的比较例1及5中制备的载体3、13、以及poros(注册商标)captureselect(注册商标)aavx(thermo scientific公司制),填充于tricorn(注册商标)5/20柱(ge healthcare公司制)。
[0343]
<制造例13:基于动物细胞的腺相关病毒(aav2)的产生、以及aav2前处理液的制备>
[0344]
使用aav载体制备试剂盒(“aavpro(注册商标)helper free system”takara bio公司制)制备了表达作为荧光蛋白质gfp的变体的venus(genbank:acq43955.1)的aav2制备用质粒。
[0345]
使用转染试剂(“polyethylenimine max”polysciences公司制、mw:40000)将制备的质粒转染至培养的hek293细胞,使其产生aav。在培养结束后,剥离细胞,回收了细胞培养液。将其离心分离,去除上清,得到了aav产生细胞。
[0346]
将上述得到的aav产生细胞悬浮于包含0.1% triton x-100的dulbecco’s磷酸盐缓冲生理盐水(sigma-aldrich公司制、以下简称为“pbs”),在冰中搅拌20分钟,进行了细胞破碎。向得到的细胞破碎液中添加7.5v/v%的1m氯化镁水溶液和0.1v/v%的250ku/ml kaneka endonuclease(株式会社钟化制),在37℃下静置30分钟,将来自于细胞的核酸分解。在反应后,向反应液添加了15v/v%的0.5m edta溶液,然后,通过深层过滤器(pall公司)进行澄清化。将得到的澄清化液作为aav2前处理液。
[0347]
<制造例14:aav2粗纯化液的制备>
[0348]
参考非专利文献(journal of virological methods 2007 140:183-192),通过阳离子交换色谱对制造例13中得到的aav2前处理液进行了纯化。
[0349]
将poros(注册商标)50hs(thermo公司制)填充于tricorn(注册商标)10/150(ge healthcare公司制),制成了阳离子交换纯化用的柱。
[0350]
制备下述a~f液,在使用前通过0.2μm过滤器。
[0351]
a液:napo4(20mmol/l),nacl(100mmol/l),ph7.4
[0352]
b液:napo4(20mmol/l),nacl(100mmol/l),sarkosyl(5mmol/l),ph7.4
[0353]
c液:napo4(20mmol/l),nacl(370mmol/l),ph7.4
[0354]
d液:nacl(1mol/l)
[0355]
e液:naoh(0.5mol/l)
[0356]
f液:用a液稀释aav2前处理液并调整为ph7.4的溶液
[0357]
将上述柱连接于akta avant 25,用d液清洗,用a液平衡化。然后,通过f液,使aav
保持于柱载体后,按照a液、b液、a液的顺序进行清洗,用c液使aav溶出。在溶出结束后,用d液及e液清洗了柱。
[0358]
对于粗纯化后的aav,通过sds-page确认了纯化程度,用定量pcr(qpcr)对液中的aav量进行了定量。
[0359]
<制造例15:基于动物细胞的腺相关病毒(aav1、5、6)的产生、以及aav1、5、6前处理液的制备>
[0360]
将aav2制备用质粒替换为aav1、5、6制备用质粒,除此以外,通过与制造例13相同的方法制备了aav1、5、6前处理液。
[0361]
<试验例1:基于无纺布载体的aav的回收试验1>
[0362]
对于实施例1及3中制备的载体1及7的3个盘,少量加入制造例14中制备的aav2粗纯化液,颠倒混合16小时,使aav2吸附于载体。
[0363]
去除上清后,加入溶出液(柠檬酸(100mmol/l)、氯化钠(500mmol/l)、ph2.1),使aav2从载体溶出。
[0364]
通过使用了quantstudio3实时pcr系统(thermo fisher scientific公司制)的定量pcr(qpcr)对液中包含的aav量进行定量。换算为单位体积的吸附量,载体1为2.7
×
10
11
vg/ml-column,载体7为1.9
×
10
12
vg/ml-column。
[0365]
<试验例2:基于无纺布载体的aav的回收试验2>
[0366]
将加入的液体从aav2粗纯化液替换为制造例15中制备的aav1前处理液,除此以外,通过与试验例1相同的方法进行了aav相对于实施例2中制备的载体2的回收试验。吸附的aav1的量为1.1
×
10
11
vg/ml-column。
[0367]
<试验例3:使用了无纺布载体的aav2的回收>
[0368]
使用制造例10中制备的载体7及10的柱,通过亲和色谱法对制造例14中得到的aav2粗纯化液进行了纯化。
[0369]
制备下述a~f液,在使用前用0.2μm过滤器进行了过滤。
[0370]
a液:tris-hcl(20mmol/l),氯化钠(500mmol/l),ph8.0
[0371]
b液:柠檬酸(100mmol/l),氯化钠(500mmol/l),ph2.1
[0372]
c液:naoh(10mmol/l)
[0373]
d液:磷酸(120mmol/l),乙酸(167mmol/l),苯甲醇(2.2%v/v)
[0374]
e液:20%乙醇
[0375]
f液:用a液稀释aav2粗纯化液并调整为ph8.0的溶液(1
×
10
12
vg左右)
[0376]
将制造例10中制备的填充有载体7及10的柱连接于akta avant 25,将流速设定为0.2ml/min,用纯水清洗,用a液平衡化。然后,通过f液,使aav保持于柱载体后,用a液清洗,用b液使aav溶出(溶出工序)。在溶出结束后,柱按照a液、c液、a液、d液的顺序进行了清洗(清洗工序)。在清洗后,将柱置换为e液进行冷藏保存。
[0377]
通过使用了quantstudio3实时pcr系统(thermo fisher scientific公司制)的定量pcr(qpcr)对各级分中包含的aav量进行了定量。另外,计算出漏出至流通液(ft)的aav量相对于溶出工序的回收aav量的比率、以及清洗工序的残留aav量相对于溶出工序的回收aav量的比率。将结果示于表5。
[0378]
[表5]
[0379][0380]
<试验例4:使用了膜载体的aav2的回收>
[0381]
使用制造例11中制备的载体4、5、6的柱,通过亲和色谱法对制造例14中得到的aav2粗纯化液进行了纯化。通过与试验例3相同的方法对各级分中包含的aav量进行定量,计算出漏出aav量、残留aav量的比率。将结果示于表5。
[0382]
<试验例5:使用了珠载体的aav2的回收>
[0383]
使用制造例12中制备的poros(注册商标)captureselect(注册商标)aavx、以及载体3的柱,通过亲和色谱法对制造例14中得到的aav2粗纯化液进行了纯化。通过与试验例3相同的方法对各级分中包含的aav量进行定量,计算出漏出aav量、残留aav量的比率。将结果示于表5。
[0384]
根据表5的结果,与poros(注册商标)captureselect(注册商标)aavx及载体3~6相比,载体7及10的残留aav量的比率低。根据这些结果可知,在高效地回收aav方面,作为载体的基材,优选使用水不溶性纤维。
[0385]
<试验例6:使用了无纺布载体的高流速条件下的aav2的回收>
[0386]
使用制造例10中制备的载体7、8、9、11、12的柱,通过亲和色谱法对制造例14中得到的aav2粗纯化液进行了纯化。将色谱的流速从0.2ml/min变更为1.2ml/min(保留时间10秒钟)或2.4ml/min(保留时间5秒钟),除此以外,通过与试验例3相同的方法对各级分中包含的aav量进行定量,计算出漏出aav量、残留aav量的比率。将结果示于表6。
[0387]
[表6]
[0388][0389]
<试验例7:使用了珠载体的高流速条件下的aav2的回收>
[0390]
使用制造例12中制备的poros(注册商标)captureselect(注册商标)aavx的柱,通过亲和色谱法对制造例14中得到的aav2粗纯化液进行了纯化。将色谱的流速从0.2ml/min变更为1.2ml/min(保留时间10秒钟)或2.4ml/min(保留时间5秒钟),除此以外,通过与试验例3相同的方法对各级分中包含的aav量进行定量,计算出漏出aav量、残留aav量的比率。将结果示于表6。
[0391]
根据表6的结果可知,对于无纺布载体7、8、9、11而言,即使将流速提高而使保留时间短于1分钟,漏出至ft的aav的比率也低,且残留aav的比率也低。另一方面,对于载体12而言,虽然残留aav的比率低,但漏出aav的比率增大。需要说明的是,载体11的漏出至ft的aav量为比其它载体更少的数值,但这是由于其为校准曲线的下限范围以外,可以预计其并不比载体7、8、9的结果逊色。
[0392]
<试验例8:基于载体7的aav2纯化>
[0393]
使用制造例10中制备的载体7的柱,通过亲和色谱法对制造例13中得到的aav2前处理液进行了纯化。
[0394]
制备下述a~f液,在使用前用0.2μm过滤器进行了过滤。
[0395]
a液:tris-hcl(20mmol/l),氯化钠(500mmol/l),ph8.0
[0396]
b液:柠檬酸(100mmol/l),氯化钠(500mmol/l),ph2.1
[0397]
c液:naoh(10mmol/l)
[0398]
d液:磷酸(120mmol/l),乙酸(167mmol/l),苯甲醇(2.2%v/v)
[0399]
e液:20%乙醇
[0400]
f液:用a液稀释aav2前处理液并调整为ph8.0的溶液(1
×
10
12
vg左右)
[0401]
将制造例10中制备的填充有载体7的柱连接于akta avant 25,将流速设定为
0.2ml/min,用纯水清洗,用a液平衡化。然后,通过f液,使aav保持于柱载体后,用a液清洗,用b液使aav溶出。在溶出结束后,柱按照a液、c液、a液、d液的顺序进行了清洗。在清洗后,将柱置换为e液进行冷藏保存。将纯化时的色谱图示于图2。
[0402]
另外,通过使用了quantstudio3实时pcr系统(thermo fisher scientific公司制)的定量pcr(qpcr)对各级分中包含的aav量进行了定量。另外,计算出清洗工序的残留aav量相对于溶出工序的回收aav量的比率。将结果示于表7。
[0403]
[表7]
[0404][0405]
<试验例9:基于珠载体的aav2纯化>
[0406]
使用了制造例12中制造的填充有poros(注册商标)captureselect(注册商标)aavx的柱来代替载体7的柱,除此以外,与试验例8同样地通过亲和色谱法对aav前处理液进行了纯化。将得到的色谱图示于图3。与试验例8同样地计算出溶出工序的回收aav及清洗工序的残留aav。将结果示于表7。
[0407]
<试验例10:基于载体14的aav5纯化>
[0408]
使用制造例10中制备的载体14的柱,通过亲和色谱法对制造例15中得到的aav5前处理液进行了纯化。制备下述a~f液,在使用前用0.2μm过滤器进行了过滤。
[0409]
a液:tris-hcl(20mmol/l),氯化钠(100mmol/l),ph8.0
[0410]
b液:柠檬酸(20mmol/l),氯化钠(100mmol/l),ph2.1
[0411]
c液:naoh(10mmol/l)
[0412]
d液:磷酸(120mmol/l),乙酸(167mmol/l),苯甲醇(2.2%v/v)
[0413]
e液:20%乙醇
[0414]
f液:用a液稀释aav5前处理液并调整为ph8.0的溶液(1
×
10
12
vg左右)
[0415]
将制造例10中制备的填充有载体14的柱连接于akta avant 25,将流速设定为0.2ml/min,用纯水清洗,用a液平衡化。然后,通过f液,使aav保持于柱载体后,用a液清洗,用b液使aav溶出。在溶出结束后,柱按照a液、c液、a液、d液的顺序进行了清洗。在清洗后,将柱置换为e液进行冷藏保存。
[0416]
另外,通过使用了quantstudio3实时pcr系统(thermo fisher scientific公司制)的定量pcr(qpcr)对各级分中包含的aav量进行了定量。另外,计算出清洗工序的残留aav量相对于溶出工序的回收aav量的比率。将结果示于表8。
[0417]
[表8]
[0418][0419]
<试验例11:基于载体13的aav5纯化>
[0420]
使用了制造例12中制造的填充有载体13的柱来代替载体14的柱,除此以外,与试验例10同样地通过亲和色谱法对aav5前处理液进行了纯化。与试验例10同样地计算出溶出工序的回收aav及清洗工序的残留aav。将结果示于表8。
[0421]
<试验例12:基于载体15的aav1、6纯化>
[0422]
使用制造例10中制备的载体15的柱,通过亲和色谱法对制造例15中得到的aav1前处理液及aav6前处理液进行了纯化。将f液变更为aav1前处理液或aav6前处理液,除此以外,通过与试验例10相同的方法对aav1、6进行了纯化。对于溶出工序中包含的aav量而言,aav1为2.2
×
10
10
vg/ml-column,aav6为4.4
×
10
10
vg/ml-column。
[0423]
根据试验例8~12的结果可知,本技术的结构物并不限定于aav的血清型,可以广泛应用。
[0424]
根据图1、2及表5~8的结果,在使用了poros(注册商标)captureselect(注册商标)aavx、载体14等珠载体的情况下,由于从溶出工序后的清洗工序中检测到一定量的aav,因此,可以认为是难以将堵塞于载体内部的aav充分回收的材料。
[0425]
另一方面,在使用了作为本发明的结构物的载体的情况下,溶出工序中显示出单一峰,溶出工序后的清洗工序中残留的aav大幅减少,可以认为是能够高效地回收而使aav不堵塞载体内部的材料。
[0426]
以上表明,本技术的结构物是溶出工序的生物粒子回收率高、且清洗工序的生物粒子残留率低的结构物。
[0427]
<试验例13:无纺布载体的aav结合容量的计算>
[0428]
使用制造例10中制备的载体7的柱,通过亲和色谱法对制造例14中得到的aav粗纯化液进行了纯化。
[0429]
制备下述a~f液,在使用前用0.2μm过滤器进行了过滤。
[0430]
a液:tris-hcl(20mmol/l),氯化钠(500mmol/l),ph8.0
[0431]
b液:柠檬酸(100mmol/l),氯化钠(500mmol/l),ph2.1
[0432]
c液:naoh(10mmol/l)
[0433]
d液:磷酸(120mmol/l),乙酸(167mmol/l),苯甲醇(2.2%v/v)
[0434]
e液:20%乙醇
[0435]
f液:用a液稀释aav粗纯化液并调整为ph8.0的溶液(2
×
10
13
vg左右)
[0436]
将柱连接于akta avant 25,将流速设定为0.2ml/min,用纯水清洗,用a液平衡化。然后,通过f液,使aav保持于柱载体后,用a液清洗,用b液使aav溶出。在溶出结束后,柱按照a液、c液、a液、d液的顺序进行了清洗。在清洗后,将柱置换为e液进行冷藏保存。通过qpcr对各级分中包含的aav量进行定量,根据溶出工序中包含的aav量计算出结合容量。将结果示于表9。
[0437]
[表9]
[0438]
[0439]
根据表9的结果可知,从病毒吸附的观点考虑,本发明的结构物显示出不逊于市售品的性能。需要说明的是,本次制备的aav粗纯化液中除了包含病毒基因的aav以外,还大量包含不含基因的衣壳。由于在qpcr测定中仅对包含病毒基因的aav进行定量,因此,可以预计实际的结合容量大于表9所示的数值。
[0440]
作为本发明的方式,可举出例如以下的方式等。
[0441]
<1>一种结构物,其具有水不溶性纤维、和连接于上述水不溶性纤维的低分子化抗体。
[0442]
<2>根据上述<1>所述的结构物,其中,上述水不溶性纤维为无纺布。
[0443]
<3>根据上述<2>所述的结构物,其中,上述无纺布包含聚烯烃及其衍生物。
[0444]
<4>根据上述<1>~<3>中任一项所述的结构物,其中,上述低分子化抗体能够吸附病毒、病毒样颗粒、或病毒载体。
[0445]
<5>根据上述<4>所述的结构物,其中,上述病毒、病毒样颗粒、或病毒载体为腺相关病毒、或腺相关病毒的衣壳、或包含腺相关病毒基因的病毒载体。
[0446]
<6>根据上述<1>~<5>中任一项所述的结构物,其中,上述低分子化抗体不具有ch2结构域及ch3结构域。
[0447]
<7>根据上述<1>~<6>中任一项所述的结构物,其中,上述低分子化抗体包含全长抗体的重链可变区或轻链可变区。
[0448]
<8>根据上述<1>~<7>中任一项所述的结构物,其中,上述低分子化抗体包含单域抗体。
[0449]
<9>根据上述<8>所述的结构物,其中,上述单域抗体包含骆驼科动物来源的重链抗体的可变区。
[0450]
<10>根据上述<1>~<9>中任一项所述的结构物,其中,上述低分子化抗体经由间隔物与上述水不溶性纤维连接在一起。
[0451]
<11>根据上述<10>所述的结构物,其中,上述间隔物包含氨基、羟基、环氧基及羧基中的任一者。
[0452]
<12>根据上述<10>或<11>中任一项所述的结构物,其中,上述间隔物包含聚合物。
[0453]
<13>一种结构物的制造方法,该方法包括:
[0454]
将水不溶性纤维与低分子化抗体进行连接的工序。
[0455]
<14>一种吸附体,其具有上述<1>~<12>中任一项所述的结构物。
[0456]
<15>一种生物粒子的纯化方法,该方法包括:
[0457]
使用上述<1>~<12>中任一项所述的结构物或上述<14>所述的吸附体。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
